

Abstract
Insig-1 and Insig-2 are regulatory proteins that restrict the cholesterol biosynthetic pathway by preventing proteolytic activation of SREBPs and by enhancing degradation of HMG-CoA reductase. Here, we created Insig–double-knockout (Insig-DKO) mice that are homozygous for null mutations in Insig-1 and Insig-2. After 18.5 days of development, 96% of Insig-DKO embryos had defects in midline facial development, ranging from cleft palate (52%) to complete cleft face (44%). Middle and inner ear structures were abnormal, but teeth and skeletons were normal. The animals were lethargic and runted; they died within 1 day of birth. The livers and heads of Insig-DKO embryos overproduced sterols, causing a marked buildup of sterol intermediates. Treatment of pregnant mice with the HMG-CoA reductase inhibitor lovastatin reduced sterol synthesis in Insig-DKO embryos and reduced the pre-cholesterol intermediates. This treatment ameliorated the clefting syndrome so that 54% of Insig-DKO mice had normal faces, and only 7% had cleft faces. We conclude that buildup of pre-cholesterol sterol intermediates interferes with midline fusion of facial structures in mice. These findings have implications for the pathogenesis of the cleft palate component of Smith-Lemli-Opitz syndrome and other human malformation syndromes in which mutations in enzymes catalyzing steps in cholesterol biosynthesis produce a buildup of sterol intermediates.
Introduction
The cholesterol biosynthetic pathway plays an essential role in mammalian development. At least 8 human malformation syndromes are caused by inherited enzyme defects in this pathway (1–4). Five of these human defects have mouse counterparts that arose through random mutagenesis or were generated by targeted recombination. The defective enzymes in these 5 human diseases and the corresponding mouse mutants are listed in Figure 1. All 5 enzymes catalyze steps in the conversion of lanosterol to cholesterol. All of the defects lead to the buildup of sterol intermediates proximal to the defective enzyme.
Figure 1. Cholesterol biosynthetic pathway (A) and its relationship to 5 human diseases that result from deficiencies of enzymes in the conversion of lanosterol to cholesterol (B).

For simplicity, the early steps from acetyl-CoA to HMG-CoA are not shown. The clinical features of the genetic diseases are reviewed in articles by Herman (4), Moebius et al. (47), and Porter (3). CHILD syndrome, congenital hemidysplasia with ichthyosiform erythroderma and limb defects; CDPX2 syndrome, X-linked dominant chondrodysplasia punctata type 2.
Bpa and Str mutant mice, identified from offspring of x-irradiated mice, harbor mutations in the C4 sterol dehydrogenase gene (a component of the C4 sterol demethylation complex), the same gene mutated in human CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) (5). The Td mouse harbors an x-irradiation–induced mutation in sterol Δ8,Δ7-isomerase, the same gene mutated in human CDPX2 syndrome (X-linked dominant chondrodysplasia punctata type 2) (6). Mouse mutants Sc5d–/– (7), Dhcr7–/– (8, 9), and Dhcr24–/– (10) were generated by targeted disruption to delete lathosterol 5-desaturase, sterol Δ7-reductase, and sterol Δ24-reductase, respectively. These defects correspond to human lathosterolosis, Smith-Lemli-Opitz syndrome (SLOS), and desmosterolosis, respectively. Deficiency in each of these 5 enzymes leads to reduced cholesterol synthesis. The cholesterol content of various tissues is reduced, and the levels of intermediate sterols upstream of the defective enzyme are elevated. For example, in human SLOS patients (11–13) and in Dhcr7–/– mice (8, 9), the plasma and tissue content of cholesterol decreases, whereas that of 7-dehydrocholesterol increases. Humans and mice with these deficiencies have multiple developmental anomalies. In 4 of 5 of these syndromes, the anomalies include cleft palate (Figure 1B).
The prototype of the 5 cholesterol synthesis syndromes and the one that has been most extensively studied is SLOS. Human SLOS patients manifest growth retardation, limb defects, and craniofacial dysmorphias as well as mental retardation (13–15). About 50% of SLOS patients (15) and 9% of the corresponding Dhcr7–/– mice have cleft palate (9). Cleft palate also occurs in humans and/or mice with mutations in sterol Δ8,Δ7-isomerase, lathosterol 5-desaturase, and sterol Δ24-reductase (Figure 1B).
The phenotypic abnormalities in the aforementioned diseases are believed to be caused, either singly or in combination, by cholesterol deficiency or the accumulation of intermediate sterols. In patients with SLOS, some investigators have attempted to correct the cholesterol deficiency and to suppress endogenous synthesis of 7-dehydrocholesterol with dietary cholesterol supplementation (16–20). Others have treated SLOS patients with simvastatin, an inhibitor of HMG-CoA reductase that reduces the synthesis of cholesterol and all intermediate sterols, while at the same time increasing expression of DHCR7 alleles encoding mutant proteins with residual enzymatic activity (21, 22). According to published reports, more than 70 patients with SLOS in 10 different studies have been fed a high-cholesterol diet (cited in refs. 16–20), and 4 children in 2 studies have been treated with simvastatin (21, 22). The degree of clinical improvement in terms of developmental progress has ranged from slight to none in the various reports.
Recent studies of cholesterol homeostasis have provided a new way to alter cellular levels of cholesterol and its biosynthetic intermediates. The key is a pair of ER membrane proteins designated Insig-1 and Insig-2, which are essential for end product–mediated feedback inhibition of cholesterol synthesis (23). When the cellular cholesterol level rises, the Insigs bind and retain Scap in the ER (24, 25). Scap is an escort protein for SREBPs, transcription factors required for transcription of all of the known genes encoding enzymes of the cholesterol biosynthetic pathway (26). In order to enter the nucleus, the SREBPs must be transported by Scap to the Golgi apparatus, where they are processed proteolytically to yield active nuclear fragments (27). Under conditions of cholesterol excess, Insigs block this transport and hence reduce cholesterol synthesis. An excess of lanosterol causes Insigs to bind to HMG-CoA reductase, an event that leads to the ubiquitination and degradation of the biosynthetic enzyme (28, 29).
Experiments in gene-targeted mice indicate that Insigs limit cholesterol synthesis even under basal conditions. In livers of mice lacking both Insig-1 and Insig-2, the hepatic content of cholesterol was elevated by 7-fold, even when the animals were consuming a low-cholesterol chow diet (30). This increase was caused by increases in all of the measured mRNAs encoding enzymes of cholesterol synthesis, owing to unrestrained processing of SREBPs. The livers also exhibited a massive increase in the amount of HMG-CoA reductase protein, owing to a failure of its rapid degradation.
In the previous studies, we noted that double-knockout (DKO) mice with germline deficiencies of both Insig-1 and Insig-2 (Insig-1–/–Insig-2–/–; hereafter referred to as Insig-DKO mice) failed to survive beyond the neonatal period (30). As a result, the aforementioned experiments were performed by creating a line of mice that harbored a germline deletion of Insig-2 plus a postnatal loss of Insig-1 in liver mediated by an inducible Cre recombinase (30).
In the current studies, we explore the causes of neonatal lethality in the germline Insig-DKO mice. We found that these mice manifest a highly selective defect in closure of facial structures, leading to cleft palate and in extreme cases to cleft face. The heads of mutant embryos overaccumulated sterol intermediates in the cholesterol biosynthetic pathway, but they had normal amounts of cholesterol. The clefting syndrome was largely alleviated by feeding pregnant females lovastatin, an HMG-CoA reductase inhibitor that lowers the levels of nearly all of the sterol intermediates in the heads of the Insig-DKO embryos. The data suggest that the clefting syndrome is caused by the accumulation of 1 or more sterol intermediates rather than by a deficiency of cholesterol itself.
Results
Mice with germline deletion in Insig-1 (Insig-1–/–) or Insig-2 (Insig-2–/–) were generated as described previously (30). Insig-1–/– or Insig-2–/– mice were first mated to generate Insig-1+/–Insig-2+/– mice, which were then interbred in an attempt to produce Insig-DKO mice. However, no Insig-DKO mice survived beyond the first day after birth. The Insig-DKO pups were born alive, as assessed by their abilities to respond to stimuli, but they failed to vocalize upon stimulation and appeared to be gasping. Shortly thereafter, the Insig-DKO pups became cyanotic and died. Many of the pups had obvious clefting of the face, and some had exencephaly. Histological examination showed that the lungs of the Insig-DKO pups were atelectatic.
To study development in Insig-DKO embryos, we mated Insig-1+/–Insig-2–/– mice with each other and obtained embryos at 18.5 days of development. Insig-1+/+Insig-2–/– or Insig-1+/–Insig-2–/– littermates, which were indistinguishable phenotypically from WT embryos (30), were used as controls. For the studies described in this article, mated females were identified by the presence of a postcopulatory vaginal plug at the beginning of the next light cycle, which is denoted as 0.5 dpc. Unless otherwise stated, embryos were collected by caesarean section at 18.5 dpc, examined for defects, and then genotyped.
Table 1 shows the genotypes and phenotypes of 341 embryos from 50 litters of intercrosses of Insig-1+/–Insig-2–/– mice. At 18.5 dpc, the observed ratio of Insig-1+/+Insig-2–/–, Insig-1+/–Insig-2–/–, and Insig-DKO embryos was 85:178:78, which is consistent with the expected Mendelian ratio of 1:2:1. Compared with control littermates, the Insig-DKO embryos were smaller (Figure 2A) and weighed about 45% less (0.67 g vs. 1.23 g or 1.19 g) (Table 1). The Insig-DKO embryos were lethargic and minimally responsive to stimuli. Whereas all 263 control embryos had grossly normal craniofacial morphology, 96% of the Insig-DKO embryos (75 of 78) had obvious craniofacial defects. The degree of the craniofacial defects ranged from cleft palate with an intact face (hereafter referred to as “cleft palate”) (middle panel of Figure 2B) to cleft palate with mid-facial cleavage defects (hereafter referred to as “cleft face”). The mid-facial clefting phenotype included splitting of the nose and lip with or without exencephaly. Among the 34 cleft face Insig-DKO embryos, 21 had the severe form of mid-facial cleavage with exencephaly (right panel of Figure 2B). Grossly, about 10% (8 of 78) of the Insig-DKO embryos exhibited unilateral or bilateral microphthalmia or anophthalmia. Three of the 78 Insig-DKO embryos exhibited normal craniofacial features without cleavage of the palate or face; however, these embryos were still 45% smaller and moved less than their control littermates.
Table 1 .
Genotype and phenotype of embryos from intercross of Insig-1+/–Insig-2–/– mice

Figure 2. Craniofacial abnormalities of Insig-DKO embryo.

Male and female Insig-1+/–Insig-2–/– mice were mated, and the resultant embryos were collected at 18.5 dpc, photographed, and genotyped. Lateral (A) and palatal views (B) of control (Insig-1+/+Insig-2–/–littermate) and Insig-DKO embryos with cleft palate or cleft face. The lower jaw was resected to reveal the oral cavity. Black arrows indicate cleft palate; white arrows indicate cleft face. pa, palate; tg, tongue. Scale bars: 2.5 mm.
Figure 3 shows histological sections of control and Insig-DKO embryos with craniofacial abnormalities at 18.5 dpc. Normal development of the mouse secondary palate occurs between 11.5 and 15.5 dpc (31). The palatal shelves start as an outgrowth of the maxillary prominences around 11.5 dpc, and they initially grow ventrally along the sides of the tongue. With continued growth, they elevate above the tongue (13.5 dpc), appose (14.5 dpc), and eventually fuse to form a complete palate (15.5 dpc). Whereas control embryos at 18.5 dpc (Figure 3, A and D) had fused palates, the cleft palate (Figure 3, B and E) and cleft face (Figure 3, C and F) Insig-DKO embryos had only rudimentary palatal shelves (arrows) with little medial growth and no fusion. In the cleft face Insig-DKO embryo (Figure 3, C and F), the nasal septum was split, and the brain was displaced rostrally, producing exencephaly. Compared with control embryos (Figure 3, G and J), Insig-DKO embryos also displayed aberrant middle and inner ear morphology (Figure 3, H, I, K, and L). In the cleft palate Insig-DKO embryo (Figure 3, H and K), the malleus was normal and properly articulated with Meckel cartilage. However, in the cleft face Insig-DKO embryos (Figure 3I), the malleus was rudimentary. The stapes was absent in both the cleft palate and cleft face embryos (compare Figure 3, K and L, with control embryo in Figure 3J). As compared with that in the control (Figure 3J), the otic capsule was abnormal in the Insig-DKO embryos (Figure 3, K and L). The pars canalicularis of the otic capsule migrated rostral-ventrally, covering the pars cochlearis (Figure 3, K and L). The stapedial arteries did not develop in either the cleft palate or cleft face Insig-DKO embryos (compare Figure 3, K and L, with Figure 3J). One other abnormality was the absence or underdevelopment of the trigeminal, facial, and vestibulocochlear ganglia.
Figure 3. Histology of control (A, D, G, andJ) andInsig-DKO embryos with cleft palate (B, E, H, andK) or cleft face (C, F, I, and L) at 18.

dpc. (A–F) Palate phenotype. The secondary palate was intact in control (A and D) but absent in Insig-DKO embryos (B, C, E, and F). The lateral margins of the palatal shelves are indicated by arrows. In Insig-DKO embryos with cleft face (C), the nasal septum was split, and the brain was displaced rostrally. (G–L) Middle and inner ear phenotype. Meckel cartilage (mc) and malleus (m) were normal in the cleft palate Insig-DKO embryo (H) but rudimentary in the cleft face embryo (I). Stapes (s) and the stapedial artery (sa) were absent or vestigial in Insig-DKO embryos (K and L). Compared with that of control embryos (J), the pars canalicularis (pca) of the otic capsule in the Insig-DKO embryos migrated rostral-ventrally (K and L). pa, palate; pco, pars cochlearis; tg, tongue. Scale bars: 0.5 mm.
The following structures were identified but were variably displaced from their normal anatomical location: thymus, thyroid, hippocampus, cerebral cortex, hypothalamus, and pituitary. Other major organs of the body, including the heart, lung, kidney, intestines, pancreas, testis, and adrenal medulla and cortex, had normal morphologies and locations. The teeth and skeleton (axial and appendicular) were also grossly and histologically normal.
As discussed above, development of the mouse secondary palate is a complex process involving the outgrowth of the maxillary prominences and the elevation, apposition, and fusion of the palatal shelves. Defects in any one of these steps can lead to cleft palate. To further examine the defects of palatogenesis in Insig-DKO embryos, we analyzed palate formation from 12.5 to 15.5 dpc in control and Insig-DKO embryos that did not have cleft face. As shown in Figure 4, early palatogenesis (12.5 to 13.5 dpc) occurred normally in Insig-DKO embryos, i.e. the palatal shelves grew and descended vertically (Figure 4, A–D). Starting at 14.5 dpc, the palatal shelves elevated, apposed, and began to fuse in the control embryo (Figure 4E). By 15.5 dpc, the palatal shelves of the control embryo (Figure 4G) were completely fused, with loss of the epithelial seam. In the Insig-DKO embryo, the palatal shelves partially elevated (Figure 4F), but they failed to appose and fuse (Figure 4, F and H).
Figure 4. Developmental study of palate formation in control andInsig -DKO embryos.

Insig-1+/–Insig-2–/– mice were mated, and the resultant embryos were collected at 12.5, 13.5, 14.5, and 15.5 dpc. The Insig-DKO embryos did not have cleft face. The embryos were fixed, sectioned, and stained with H&E. pa, palate; ps, palatal shelves; tg, tongue. Scale bars: 0.5 mm.
To assess the overall skeletal development and specifically that of the cranium, control and cleft face Insig-DKO embryos at 18.5 dpc were stained with Alizarin red and Alcian blue for examination of bone and cartilage structures, respectively. As shown in Figure 5, the skull of the Insig-DKO embryo was smaller than that of the control. In the dorsal view, the cleft face Insig-DKO embryo (Figure 5B) exhibited a median facial defect including a complete splitting of the frontal and nasal bones along the metopic suture. In the ventral view of the Insig-DKO embryo (Figure 5D), the palatal bone was absent and the basisphenoid and hyoid were poorly developed. In addition, Meckel cartilage was discontinuous, and the tympanic ring was absent or rudimentary. No other bone and cartilage abnormalities were noted in the axial or appendicular skeletons of the Insig-DKO embryos.
Figure 5. Alizarin red– and Alcian blue–stained skull preparations of control embryo andInsig -DKO embryo with cleft face.
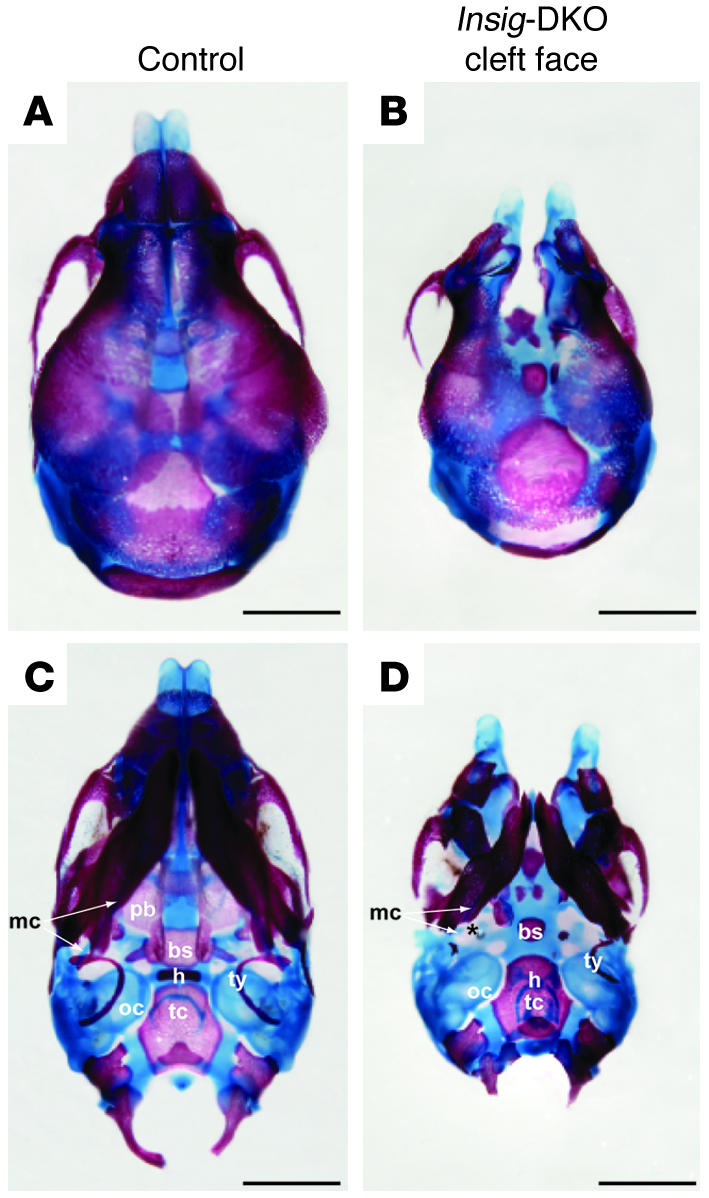
(A and B) Dorsal view. (C and D) Ventral view. Compared with that in the control embryo (A and C), the median facial cleft in Insig-DKO embryo was clearly visible (B and D). The palatal bone (pb), basisphenoid (bs), hyoid (h), Meckel cartilage (mc), and tympanic ring (ty) all showed various degrees of abnormality in the Insig-DKO embryo (C and D). oc, otic capsule; tc, thyroid cartilage. The asterisk indicates a breakpoint in Meckel cartilage. Scale bars: 1 mm.
Inasmuch as Insig proteins play crucial roles in feedback inhibition of cholesterol synthesis, we next explored the possibility that the craniofacial abnormalities in the Insig-DKO embryos are caused by sterol overproduction. To study sterol synthesis in the embryos at 18.5 dpc, we injected pregnant females intravenously with 3H-labeled water, a tracer whose incorporation into sterols reflects the relative rate of cholesterol synthesis (32). After 1 hour, the embryos were removed by caesarean section, tissue was taken for genotyping, and the livers and heads were dissected separately and hydrolyzed in ethanolic KOH. The rest of the body was also hydrolyzed. Total sterols were precipitated with digitonin, and the radioactivity was measured. One group of pregnant females was maintained on normal chow. Beginning on 5.5 dpc, another group was fed normal chow supplemented with 0.2% lovastatin, an inhibitor of HMG-CoA reductase (33). When the mothers were fed normal chow, the livers of the Insig-DKO embryos exhibited a 9-fold increase in sterol synthesis as compared with the control littermates (Figure 6A). The lovastatin diet had no effect on sterol synthesis in livers of the control embryos, but it did reduce the elevated sterol synthesis in livers of the Insig-DKO embryos. Sterol synthesis was also elevated in the heads of the Insig-DKO embryos, although not as dramatically as in the liver. Synthesis in the head was reduced by the lovastatin diet (Figure 6B). The remainder of the carcass (“Other tissues” in Figure 6C) also showed an increased synthesis of [3H]sterols in the Insig-DKO embryos, and again this was reduced by lovastatin.
Figure 6. In vivo synthesis rates of sterols in tissues ofInsig -DKO embryos from pregnant females fed a chow diet with or without lovastatin.

Male and female Insig-1+/–Insig-2–/– mice were mated, and mated females were identified and fed ad libitum a regular chow diet or a chow diet containing 0.2% (wt/wt) lovastatin from 5.5 to 18.5 dpc. At 18.5 dpc, pregnant female mice were injected intraperitoneally with 3H-labeled water (50 mCi of in 0.25 ml isotonic saline). One hour later, the embryos were collected by caesarean section for measurement of sterol synthesis in liver (A), head (B), and other tissues (i.e., the whole body minus the liver and head; C). Sterol synthesis was calculated as μmoles of 3H-labeled water incorporated into digitonin-precipitable sterols per hour per gram of tissue. Each bar represents mean ± SEM of values from 16 to 18 embryos.
To determine whether lovastatin reduced the tissue content of individual sterols, we sacrificed a group of pregnant females on 18.5 dpc and measured the sterols in embryonic livers and heads using coupled gas chromatography–mass spectroscopy (GC-MS). Half of the pregnant females were fed 0.2% lovastatin beginning on 5.5 dpc (Table 2). When the mothers were fed a chow diet, the Insig-DKO embryonic livers had a 6.2-fold increase in cholesterol content as compared with their control littermates (compare columns iii and i in Table 2). This increase was largely, but not completely, abolished when the mothers were fed lovastatin (column iv). In the Insig-DKO embryonic livers, the measured content of sterol intermediates in the cholesterol biosynthetic pathway was elevated even more than that of cholesterol. The fold elevations ranged from 7.8 for lathosterol to 105 for desmosterol (compare columns iii and i). Lovastatin decreased the levels of sterol intermediates in the livers of the control as well as the Insig-DKO embryos. The relative reduction in the Insig-DKO livers was greater than that in the control livers.
Table 2 .
Sterol content of livers and heads of Insig-DKO embryos (18.5 dpc) from pregnant females fed a diet with or without lovastatin

Surprisingly, the heads of the Insig-DKO embryos did not have an elevation in cholesterol level, but they did show clear elevations in the levels of intermediates (Table 2; compare columns vii and v). The range was 1.4-fold for 7-dehydrocholesterol to 4-fold for lanosterol. The total of all the measured sterol intermediates increased by 3.1-fold. In the heads of the control embryos, lovastatin treatment lowered the levels of all of the intermediates significantly (compare columns vi and v). An even greater relative reduction was observed in the heads of the Insig-DKO embryos, with the exception of 7-dehydrocholesterol, the level of which rose slightly after lovastatin treatment (compare columns viii and vii). Similar results were observed in 2 other experiments.
The measurements in Table 2 were made at 18.5 dpc, which is after palatal fusion has occurred in WT embryos. To confirm that sterol intermediates accumulate in Insig-DKO embryos prior to palatal fusion and to localize the accumulation to palatal structures, we dissected the palate and maxillas from 13.5-dpc control and Insig-DKO embryos (31). We also excised the livers of these embryos. As expected, even at 13.5 dpc, the livers of the Insig-DKO embryos had elevated levels of sterol intermediates (Table 3). The fold increases ranged from 1.2 for 24-dihydrolanosterol to 22 for desmosterol. In the dissected palate/maxilla (which includes the palate, maxilla, and eyes, but not the brain), the sterol intermediate levels were elevated from 1.3-fold for 7-dehydrocholesterol to 20-fold for lanosterol. The total of all measured sterol intermediates increased by 5.5- and 7.3-fold in the 13.5-dpc Insig-DKO embryonic liver and palate/maxilla, respectively. The cholesterol content of liver and palate/maxilla was not dramatically different between the control and Insig-DKO embryos at 13.5 dpc (Table 3).
Table 3 .
Sterol content of liver and palate/maxilla tissues of control and Insig-DKO embryos (13.5 dpc)

Figure 7A shows the distribution of craniofacial abnormalities in Insig-DKO embryos derived from pregnant females fed a chow diet or a chow diet containing 0.2% lovastatin. Embryos from 50 litters in both dietary groups were analyzed at 18.5 dpc. The 50 litters in the control chow-fed group contained a total of 341 embryos, of which 78 were Insig-DKO embryos (see Table 1 for details). Among these 78 Insig-DKO embryos, 75 (96.2%) had craniofacial defects, and only 3 (3.8%) had normal craniofacial morphology. The 50 litters in the 0.2% lovastatin-fed group contained a total of 361 embryos, of which 76 were Insig-DKO embryos. Among these 76 Insig-DKO embryos, 41 (53.9%) had normal craniofacial morphology. Only 5 of 76 (6.6%) from the lovastatin-fed group had the severe cleft face defect, whereas 34 of 78 (43.6%) from the chow-fed group had cleft face. The percentage of Insig-DKO embryos with cleft palate was reduced slightly by lovastatin. Taken together, the data suggest that lovastatin feeding reduced the severity of the craniofacial defects of Insig-DKO embryos. It is important to point out that the Insig-DKO embryos (with or without craniofacial defects) from the lovastatin-fed group were still 36% smaller than their littermate controls (0.69 g vs. 1.08 g). Despite their normal facial structures, no Insig-DKO mice from lovastatin-fed mothers survived beyond the first day after birth. Thus, although feeding lovastatin to pregnant females reduced the craniofacial defects in Insig-DKO embryos, it did not prevent growth retardation or neonatal lethality even when the facial structures were normal.
Figure 7. Prevention of craniofacial defects inInsig -DKO embryos after feeding of lovastatin to pregnant females.

Insig-1+/–Insig-2–/– mice were mated with each other, and mated females were identified by the presence of a postcopulatory vaginal plug. Mated females were fed ad libitum a regular chow diet (red bars) or a chow diet containing 0.2% (wt/wt) lovastatin (blue bars) from 5.5 to 18.5 dpc. (A) Embryos from 50 litters in both dietary groups were collected by caesarean section at 18.5 dpc and analyzed for craniofacial morphology by 2 independent observers prior to genotyping by PCR. The 50 litters in the control chow diet group contained a total of 341 embryos, of which 78 had the Insig-DKO genotype (same data as in Table 1). The 50 litters in the lovastatin-fed group contained a total of 361 embryos, of which 76 had the DKO genotype. The embryos from both groups were harvested in 5 different experiments over a 6-month period in which 5–19 pregnant females in both dietary groups were studied concurrently. The mean ± SEM for body weights of lovastatin-treated DKO embryos was 0.69 ± 0.01 g as compared with 1.08 ± 0.01 g for lovastatin-treated control embryos. (B–E) Three Insig-DKO embryos from lovastatin-fed females represented in A were processed for histological analysis. One embryo (B and D) showed a cleft palate phenotype (not corrected) with middle and inner ear abnormalities, and 2 embryos showed normal craniofacial phenotype (corrected; 1 of which is shown in C and E). Arrows indicate the lateral margins of the palatal shelves. pa, palate; pca, pars canalicularis; pco, pars cochlearis; s, stapes; sa, stapedial artery. Scale bars: 0.5 mm.
To determine whether lovastatin feeding corrected the middle and inner ear defects, we examined histologically 3 Insig-DKO embryos from the lovastatin-fed group in Figure 7A. Two had normal facial structures. We designate these as “corrected.” One had a “not-corrected” cleft palate. Shown in Figure 7, B–E, is a histological analysis of 1 of the 2 corrected embryos (Figure 7, C and E). For comparison, the not-corrected Insig-DKO embryo with cleft palate from a lovastatin-fed female is also shown (Figure 7, B and D). Compared with the not-corrected Insig-DKO embryo (Figure 7B), the corrected Insig-DKO embryo had an intact palate (Figure 7C). Moreover, the corrected Insig-DKO embryo showed normal middle and inner ear structures (compare Figure 7, E and D). The stapes and the stapedial artery formed in their correct anatomical positions, and the otic capsule, consisting of the pars canalicularis and the pars cochlearis, had normal structure and morphology.
Lovastatin feeding in utero also corrected the ocular abnormalities observed in the Insig-DKO embryos. In the chow-fed group, 8 of 78-DKO embryos showed microphthalmia or anophthalmia, whereas only 1 of the 76 Insig-DKO embryos from the lovastatin-fed group had any gross eye defects.
Discussion
The current experiments reveal that accumulation of intermediates in the cholesterol biosynthetic pathway during embryonic life interferes with normal fusion of midline facial structures, producing cleft palate, and even cleft face, accompanied by bony abnormalities of the middle and inner ear. Accumulation of sterol intermediates was achieved by germline elimination of the genes encoding Insig-1 and Insig-2, redundant proteins that are necessary for end-product feedback repression of the cholesterol biosynthetic pathway (30). The teratogenic role of sterol intermediates was supported by the observation that clefting decreased when the pregnant mothers were treated with lovastatin, an inhibitor of sterol synthesis that lowers the levels of the sterol intermediates. Unlike in previous studies of cleft palate in animals with blocks in the cholesterol biosynthetic pathway (Figure 1B), the accumulation of sterol intermediates in the Insig-DKO mice was not accompanied by cholesterol deficiency. Indeed, at both 13.5 and 18.5 dpc, the tissue cholesterol content was either normal or elevated in the Insig-DKO embryos (Tables 2 and 3). If the current model applies to clefting defects arising from enzymatic blocks in cholesterol biosynthesis, the evidence indicates that the clefting defects are caused by accumulation of sterol intermediates prior to the block rather than to deficiencies of products following the block.
The defective craniofacial structures in the Insig-DKO embryos are derived from cranial neural crest cells (34). It is noteworthy that several other structures derived from cranial neural crest cells, i.e., the teeth and certain bones (maxilla and mandible), were grossly normal. No gross defects were seen in structures derived from trunk neural crest cells, such as the adrenal medulla and dorsal root ganglia. These findings suggest that cranial neural crest cells may be more sensitive than trunk neural crest cells to excess sterol intermediates caused by Insig deficiency.
The effect of lovastatin treatment on facial clefting in the Insig-DKO embryos was clear-cut but incomplete. In a total of 76-DKO embryos exposed in utero to lovastatin (Figure 6), the number of mice with normal faces rose from 3.8% without treatment to 54% with treatment, and there was a corresponding 85% reduction in the number with cleft face. However, 40% of the statin-treated embryos continued to exhibit cleft palate, a proportion that was only slightly lower than the 53% in the untreated group. We explain these data by hypothesizing that statins decreased the severity of the clefting defect in nearly all of the Insig-DKO embryos. Those that were destined to have cleft face now had cleft palate, whereas those that were destined to have cleft palate now had normal midline development. Why some Insig-DKO embryos have more severe clefting defects than others is not known, but such variability is the rule in genetic developmental defects.
Although statins had a dramatic effect on the clefting syndrome, they did not ameliorate the other abnormalities in the Insig-DKO mice. Even though their palates were normal at birth, these animals remained runted, moved sluggishly, and did not feed. All of them died within 24 hours after birth. The failure of these characteristics to improve with statins may be attributable to the fact that lovastatin did not reduce the levels of these intermediates sufficiently to avoid these consequences (Table 2). Alternatively, it is possible that the noncorrected abnormalities are not caused by the accumulation of sterol intermediates per se, but reflect an unidentified action of Insig that is disrupted in these mice.
In mouse embryos, the palate develops between 11.5 and 15.5 dpc (31). This complex process involves the outgrowth of the maxillary prominences and the elevation, apposition, and fusion of the palatal shelves. Defects in any one of these steps can lead to cleft palate. The cleft palate in the Insig-DKO embryos appears to be caused by the failure of the palatal shelves to grow toward each other and to fuse (Figure 3F). Growth and fusion of the palate can be studied in vitro using palatal shelves dissected from 13.5-dpc mouse embryos (35). In the future, the use of this organ culture system may permit identification of the intermediate sterol or combination of sterols that interferes with palatal fusion.
The molecular mechanism by which a buildup of sterol intermediates causes facial clefting is unknown, in part because the complex pathway of facial development is incompletely understood and in part because cleft palate is a component of many developmental syndromes, genetic as well as environmental (36, 37). Despite this uncertainty, it is tempting to postulate that sterol intermediates alter the pathway regulated by Sonic hedgehog (Shh), a morphogenic protein that possesses a covalently attached cholesterol moiety (38). Shh plays a major role in development of the face and brain (39). Shh binds to its receptor, Patched, on cell surfaces, thereby relieving inhibition of Smoothened, a heptahelical protein that resembles G protein–coupled receptors. Patched contains a sterol-sensing domain similar to the domains in Scap and HMG-CoA reductase that bind to Insigs (40), but Patched has not been shown to interact with Insigs.
The phenotype of Shh deficiency differs from that seen in the Insig-DKO mice. Insig deficiency produces midline fission, leading to cleft palate and cleft face. Shh deficiency produces midline fusion, leading to cyclopia and holoprosencephaly (41). The differences between these effects raise the possibility that the sterol buildup in Insig-DKO mice leads not to Shh deficiency but rather to Shh overactivity, which would produce excessive activity of Smoothened. Overactivity of Smoothened might result if the buildup of a sterol interfered with the inhibitory action of Patched or if it directly activated Smoothened. In view of the presence of a sterol-sensing domain in Patched (and the related protein Dispatched), we cannot rule out the possibility that one of these proteins is regulated by Insigs and that Insig deficiency directly alters its function. Arguing against this notion is the observation that the clefting syndrome in Insig-DKO mice is ameliorated when levels of sterol biosynthetic intermediates are reduced by lovastatin treatment. This finding indicates that the clefting syndrome is caused indirectly by sterol excess rather than directly by Insig deficiency.
As a first step in analyzing the Shh signaling pathway in the Insig-DKO embryos, we isolated total RNA from the palate/maxilla tissues of 13.5-dpc control and Insig-DKO embryos. Expression levels of Shh, Smoothened, Patched-1, and Gli-1 were measured by quantitative real-time PCR. The latter 2 genes are positively regulated by the Shh pathway, and their expression of mRNAs would be expected to be increased if Smoothened were overactive (42). We observed no differences in mRNA levels for any of these genes between the control and Insig-DKO embryos in 2 independent experiments (data not shown). Inasmuch as real-time PCR can only quantify the total amount of mRNA but cannot reveal changes in spatial and temporal expression patterns, we plan to conduct more detailed analyses using in situ hybridization to determine whether the Shh signaling pathway is perturbed in the Insig-DKO embryos.
From the standpoint of cholesterol homeostasis, a fascinating aspect of the current study emerged in the experiment in which sterol synthesis was measured in embryonic liver after injection of the mothers with 3H-labeled water (Figure 6A). In this experiment, lovastatin failed to inhibit cholesterol synthesis in livers of the WT embryos, whereas the drug caused a 70% reduction in the Insig-DKO mice. An explanation is suggested by previous studies of the response to HMG-CoA reductase inhibitors in cultured cells (43) and in mouse liver (33). When normal cells are exposed to a reductase inhibitor, the level of cholesterol falls initially, and this decrease releases the partial inhibition of SREBP processing that is normally mediated by Insigs in the steady state (23). The increased nuclear SREBP activates the gene for reductase. In addition, reductase protein becomes more stable because there is less lanosterol, which is required for rapid degradation. As a result of these homeostatic responses, the amount of reductase protein increases. In the new steady state, the rate of cholesterol synthesis is restored nearly to normal, and this compensation is sustained as long as cellular sterol levels remain sufficiently low to maintain the accumulation of reductase. Both of these homeostatic responses are dependent on Insigs (30). In the Insig-deficient embryos, the steady-state inhibitory effects of sterols are lost. As a result, in the untreated embryos, the amount of HMG-CoA reductase protein is maximal. When lovastatin is administered, there is no further increase in the amount of reductase protein, so the full effect of the inhibitor is seen.
We can illustrate this point with arbitrary numbers. Let us suppose that normal cells in the steady state have 10 units of reductase activity. A certain dose of lovastatin inhibits the enzyme by 70%. Through the homeostatic mechanism described above, the amount of reductase protein increases 3-fold. There are now 30 units of reductase. Lovastatin inhibits this activity by 70%, so the net activity is 9 units, which is nearly the same as the initial activity. As a result, the incorporation of 3H-labeled water into sterols is not reduced by lovastatin. The Insig-deficient embryo begins with a maximum activity (let’s say 100 units). Lovastatin inhibits it by 70%. Since there can be no further increase in the amount of reductase, the full inhibition is seen, and the overall activity falls to 30 units instead of 100 units. As a result, the synthesis of sterols from 3H-labeled water in liver falls by 70%, as seen in Figure 6A.
The current results give further emphasis to the emerging concept that Insig proteins are central to sterol homeostasis, and they indicate that this role extends to embryonic development as well as to adult physiology.
Methods
Generation of Insig-deficient mice.
Mice with germline deletion in Insig-1 (Insig-1–/–) or Insig-2 (Insig-2–/–) were generated as described previously (30). Insig-1–/– mice and Insig-2–/– mice were first mated to each other to generate Insig-1+/–Insig-2+/– mice, which were then interbred to produce Insig-1–/–Insig-2–/– mice. Inasmuch as no Insig-1–/–Insig-2–/– offspring survived beyond the first day after birth, we mated Insig-1+/–Insig-2–/– mice with each other to produce Insig-DKO embryos. Insig-1+/–Insig-2–/– or Insig-1+/+Insig-2–/– littermates, which were indistinguishable from WT embryos, were used as controls. Mated females were identified by the presence of a postcopulatory vaginal plug at the beginning of the next light cycle, which is denoted as 0.5 dpc. Unless otherwise stated, embryos were collected by caesarean section at 18.5 dpc. Genomic DNA was prepared from embryonic tissues and used for genotyping by PCR as previously described (30).
All mice were housed in colony cages with a 12-hour light/12-hour dark cycle and fed Teklad Mouse/Rat Diet no. 7002 (Harlan Teklad Global Laboratory Diets; Harlan Teklad). Unless otherwise stated, adult mice were fed a chow diet ad libitum. All animal experiments were performed with the approval of the Institutional Animal Care and Research Advisory Committee at University of Texas Southwestern Medical Center at Dallas.
Feeding lovastatin to pregnant female mice.
To examine the effects of statins on the developmental defects of Insig-DKO embryos, pregnant female mice were fed ad libitum from 5.5 to 18.5 dpc with a chow diet containing 0.2% (wt/wt) lovastatin (obtained from Merck). The diet was prepared by grinding the drug in a small amount of Teklad Mouse/Rat Diet no. 7001 (powder form) with a mortar and pestle and then mixing this fine powder with the appropriate amount of Diet no. 7001 to obtain a final lovastatin concentration of 0.2%.
Determination of tissue sterol composition.
Sterol levels in mouse tissues were measured by GC-MS as described previously (44). Livers or heads from 3 embryos were pooled, weighed, and placed into the sample tube. An aliquot of ethanol containing the internal standards 5α-cholestane (50 μg) and epicoprostanol (2.5 μg) was added to each sample tube, and tissues were hydrolyzed by heating (100°C) in ethanolic KOH (100 mM) for 2 hours. Lipids were extracted in petroleum ether (final volume, 6 ml), 1/30 of which (0.2 ml) was dried under nitrogen, and derivatized with hexamethyldisilazane-trimethylchlorosilane (HMDS-TMCS). GC-MS analysis was performed using an Agilent 6890N gas chromatograph coupled to an Agilent 5973 mass selective detector (Agilent Technologies). The trimethylsilyl-derived sterols were separated on an HP-5MS (5%-phenyl)-methylpolysiloxane capillary column (30 m × 0.25 mm internal diameter × 0.25 μm film) (Agilent Technologies) with carrier gas helium at the rate of 1 ml/min. The temperature program was 150°C for 2 minutes, followed by increases of 20°C/min up to 280°C, which was then held for 13 minutes. For cholesterol, the injector was operated in a 1:10 split mode. For all other sterols, the injector was operated in splitless mode at 280°C. The mass spectrometer was operated in selective ion monitoring mode. The mass to charge ratios for the extracted ions were: 393.4 (lanosterol), 395.0 (24-dihydrolanosterol), 456.4 (zymosterol), 458.4 (lathosterol), 350.4 (7-dehydrocholesterol), 343.3 (desmosterol), and 458.4 (cholesterol).
Standard curves were generated by MS analysis of various amounts of each sterol in the following ranges: lanosterol, 0.5 to 800 ng; 24-dihydrolanosterol, 1 to 800 ng; zymosterol, 2.5 to 2,000 ng; lathosterol, 2.5 to 1,500 ng; 7-dehydrocholesterol, 1 to 2,000 ng; desmosterol, 2 to 10,000 ng; and cholesterol, 0.1 to 100 μg.
Histology and skeletal analysis.
For histological analysis, 18.5-dpc embryos were skinned, fixed in 10% neutral buffered formalin (catalog HT50-1-320; Sigma-Aldrich), embedded in paraffin, and sectioned at 5 μm. Paraffin-embedded sections were stained with H&E. The 12.5- to 15.5-dpc embryos represented in Figure 4 were not skinned prior to fixation. For skeletal analysis, 18.5-dpc embryos were collected, prepared, and stained with Alizarin red and Alcian blue for examination of bone and cartilage structure, respectively. Briefly, embryos were partially skinned (the skins were used to prepare genomic DNA for genotyping by PCR) and fixed in 100% ethanol overnight. The following day, embryos were completely skinned and fixed again in 100% ethanol overnight. The cartilage was stained for 40 hours with an Alcian blue/ethanol solution containing 0.015% (wt/vol) Alcian Blue 8GX (catalog A5268-10G; Sigma-Aldrich) and 20% (vol/vol) glacial acetic acid. The stained embryos were washed in 100% ethanol for 3 hours and transferred to a 2% (wt/vol) KOH solution for 24 hours. The bone was then stained for 4–5 hours with an Alizarin red solution containing 0.005% (wt/vol) Alizarin red (catalog A5533-25G; Sigma-Aldrich) and 1% KOH. The stained skeletons were cleared for 2 days in a solution containing 1% KOH and 20% glycerol that was replaced daily with fresh solution. Thereafter, the stained skeletons were stored in a solution containing 50% glycerol and 50% ethanol.
Sterol synthesis rates in vivo.
Rates of sterol synthesis were measured in 18.5-dpc embryos using 3H-labeled water as previously described, with slight modification (45, 46). Mated female mice were injected intraperitoneally with 3H-labeled water (50 mCi in 0.25 ml isotonic saline), and 1 hour later embryos were collected by caesarean section. The maternal blood and embryonic head, liver, and all remaining tissues (referred to as “other tissues”) were used for measurement of sterol synthesis, which was calculated as micromoles of 3H-labeled water incorporated into digitonin-precipitable sterols per hour per gram of tissue.
Acknowledgments
We thank the following colleagues for invaluable assistance in these studies: Fang Xu and Jonathan Cohen (GC-MS measurements); Jay Horton (3H-labeled water experiments); Brian Shirley (histological sections); John Shelton (photography); and Monica Mendoza and Isis Soto (animal studies). We also thank Luis Parada for critical review of the manuscript. This research was supported by grants from the NIH (HL-20948), the Perot Family Foundation, and the Moss Heart Foundation. Luke J. Engelking and Bret M. Evers are supported by Medical Scientist Training Grant GM08014.
Footnotes
Nonstandard abbreviations used: DKO, double-knockout; GC-MS, gas chromatography–mass spectroscopy; Shh, Sonic hedgehog; SLOS, Smith-Lemli-Opitz syndrome.
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J. Clin. Invest. 116:2356–2365 (2006). doi:10.1172/JCI28988.
Luke J. Engelking and Bret M. Evers contributed equally to this work.
See the related Commentary beginning on page 2322.
References
- 1.Kelley R.I., Herman G.E. Inborn errors of sterol biosynthesis. Annu. Rev. Genomics Hum. Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- 2.Nwokoro N.A., Wassif C.A., Porter F.D. Genetic disorders of cholesterol biosynthesis in mice and humans. Mol. Genet. Metab. 2001;74:105–119. doi: 10.1006/mgme.2001.3226. [DOI] [PubMed] [Google Scholar]
- 3.Porter F.D. Human malformation syndromes due to inborn errors of cholesterol synthesis. . Curr. Opin. Pediatr. 2003;15:607–613. doi: 10.1097/00008480-200312000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Herman G.E. Disorders of cholesterol biosynthesis: prototypic metabolic malformation syndromes. . Hum. Mol. Genet. 2003;12:R75–R88. doi: 10.1093/hmg/ddg072. [DOI] [PubMed] [Google Scholar]
- 5.Liu X.Y., et al. The gene mutated in bare patches and striated mice encodes a novel 3β-hydroxysteroid dehydrogenase. Nat. Genet. 1999;22:182–187. doi: 10.1038/9700. [DOI] [PubMed] [Google Scholar]
- 6.Derry J.M.J., et al. Mutations in a delta8 -delta7 sterol isomerase in the tattered mouse and X-linked dominant chondrodysplasia punctata. . Nat. Genet. 1999;22:286–290. doi: 10.1038/10350. [DOI] [PubMed] [Google Scholar]
- 7.Krakowiak P.A., et al. Lathosterolosis: an inborn error of human and murine cholesterol synthesis due to lathosterol 5-desaturase deficiency. Hum. Mol. Genet. 2003;12:1631–1641. doi: 10.1093/hmg/ddg172. [DOI] [PubMed] [Google Scholar]
- 8.Fitzky B.U., et al. 7-Dehydrocholesterol-dependent proteolysis of HMG-CoA reductase suppresses sterol biosynthesis in a mouse model of Smith-Lemli-Opitz/RSH syndrome. J. Clin. Invest. 2001;108:905–915. doi: 10.1172/JCI200112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wassif C.A., et al. Biochemical, phenotypic and neurophysiological characterization of a genetic mouse model of RSH/Smith-Lemli-Opitz syndrome. Hum. Mol. Genet. 2001;10:555–564. doi: 10.1093/hmg/10.6.555. [DOI] [PubMed] [Google Scholar]
- 10.Wechsler A., et al. Generation of viable cholesterol-free mice. Science. 2003;302:2087. doi: 10.1126/science.1090776. [DOI] [PubMed] [Google Scholar]
- 11.Irons M., Elias E.R., Salen G., Tint G.S., Batta A.K. Defective cholesterol biosynthesis in Smith-Lemli-Opitz syndrome. Lancet. 1993;341:1414. doi: 10.1016/0140-6736(93)90983-n. [DOI] [PubMed] [Google Scholar]
- 12.Tint G.S., et al. Markedly increased tissue concentrations of 7-dehydrocholesterol combined with low levels of cholesterol are characteristic of the Smith-Lemli-Opitz syndrome. J. Lipid Res. 1995;36:89–95. [PubMed] [Google Scholar]
- 13.Porter F.D. RSH/Smith-Lemli-Opitz syndrome: a multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol. Genet. Metab. 2000;71:163–174. doi: 10.1006/mgme.2000.3069. [DOI] [PubMed] [Google Scholar]
- 14.Opitz J.M. RSH (so-called Smith-Lemli-Opitz) syndrome. Curr. Opin. Pediatr. 1999;11:353–362. doi: 10.1097/00008480-199908000-00015. [DOI] [PubMed] [Google Scholar]
- 15.Kelley R.I., Hennekam R.C.M. The Smith-Lemli-Opitz syndrome. J. Med. Genet. 2000;37:321–335. doi: 10.1136/jmg.37.5.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elias E.R., Irons M.B., Hurley A.D., Tint G.S., Salen G. Clinical effects of cholesterol supplementation in six patients with the Smith-Lemli-Opitz syndrome (SLOS). Am. J. Med. Genet. 1997;68:305–310. doi: 10.1002/(sici)1096-8628(19970131)68:3<305::aid-ajmg11>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 17.Irons M., et al. Treatment of Smith-Lemli-Opitz syndrome: results of a multicenter trial. Am. J. Med. Genet. 1997;68:311–314. [PubMed] [Google Scholar]
- 18.Starck L., Lovgren-Sandblom A., Bjorkhem I. Cholesterol treatment forever? The first Scandinavian trial of cholesterol supplementation in the cholesterol-synthesis defect Smith-Lemli-Opitz syndrome. J. Intern. Med. 2002;252:314–321. doi: 10.1046/j.1365-2796.2002.01037.x. [DOI] [PubMed] [Google Scholar]
- 19.Merkens L.S., et al. Effects of dietary cholesterol on plasma lipoproteins in Smith-Lemli-Opitz syndrome. Pediatr. Res. 2004;56:726–732. doi: 10.1203/01.PDR.0000141522.14177.4F. [DOI] [PubMed] [Google Scholar]
- 20.Sikora D.M., et al. Cholesterol supplementation does not improve developmental progress in Smith-Lemli-Opitz syndrome. J. Pediatr. 2004;144:783–791. doi: 10.1016/j.jpeds.2004.02.036. [DOI] [PubMed] [Google Scholar]
- 21.Jira P.E., et al. Simvastatin: a new therapeutic approach for Smith-Lemli-Opitz syndrome. J. Lipid Res. 2000;41:1339–1346. [PubMed] [Google Scholar]
- 22.Starck L., Lovgren-Sandblom A., Bjorkhem I. Simvastatin treatment in the SLO syndrome: a safe approach? Am. J. Med. Genet. 2002;113:183–189. doi: 10.1002/ajmg.10722. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein J.L., DeBose-Boyd R.A., Brown M.S. Protein sensors for membrane sterols. Cell. 2006;124:35–46. doi: 10.1016/j.cell.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 24.Yabe D., Brown M.S., Goldstein J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U. S. A. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang T., et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 26.Horton J.D., Goldstein J.L., Brown M.S. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. . J. Clin. Invest. 2002;109:1125–1131. doi: 10.1172/JCI200215593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown M.S., Goldstein J.L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 28.Sever N., et al. Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J. Biol. Chem. 2003;278:52479–52490. doi: 10.1074/jbc.M310053200. [DOI] [PubMed] [Google Scholar]
- 29.Sever N., Yang T., Brown M.S., Goldstein J.L., DeBose-Boyd R.A. Accelerated degradation of HMG CoA reductase mediated by binding of Insig-1 to its sterol-sensing domain. Mol. Cell. 2003;11:25–33. doi: 10.1016/s1097-2765(02)00822-5. [DOI] [PubMed] [Google Scholar]
- 30.Engelking L.J., et al. Schoenheimer effect explained — feedback regulation of cholesterol synthesis in mice mediated by Insig proteins. J. Clin. Invest. 2005;115:2489–2498. doi: 10.1172/JCI25614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferguson M.W. Palate development. Development. 1988;103(Suppl.):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- 32.Dietschy J.M., Spady D.K. Measurement of rates of cholesterol synthesis using tritiated water. J. Lipid Res. 1984;25:1469–1476. [PubMed] [Google Scholar]
- 33.Kita T., Brown M.S., Goldstein J.L. Feedback regulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase in livers of mice treated with mevinolin, a competitive inhibitor of the reductase. J. Clin. Invest. 1980;66:1094–1100. doi: 10.1172/JCI109938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Le Douarin, N.M., and Kalcheim, C. 1999.The neural crest. Cambridge University Press. Cambridge, United Kingdom. 469 pp. [Google Scholar]
- 35.Taya Y., O’Kane S., Ferguson M.W.J. Pathogenesis of cleft palate in TGF-β3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- 36.Murray J.C. Developmental biology: frontiers for clinical genetics. Clin. Genet. 2002;61:248–256. [Google Scholar]
- 37.Stanier P., Moore G.E. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum. Mol. Genet. 2004;13:R73–R81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- 38.Porter J.A., Young K.E., Beachy P.A. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274:255–259. doi: 10.1126/science.274.5285.255. [DOI] [PubMed] [Google Scholar]
- 39.Tabin C.J., McMahon A.P. Recent advances in Hedgehog signalling. Trends Cell Biol. 1997;7:442–446. doi: 10.1016/S0962-8924(97)01159-8. [DOI] [PubMed] [Google Scholar]
- 40.Nohturfft A., Brown M.S., Goldstein J.L. Sterols regulate processing of carbohydrate chains of wild-type SREBP cleavage-activating protein (SCAP), but not sterol-resistant mutants Y298C or D443N. Proc. Natl. Acad. Sci. U. S. A. 1998;95:12848–12853. doi: 10.1073/pnas.95.22.12848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang C., et al. Cyclopia and defective axial patterning in mice lackingSonic hedgehog gene function. . Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- 42.Villavicencio E.H., Walterhouse D.O., Iannaccone P.M. The Sonic Hedgehog-Patched-Gli pathway in human development and disease. Am. J. Hum. Genet. 2000;67:1047–1054. doi: 10.1016/s0002-9297(07)62934-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown M.S., Faust J.R., Goldstein J.L., Kaneko I., Endo A. Induction of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity in human fibroblasts incubated with compactin (ML-236B), a competitive inhibitor of the reductase. J. Biol. Chem. 1978;253:1121–1128. [PubMed] [Google Scholar]
- 44.Xu F., et al. Dual roles for cholesterol in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2005;102:14551–14556. doi: 10.1073/pnas.0503590102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Belknap W.M., Dietschy J.M. Sterol synthesis and low density lipoprotein clearance in vivo in the pregnant rat, placenta, and fetus. J. Clin. Invest. 1988;82:2077–2085. doi: 10.1172/JCI113829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimano H., et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J. Clin. Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moebius F.F., Fitzky B.U., Glossmann H. Genetic defects in postsqualene cholesterol biosynthesis. Trends Endocrinol. Metab. 2000;11:106–114. doi: 10.1016/s1043-2760(00)00235-6. [DOI] [PubMed] [Google Scholar]
- 48. Abbott, B.D. 2000. Palatal dysmorphogenesis: palate organ culture. InDevelopmental biology protocols. R.S. Tuan and C.W. Lo, editors. Humana Press Inc. Totowa, New Jersey, USA. 195–201 . [DOI] [PubMed] [Google Scholar]