

Abstract
A novel, constitutively expressed and secreted IL-18 binding protein (IL-18BP) neutralizes IL-18 and thereby suppresses the production of IFN-γ, resulting in reduced T-helper type 1 immune responses. In the present study, four human and two mouse isoforms, resulting from mRNA splicing and found in various cDNA libraries, were expressed, purified, and assessed for binding and neutralization of IL-18 biological activities. Human IL-18BP isoform a (IL-18BPa) exhibited the greatest affinity for IL-18 with a rapid on-rate, a slow off-rate, and a dissociation constant (Kd) of 399 pM. IL-18BPc shares the Ig domain of IL-18BPa except for the 29 C-terminal amino acids; the Kd of IL-18BPc is 10-fold less (2.94 nM). Nevertheless, IL-18BPa and IL-18BPc neutralize IL-18 >95% at a molar excess of two. IL-18BPb and IL-18BPd isoforms lack a complete Ig domain and lack the ability to bind or neutralize IL-18. Murine IL-18BPc and IL-18BPd isoforms, possessing the identical Ig domain, also neutralize >95% murine IL-18 at a molar excess of two. However, murine IL-18BPd, which shares a common C-terminal motif with human IL-18BPa, also neutralizes human IL-18. Molecular modeling identified a large mixed electrostatic and hydrophobic binding site in the Ig domain of IL-18BP, which could account for its high affinity binding to the ligand. It is likely that preferential secretion of functional and nonfunctional isoforms of IL-18BP affect the immune response.
Immune responses to antigens are divided into T-lymphocyte helper types 1 (Th1) and 2 (Th2) (1). Th1 responses include the secretion of cytokines IL-2, IL-12, and IFN-γ and the generation of specific cytotoxic T-lymphocytes recognizing specific antigen. The Th2 response is characterized by the cytokine IL-4 and the production of specific antibodies to the antigen. The Th1 response is a vital arm of host defense against many organisms; however, the Th1 response also is associated with several autoimmune diseases as well as organ transplant rejection. Therefore, natural regulation of the Th1 cytokines likely affects the intensity of the Th1 response.
IL-18, originally termed IFN-γ inducing factor (2), is a Th1 cytokine. Mice deficient in IL-18 have a blunted natural killer cell response and markedly reduced IFN-γ production (3). IL-12 up-regulates the IL-18 receptor alpha chain (4), a member of the IL-1 receptor (IL-1R) family (5, 6). In fact, this regulation of the IL-18 receptor may explain the well-established synergism of IL-18 plus IL-12 in the production of IFN-γ. Although an inducible homodimer of the IL-12 p40 chain can act as a receptor antagonist and inhibit IL-12, there is no evidence that this dimer is expressed in humans, and therefore there is no constitutive inhibitor of IL-12 described to date. On the other hand, a novel IL-18 binding protein (IL-18BP) has been described (7), and being constitutively expressed and secreted, binds IL-18, acts as a natural inhibitor of IL-18-induced IFN-γ, and suppresses the Th1 response.
With a single Ig domain, IL-18BP resembles the extracellular segment of cytokine receptors with Ig-like structures; however, IL-18BP is a novel protein distinct from the IL-1 and IL-18 receptor families (7). Located in chromosome 11q13 at the inverted position of the nuclear mitotic apparatus protein-1, the human IL-18BP gene encodes for at least four distinct isoforms derived from mRNA splice variants isolated from several cDNA libraries. Two isoforms of murine IL-18BP were similarly cloned. Little is known about the ability of these six isoforms of IL-18BP to bind and neutralize IL-18. They differ primarily in their carboxyl termini whereas the N-terminal one-third to two-thirds of the amino acids are identical. It is unclear whether the N terminus or C terminus contributes the structural requirements for binding and neutralization. Moreover, if these isoforms vary significantly in their ability to neutralize the biological activity of IL-18, one may conclude that the Th1 response will be affected in individuals expressing one isoform preferentially compared with another isoform.
Materials and Methods
Cells and Reagents.
COS cells were purchased from the American Type Culture Collection (Rockville, MD), and the human NKO cell line was provided by Tomoaki Hoshino and Howard Young (National Cancer Institute, Frederick, MD). The original NK cell line was isolated by Hans Klingerman (Rush Medical Center, Chicago). Cells were maintained in DMEM or RPMI medium 1640 (GIBCO/BRL), respectively, containing 10% FBS with 100 units/ml of penicillin, and 100 μg/ml of streptomycin. Recombinant human mature IL-18 was provided by Michael Su and Yong Gu (Vertex Pharmaceuticals, Cambridge, MA) (8), and the precursor of human IL-18 was a gift of Monica Tsang (R & D Systems). IL-2 was purchased from R & D Systems. Recombinant human IL-12 and murine IL-18 were a kind gift from Peprotech (Rocky Hill, NJ). Lipopolysaccharide (Escherichia coli 055:B5) was obtained from Sigma, dissolved in water at 1 mg/ml, and stored at −20°C. Talon affinity columns were obtained from CLONTECH and used as recommended. Human IL-18BPa-his6 was purified to homogeneity from a stable Chinese hamster ovary (CHO) cell line (InterPharm Laboratories, Nes Ziona, Israel).
Human and Mouse IL-18 Assays.
Human NKO cells were maintained in RPMI containing 10% FBS with IL-2 (50 pg/ml). For IL-18 assays, NKO cells were suspended at 1 × 106/ml in RPMI and stimulated in 200-μl vol in 96-well plates with IL-12 (2 ng/ml) together with IL-18 (20 ng/ml). These concentrations were optimal for IL-18-induced IFN-γ production. IL-18 was premixed with IL-18BP for 20 min at room temperature before being added to cells. After 24 h at 37°C, the culture supernatants were collected. Human IFN-γ was measured by an electrochemiluminescence assay (Igen, Gaithersburg, MD) as described (9).
Peripheral blood mononuclear cells (PBMC) were isolated from human subjects after they gave informed consent as described (8). The study was approved by the Colorado Multiple Institutional Review Board. PBMC were isolated by centrifugation of heparinized blood over Ficoll-Hypaque gradients or histopaque cushions. PBMC were washed with pyrogen-free saline, resuspended at 5 × 106 cells/ml in RPMI containing 10% FBS, and stimulated as above with IL-12 plus IL-18 in 1-ml vol in 24-well plates. After 24 h at 37°C, the culture's supernatants were collected and IFN-γ was measured.
Mouse spleen cells were used to assay murine IL-18 as described (10). Mouse spleen cells were cultured at 5 × 106/ml in RPMI containing 10% FBS in 96-well plates. Murine IL-18 (20 ng/ml) was preincubated with various preparations of human or murine IL-18BP for 1 h at room temperature before being added to murine spleen cells and then stimulated with lipopolysaccharide (0.5 μg/ml). After 24 h at 37°C, supernatants were assayed for murine IFN-γ by using an immunoassay as described (10).
Transient Expression and Purification of IL-18BP-his6.
The coding region of each human and mouse IL-18BP cDNA (Fig. 1) was amplified by PCR from cDNA clones originally obtained from human monocyte (11), a Jurkat cell line cDNA library (CLONTECH), and a mouse spleen cDNA library (CLONTECH). The sense primer for each human isoform was 5′-TATATCTAGAGCCACCATGAGACACAACTGGACACCA. The reverse primer for human IL-18BPa was 5′- ATATCTAGATTAATGATGATGATGATGATGACCCTGCTGCTGTGGACTGC. The reverse primer for human IL-18BPc was 5′- ATATCTAGATTAATGATGATGATGATGATGAGGTTGTGCTGCTGCTGGCC. The reverse primer for human IL-18BPb and IL-18BPd was 5′- ATATCTAGATTAATGATGATGATGATGATGCAGGCTGCTCTGGCAGAGC. The sense primer for both mouse IL-18BP isoforms was 5′-TATATCTAGAGCCACCATGAGACACTGCTGGACAGC. The reverse primer for mouse IL-18BPc was 5′-ATATCTAGATTAATGATGATGATGATGATGGCTTACAGGTTGTACTGGAAA. The reverse primer for mouse IL-18BPd was 5′- ATATCTAGATTAATGATGATGATGATGATGTGCAACCCCTGGGCCTGCT.
Figure 1.

Alignment of IL-18BP isoforms amino acids. (A) Four human isoforms, IL-18BPa, IL-18BPc, IL-18BPd, and IL-18BPb, are depicted. * above the asparagine residues (nos. 77, 101, and 145) locate glycosylation sites. The inverted carets indicate arrest sites of the amino acid sequence resulting from mRNA splicing followed by the amino acid sequence encoded by the new ORF. The Ig domain is indicated in green. The color blue represents identical amino acids in the new ORF of inactive isoforms. The color red represents closely related carboxyl termini of active isoforms. The color yellow represents a potential binding motif. (B) Mouse IL-18BPc and IL-18BPd isoforms are shown. The colors represent similar indications as shown for human IL-18BP isoforms.
The sense primer pertains to the XbaI site and the Kozak consensus sequence before the ORF. The reverse primers included a cassette coding for a C-terminal his6-tag to facilitate purification. The resulting PCR products were excised with XbaI and cloned into the XbaI site of the pEF-BOS mammalian expression vector (12). The construct was confirmed by DNA sequencing.
Batches of 6 × 107 COS cells were transfected with plasmid encoding pEF-BOS-IL-18BPs as described (7). Ten micrograms of DNA and DEAE-dextran (1.2 mg in 140 mM NaCl, 5 mM KCl, 0.7 mM K2HPO4, 25 mM Tris⋅HCl, pH 7.4) were incubated for 30 min at room temperature as described (13). The cells then were washed with DMEM/10% FBS, plated in DMEM/10% for 4 h, washed with serum-free DMEM, and incubated for 4 days in serum-free DMEM. Culture supernatants were concentrated 20-fold in boiled dialysis bags submerged in saturated polyethylene glycol (molecular weight 8,000, Sigma) followed by dialysis in Talon loading buffer. Concentrated COS supernatants were applied to Talon column and IL-18BP-his6-tag was eluted with imidazole buffer according to the manufacturer's instructions. Eluted IL-18BPs then were concentrated in boiled dialysis bags against saturated polyethylene glycol and dialyzed against PBS, pH 7.4.
Immunoblotting.
Polyclonal anti-human IL-18BPa was generated in rabbits against purified IL-18BPa from CHO cells. A mAb against a covalent tag of six histidines (his6-tag) was purchased from R & D Systems. Preparations containing each isoform of IL-18BP were resolved by SDS/PAGE (10%) under reducing conditions. Immunoblotting was performed with primary antisera to IL-18BP (1:500) or the monoclonal his6-tag (500 ng/ml) followed by developing antisera to rabbit IgG or mouse IgG (Jackson ImmunoResearch).
BIAcore Affinity Determinations.
Mature human IL-18 and the precursor form of human IL-18 were immobilized to individual channels in a BIAcore chip as recommended by the manufacturer (Amersham Pharmacia). Various isoforms of IL-18BP were analyzed on the chip, and the binding constants and kinetics were determined.
Molecular Modeling of the IL-18BP-IL-18 Complex.
Modeling of the complex between IL-18 and IL-18BP is based on the x-ray structure of the IL-1β/IL-1R type I complex (14). Sequence alignments were done against structures in the Protein Data Bank (PDB), using the Smith Waterman algorithm (15) and the blast program (16). Secondary structure prediction was made by the phd program (17). The homology module in the Molecular Simulations (Waltham, MA) set of programs was used for model construction and structure verification. Severe clashes in the initial model were corrected manually by choosing an alternative side-chain conformation, and then the model was energy-minimized to relieve other clashes and to regularize the backbone conformation. The Cα atoms were constrained to their initial position during this minimization so that the overall fold of the molecules was not disrupted. The structure was verified by testing the one-dimensional–three-dimensional compatibility (18).
Results
Neutralization of IL-18 Activity by Different IL-18BP Isoforms.
Supernatants from COS cells transiently transfected with empty BOS vector or BOS-cDNA coding for different isoforms was concentrated 20-fold, dialyzed, and added to various assays of IL-18 biological activity. In Fig. 2, IL-18-induced IFN-γ from human PBMC, NKO cells, and murine spleen cells are shown. In each of the three assays, IL-18 activity in the presence of supernatants from COS transfected with the empty vector (mock) was set at 100%. Human IL-18BPa (GenBank accession no. AF110799), human IL-18BPc (GenBank accession no. AF110801), and mouse IL-18BPd (GenBank accession no. AF110803) consistently inhibited more than 75% of the biological activity of human IL-18 in each of the assays. Supernatants from COS cells expressing human IL-18BPa were the most effective in inhibiting IL-18 with >90% reduction in human cells and 80% in murine spleen cells. Although mouse IL-18BPc (GenBank accession no. AF110802) and IL-18BPd reduced mouse IL-18 activity in mouse spleen cells (Fig. 2C), mouse IL-18BPd also inhibited human IL-18 in human cells (Fig. 2 A and B). However, mouse IL-18BPc was not effective in inhibiting human IL-18. In contrast, human IL-18BPb (GenBank accession no. AF110800) and IL-18BPd (GenBank accession no. AF215907) were ineffective in reducing human IL-18 activity in human cells, and IL-18BPb also had no effect on mouse IL-18.
Figure 2.

Ability of recombinant human and mouse IL-18BP isoforms to inhibit IL-18 biological activities. Supernatants from transiently transfected COS cells were assayed on (A) human PBMC, (B) human NKO cell line, and (C) mouse spleen cells. Supernatants were removed and dialyzed against PBS and added to each cell culture undiluted. Data are mean ±SEM IFN-γ levels of three experiments. Mock indicates supernatants from COS cells transiently transfected with the empty vector.
Purification of Recombinant Isoforms of Human and Murine IL-18BP.
The supernatants from transient transfection of COS cells were purified on metal affinity columns. As shown in Fig. 3A, Western blotting reveals the different molecular sizes using antibodies directed to the his6-tag. As anticipated, human IL-18BPa, IL-18BPc, and IL-18BPd have approximately the same size (35–45 kDa) as the IL-18BPa purified from CHO cells. The calculated size of nonglycosylated IL-18BPb is 9 kDa but on Western blotting, the varying sizes suggest aggregates. In Fig. 3B, the antibody used in Western blotting was raised against the IL-18BPa isoform purified from CHO cells. This antibody recognized human IL-18BPa and IL-18BPc but not the human IL-18BPb or IL-18BPd isoforms. Murine IL-18BPc and IL-18BPd were recognized by the anti-human IL-18BPa. This finding was not unexpected because of the amino acids identity of human IL-18BPa.
Figure 3.

Western blotting of purified IL-18BP isoforms. COS supernatants purified over Talon columns were subjected to 10% SDS/PAGE under reducing (50 mM DTT) conditions and transferred to nitrocellulose. (A) Blot was probed with mAb anti-his6-tag. (B) Blot was probed with rabbit anti-human IL-18BPa. Molecular weights are indicated on the vertical axis. Mock represents supernatants from COS cells transfected with the empty pEF-BOS vector and subjected to the same purification as the IL-18BP isoforms. CHO-derived IL-18BPa is shown on the right and the lane numbers indicate IL-18BPa in ng.
Titration of IL-18BP Activity.
Six purified isoforms of IL-18BP were tested for inhibition of human IL-18 at different molar ratios of IL-18BP to human IL-18. The protein concentration of each isoform eluting from the Talon column was determined by using silver staining of SDS/PAGE and normalized to the IL-18BP CHO standard. In addition, protein concentration was estimated by Western immunoblotting using anti-his6-tag antibody. As shown in Fig. 4, complete inhibition of IL-18 activity on NKO cells was observed at a 2-fold molar excess of IL-18BPa over IL-18. The same extent of inhibition was observed by using IL-18BPa purified from CHO cells (data not shown). At equimolar ratios, inhibition by IL-18BPa was approximately 50%. Equimolar and molar excess of two human IL-18BPc exhibited similar potency as IL-18BPa. In contrast, there was no inhibition of IL-18 activity by IL-18BPb or IL-18BPd at a molar excess of two. Higher ratios of 6-fold molar excess of these isoforms also lacked inhibitory activity. Murine IL-18BPd inhibited human IL-18 similar to IL-18BPa whereas murine IL-18BPc was ineffective. The inhibition of IL-18 using purified isoforms of IL-18BP (Fig. 4) are similar to those observed using unfractionated supernatants from COS cells (Fig. 2).
Figure 4.

Titration of human and murine IL-18BP isoforms on human IL-18 activity. Human and murine isoforms were incubated with human IL-18 (20 ng/ml) for 30 min at increasing concentrations using 2-fold dilutions of IL-18BP. After this incubation, the mixtures were added to NKO cells cocultured with IL-12 (2 ng/ml). After 24 h IFN-γ was measured. The stimulation control of IL-12 (2 ng/ml) plus IL-18 (20 ng/ml) with purified supernatants from mock-transfected COS cells was set at 100%, and the percent change was calculated for each concentration of IL-18BP isoforms tested.
Determination of IL-18BP Affinity for IL-18.
A BIAcore sensor chip with immobilized human precursor IL-18 was unable to bind any of the isoforms of IL-18BP. In contrast, mature human IL-18 bound IL-18BPa and IL-18BPc. The mean dissociation constant for human IL-18BPa was 0.399 ± 0.034 nM (n = 6) whereas the affinity of human IL-18BPc was approximately 10-fold lower (2.94 ± 0.86 nM, n = 4). As shown in Fig. 5, the binding of increasing amounts of IL-18BPa to immobilized human mature IL-18 reveals a rapid association rate (1.38 ± 0.11 mol⋅sec−1) and a markedly slow dissociation rate (6.43 ± 0.4 sec−4).
Figure 5.

Kinetics of human IL-18BPa binding to human IL-18. Data are derived from a BIAcore chip of immobilized human IL-18. The numbers at the right of each line indicate the concentration of IL-18BPa in nM.
Modeling of IL-18BP.
Sequence alignment of IL-18BPa against the PDB identified two possible modeling templates: domain III of IL-1R (PDB entry 1itb), which shows 25.3% sequence identity with residues 72–163 in IL-18BP (see below), and domain 1 of CD4 (PDB entry 3cd4) (11), which shows 27.6% sequence identity with the same fragment in IL-18BP (not shown). Sequence alignment with the blast program also identified the constant domain of the light chain anti-T-cell receptor antibody H57 (PDB entry 1nfd). The latter molecule shows 25% sequence identity with residues 51–105 of IL-18BP, i.e., the N-terminal end of the Ig-like domain. The templates and IL-18BP carry several established identifiers of the Ig fold, including the conserved cysteines and tryptophan in β-strands B, F, and C.
The N-terminal and C-terminal fragments of IL-18BP could not be aligned with a known structure. The N-terminal fragment, residues 29–59, was predicted to contain a single β-strand followed by a long loop. The C-terminal fragment, residues 165–197, was predicted to be a loop followed by a single β-strand. The final alignment of IL-18BP against IL-1R type I is shown below.
 |
The one-dimensional–three-dimensional compatibility score for the IL-18BP model was 31.2 (expected score 47.4), and there was only one segment with negative scores, i.e., possibly misfolded. This segment corresponds to the A-B loop that is found distant from the IL-18BP/IL-18 interface. No attempt was made to modify this segment.
Modeling of IL-18.
IL-18 was aligned to IL-1β as described (19), but with modifications, in particular at the N and C termini. The final sequence alignment of IL-18 against human-IL-β (chain A in PDB entry 1itb) and mouse-IL-1β (PDB entry 8i1b) is depicted below. The model was constructed based on the alignment above, as described in Materials and Methods. The one-dimensional–three-dimensional compatibility score for this model is 48.9 (expected value 69.7). There are two zones where the score is negative (residues 70–74 and 147–148). Both are found in loops, where inserts/deletions with respect to IL-1 occur and where the model is inaccurate.
 |
The Interface in the Complex of IL-18 with IL-18BP.
IL-18BP interacts with IL-18 through β-strands C and D, the loop connecting them and the F-G loop. The model of IL-18BP is reliable for most of the sequence including the fragments that interact with IL-18, provided that β-strand D was correctly identified and aligned. IL-18 interacts with IL18BP via residues 85, 87, 89, 130, 132, 143, and 149, which are found in the sequence fragment that could be reliably aligned with IL-1β, but also with the N- and C-terminal β-strands (residues 42 and 189), which were difficult to align, thus limiting the accuracy of the prediction. The model suggests that the interaction of IL1–8BP and IL-18 is stabilized, on the one hand, by strong electrostatic interactions (buried ion pairs between E42 in IL18 and K130 in IL18BP and between K89 in IL18 and E114 in IL18BP) and also by the burial of large hydrophobic patches. The latter consist of residues I85, M87, M96, P143, M149, and L189 in IL-18 and F93, I95, Y97, V151, and P153 in IL-18BP. Notably, one of the ion pairs is less reliable because it involves E42 in IL-18, which is found in the N-terminal β-strand, where the alignment is least accurate.
Mouse IL-18BPd neutralizes the activity of human IL-18, hence the interacting surface should be similar. Alignment of the mouse and human sequences of IL-18BP reveals that nearly all the residues involved in IL-18 binding are conserved. There are two differences: in position 116 Ser is replaced by His and in position 130 Lys is replaced by Arg. Both replacements are not likely to disrupt the binding of human IL-18. The predicted structure of the complex is shown in Fig. 6.
Figure 6.
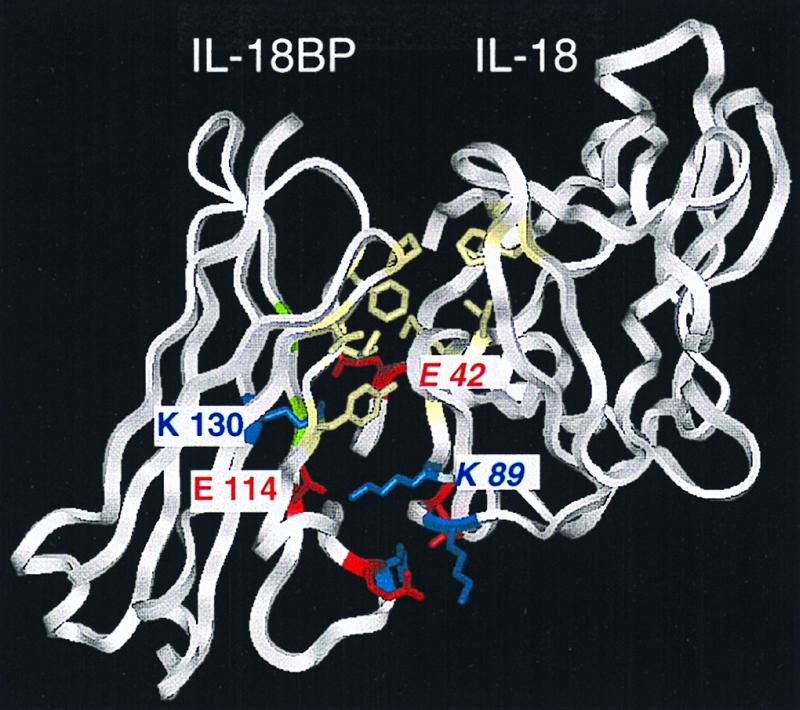
A predicted ribbon model of the IL-18/IL-18BP complex. This model is based on the x-ray structure of the IL-1β/IL-1R type I complex. The model depicts the Ig domain of precursor IL-18BPa (residues 60–164). Hydrophobic amino acid residues of the two proteins are shown in yellow. Positively and negatively charged residues are shown in blue and red, respectively. The amino acid residues of IL-18 are in italics.
Discussion
These studies reveal that the ability of different isoforms of IL-18BP to bind and inhibit the biological activity of IL-18 primarily resides in the Ig domain with a possible contribution of the carboxyl end of the molecule. The ability of human IL-18BPa and IL-18BPc and murine IL-18BPc and IL-18BPd to neutralize human IL-18 and murine IL-18, respectively, at equimolar concentrations supports the concept that the Ig domain possesses the essential binding requirements for IL-18. Although the first 97 N-terminal mature amino acids (58%) of human IL-18BPd are identical to human IL-18BPa, IL-18BPd lacks a complete Ig domain and is inactive in inhibiting IL-18 as well as binding to immobilized IL-18. The most relevant crystallographic analysis to the present study is of the binding of IL-1β to IL-1R type I and the importance of the third Ig domain (14). In fact, antibodies directed to the third Ig domain of the IL-1R type I block binding of IL-1β whereas antibodies to the N-terminal domains have no effect (20).
Both the human IL-18BPa and IL-18BPc as well as the murine IL-18BPd isoform inhibited 50% human IL-18BP at equimolar ratios. This is an unusually low molar ratio to observe inhibition of biological activity in a 24-h bioassay. With soluble receptors such as the IL-1R type I and II (21) and the tumor necrosis factor receptor p55 and p75 (22), the molar excess required to inhibit 50% of biological activity of the respective ligands is at least 5-fold. One likely explanation for this potency of IL-18BPa and IL-18BPc is the high affinity binding and slow off-rate of both forms. Molecular modeling identified a large interaction site with IL-18, which buries 1,170 Å2 for both molecules. The site includes two strong electrostatic ion pair interactions (E42 in IL-18 and K130 in IL-18BP and K89 in IL-18 and E114 in IL-18BP), which are buried near the center of the interface and are unlikely to be surrounded by water. The binding is further strengthened by the burial of matching hydrophobic patches found on the surfaces of the two molecules.
Although the BIAcore binding of IL-18BPc was approximately 10-fold less than that of IL-18BPa, the Kd of IL-18BPa remains sufficiently high to inhibit IL-18 at a molar excess of two. The lower BIAcore affinity may be related to the fact that the terminal 29 aa of IL-18BPc are not identical to those of IL-18BPa. At present, the contribution of the C-terminal amino acids after the Ig domain is unclear. Mouse IL-18BPc, which shares the Ig domain with human IL-18BPa and IL-18BPc, consistently did not affect the biological activity of human IL-18. However, the C-terminal amino acids of human IL-18BPa shares limited identity with murine IL-18BPd. In fact, as shown in Fig. 7, there is a stretch of amino acids that are similar in these isoforms (QEAL, QEEL, respectively above). Secondary structure prediction suggests that these amino acids contribute to the formation of a β-strand. Mouse IL-18BPc, possessing the identical Ig domain with mouse IL-18BPd but lacking homology in this putative strand, inhibits mouse but not human IL-18. Although in-exon splicing results in frame shifts with isoforms possessing different C-terminal sequences, the frame shifts do not appear to be haphazard because the three isoforms with the homogous C-terminal amino acids share the ability to neutralize human IL-18 in two distinct bioassays (Fig. 2).
Figure 7.

Homology between C-terminal sequences of human and mouse IL-18BP. Multiple sequence alignment suggests that the human IL-18BPa (indicated as hIL-18BPa) and mouse IL-18BPd (mIL-18BPd) are structurally related, whereas mouse IL-18BPc is more related to human IL-18BPc.
The first three potential glycosylation sites derived from the primary amino acid sequences of the IL-18BP isoforms were found to be exposed in model structure. Glycosylation of IL-18BP may play a role in its function as an inhibitor of IL-18 but whether this role is one of solubility or binding remains unclear. Human IL-18BPa and IL-18BPc have identical glycosylation sites but differ in BIAcore binding and the ability to inhibit IL-18 activity. Similarly, mouse IL-18BPc and IL-18BPd have identical potential glycosylation sites but differ in their ability to neutralize human IL-18. In each of the experiments, ligand passing by IL-18BPs as evidence by increased bioactivity was not observed as has been observed with cytokine soluble receptors (22, 23).
Molluscum contagiosum viral proteins MC53 and MC54 share a significant homology to mammalian IL-18BP (7). Recently Xiang and Moss (24) reported that M. contagiosum proteins MC53 and MC54 possess the ability to bind and neutralize human IL-18 in a fashion similar to that of IL-18BP. As proposed above, the data from the present studies on the mammalian IL-18BP isoforms support the hypothesis that the binding and neutralizing properties of IL-18BP reside in the Ig domain. Nevertheless, there is homology in the stretch from 174 to 191; between 174 and 189 of the human IL-18BPa there is 83% homology with viral MC53 and between IL-18BPa 176 and 192, 68% with viral MC54.
Diseases associated with cytokine polymorphisms are mostly defects in the receptors and not the ligands. The finding of the different isoforms of IL-18BP in the various libraries may be tissue specific or related to events during cell stimulation. The IL-18BPa cDNA was the most abundant clone in each of the human libraries. IL-18BPc was present in Jurkat and spleen libraries. IL-18BPb was found in monocytes and Jurkat libraries and IL-18BPd in Jurkat cells only. The findings of the different isoforms in the various libraries may have significance for the Th1 immune response. The clinical significance of the present study involves natural regulation of the Th1 response. At present, it appears that IL-18BPa is the major form constitutively expressed in human spleen (7). It is unknown whether the other human isoforms are expressed and secreted in vivo. The 40 N-terminal amino acids of the naturally occurring urinary IL-18BP (7) are common to each of the four isoforms. Because IL-18BPb and IL-18BPd do not bind to immobilized IL-18, the existence of these isoforms in human urine is possible.
There are many examples of exon splicing events for cytokines and cytokine receptors, but there are few clinical examples of diseases caused by these splicing events. Because IL-18BPb and IL-18BPd were unable to neutralize IL-18, their presence in vivo would likely not have a clinical impact on the Th1 response. However, if some individuals preferentially undergo the IL-18BPb and IL-18BPd splicing events over those of IL-18BPa and IL-18BPc, then one would expect a reduced ability to inhibit the activity of IL-18 during a Th1 response compared with individuals who secrete the IL-18BPa and IL-18BPc isoforms. In the case of IL-18BP, two events can result in isoform formation. For example, during routine intron excision, exon skipping takes place resulting in a frame shift and a new carboxyl terminus. The second example also results in a new carboxyl terminus after an in-exon splicing event. In both cases, an early stop codon may contribute to the size of the isoform. In IL-18BPb and IL-18BPd, in-exon splicing takes place in exon 4, resulting in a new C-terminal amino acid sequence because of a frame shift. Because cDNAs for the IL-18BPa as well as IL-18BPb were found in a human blood monocyte library, it is possible that some individuals may preferentially produce one isoform over the other. Control of in-exon splicing is poorly understood, but in the case of IL-18BP, the mechanism for in-exon splicing may have significant clinical impact.
Acknowledgments
We thank Natan Tal and Aharon Rabinkov of the Weizmann Institute Department of Biological Services for performing the BIAcore analysis and Dr. Ron Pinkus of InterPharm Laboratories, Nes Ziona, Israel, for providing the purified CHO-derived IL-18BPa-his6-tag. M.R. is the Maurice and Edna Weiss Professor of cytokine research. These studies are supported by National Institutes of Health Grant A-15614, AI-2532359, Colorado Cancer Center CA 46934 (to C.A.D.) and the Advances Research Systems/Serono Group (to M.R.).
Abbreviations
- IL-18BP
IL-18 binding protein
- Th
T-lymphocyte helper cell
- IL-1R
IL-1 receptor
- PDB
Protein Data Bank
- CHO
Chinese hamster ovary
- PBMC
peripheral blood mononuclear cells
Footnotes
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF215907).
References
- 1.Seder R A, Paul W E. Annu Rev Immunol. 1994;12:635–673. doi: 10.1146/annurev.iy.12.040194.003223. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura K, Okamura H, Wada M, Nagata K, Tamura T. Infect Immun. 1989;57:590–595. doi: 10.1128/iai.57.2.590-595.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto K, Okamura H, Nakanishi K, Akira S. Immunity. 1998;8:383–390. doi: 10.1016/s1074-7613(00)80543-9. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, Akira S, Nakanishi K. J Immunol. 1998;161:3400–3407. [PubMed] [Google Scholar]
- 5.Parnet P, Garka K E, Bonnert T P, Dower S K, Sims J E. J Biol Chem. 1996;271:3967–3970. doi: 10.1074/jbc.271.8.3967. [DOI] [PubMed] [Google Scholar]
- 6.Torigoe K, Ushio S, Okura T, Kobayashi S, Taniai M, Kunikate T, Murakami T, Sanou O, Kojima H, Fuji M, et al. J Biol Chem. 1997;272:25737–25742. doi: 10.1074/jbc.272.41.25737. [DOI] [PubMed] [Google Scholar]
- 7.Novick D, Kim S-H, Fantuzzi G, Reznikov L, Dinarello C A, Rubinstein M. Immunity. 1999;10:127–136. doi: 10.1016/s1074-7613(00)80013-8. [DOI] [PubMed] [Google Scholar]
- 8.Puren A J, Fantuzzi G, Gu Y, Su M S-S, Dinarello C A. J Clin Invest. 1998;101:711–724. doi: 10.1172/JCI1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Puren A J, Razeghi P, Fantuzzi G, Dinarello C A. J Infect Dis. 1998;178:1830–1834. doi: 10.1086/314481. [DOI] [PubMed] [Google Scholar]
- 10.Fantuzzi G, Puren A J, Harding M W, Livingston D J, Dinarello C A. Blood. 1998;91:2118–2125. [PubMed] [Google Scholar]
- 11.Miki T, Matsui T, Heidaran M, Aaronson S A. Gene. 1989;83:137–146. doi: 10.1016/0378-1119(89)90411-3. [DOI] [PubMed] [Google Scholar]
- 12.Mizushima S, Nagata S. Nucleic Acids Res. 1990;18:5322. doi: 10.1093/nar/18.17.5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sompayrac L H, Danna K L. Proc Natl Acad Sci USA. 1981;78:7575–7578. doi: 10.1073/pnas.78.12.7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vigers G P A, Anderson L J, Caffes P, Brandhuber B J. Nature (London) 1997;386:190–194. doi: 10.1038/386190a0. [DOI] [PubMed] [Google Scholar]
- 15.Hale K K, Smith C G, Baker S L, Vanderslice R W, Squires C H, Gleason T M, Tucker K K, Kohno T, Russel D A. Cytokine. 1995;7:26–38. doi: 10.1006/cyto.1995.1004. [DOI] [PubMed] [Google Scholar]
- 16.Kusters S, Tiegs G, Alexopoulou L, Pasparakis M, Douni E, Kunstle G, Bluethmann H, Wendel A, Pfizenmaier K, Kollias G, Grell M. Eur J Immunol. 1997;27:2870–2875. doi: 10.1002/eji.1830271119. [DOI] [PubMed] [Google Scholar]
- 17.Luthy R, Bowie J U, Eisenberg D. Nature (London) 1992;356:83–85. doi: 10.1038/356083a0. [DOI] [PubMed] [Google Scholar]
- 18.Fischer D, Eisenberg D. Protein Sci. 1996;5:947–955. doi: 10.1002/pro.5560050516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bazan J F, Timans J C, Kaselein R A. Nature (London) 1996;379:591. doi: 10.1038/379591a0. [DOI] [PubMed] [Google Scholar]
- 20.Clark B D, Ikejima T, Mancilla J, Sirko S, Orencole S F, Isji N, Okuda K, Dinarello C A. J Interferon Cytokine Res. 1996;16:1079–1088. doi: 10.1089/jir.1996.16.1079. [DOI] [PubMed] [Google Scholar]
- 21.Arend W P, Malyak M, Smith M F, Whisenand T D, Slack J L, Sims J E, Giri J G, Dower S K. J Immunol. 1994;153:4766–4774. [PubMed] [Google Scholar]
- 22.Terlizzese M, Simoni P, Antonetti F. J Interferon Cytokine Res. 1996;16:1047–1053. doi: 10.1089/jir.1996.16.1047. [DOI] [PubMed] [Google Scholar]
- 23.Aderka D, Engelmann H, Maor Y, Brakebusch C, Wallach D. J Exp Med. 1992;175:323–329. doi: 10.1084/jem.175.2.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiang Y, Moss B. Proc Natl Acad Sci USA. 1999;96:11537–11542. doi: 10.1073/pnas.96.20.11537. [DOI] [PMC free article] [PubMed] [Google Scholar]