

Abstract
The ciliate Tetrahymena thermophila is a model organism for molecular and cellular biology. Like other ciliates, this species has separate germline and soma functions that are embodied by distinct nuclei within a single cell. The germline-like micronucleus (MIC) has its genome held in reserve for sexual reproduction. The soma-like macronucleus (MAC), which possesses a genome processed from that of the MIC, is the center of gene expression and does not directly contribute DNA to sexual progeny. We report here the shotgun sequencing, assembly, and analysis of the MAC genome of T. thermophila, which is approximately 104 Mb in length and composed of approximately 225 chromosomes. Overall, the gene set is robust, with more than 27,000 predicted protein-coding genes, 15,000 of which have strong matches to genes in other organisms. The functional diversity encoded by these genes is substantial and reflects the complexity of processes required for a free-living, predatory, single-celled organism. This is highlighted by the abundance of lineage-specific duplications of genes with predicted roles in sensing and responding to environmental conditions (e.g., kinases), using diverse resources (e.g., proteases and transporters), and generating structural complexity (e.g., kinesins and dyneins). In contrast to the other lineages of alveolates (apicomplexans and dinoflagellates), no compelling evidence could be found for plastid-derived genes in the genome. UGA, the only T. thermophila stop codon, is used in some genes to encode selenocysteine, thus making this organism the first known with the potential to translate all 64 codons in nuclear genes into amino acids. We present genomic evidence supporting the hypothesis that the excision of DNA from the MIC to generate the MAC specifically targets foreign DNA as a form of genome self-defense. The combination of the genome sequence, the functional diversity encoded therein, and the presence of some pathways missing from other model organisms makes T. thermophila an ideal model for functional genomic studies to address biological, biomedical, and biotechnological questions of fundamental importance.
The macronuclear genome ofTetrahymena thermophila is sequenced and analyzed. Conservation in this single-celled ciliate of some features normally observed in only multicellular organisms sheds light on early eukaryotic evolution.
Introduction
Tetrahymena thermophila is a single-celled model organism for unicellular eukaryotic biology [1]. Studies of T. thermophila (referred to as T. pyriformis variety 1 or syngen 1 prior to 1976 [2]) have contributed to fundamental biological discoveries such as catalytic RNA [3], telomeric repeats [4,5], telomerase [6], and the function of histone acetylation [7]. T. thermophila is advantageous as a model eukaryotic system because it grows rapidly to high density in a variety of media and conditions, its life cycle allows the use of conventional tools of genetic analysis, and molecular genetic tools for sequence-enabled experimental analysis of gene function have been developed [8,9]. In addition, although it is unicellular, it possesses many core processes conserved across a wide diversity of eukaryotes (including humans) that are not found in other single-celled model systems (e.g., the yeasts Saccharomyces cerevisiae and Schizosaccharomyces pombe).
T. thermophila is a member of the phylum Ciliophora, which also includes the genera Paramecium, Oxytricha, and Ichthyophthirius. A cartoon showing the phylogenetic position of T. thermophila relative to other eukaryotes for which the genomes have been sequenced is shown in Figure 1. The ciliates are one of three major evolutionary lineages that make up the alveolates. The other two lineages are dinoflagellates and the exclusively parasitic apicomplexa, which includes the Plasmodium species that cause malaria. Although experimental tools are improving for the apicomplexa [10–12], they can still be challenging to work with, and in some situations T. thermophila can serve as a useful “distant cousin” model for this group [13].
Figure 1. Unrooted Consensus Phylogeny of Major Eukaryotic Lineages.

Representative genera are shown for which whole genome sequence data are either in progress (marked with asterisks *) or available. The ciliates, dinoflagellates, and apicomplexans constitute the alveolates (lighter yellow box). Branch lengths do not correspond to phylogenetic distances. Adapted from the more detailed consensus in [197].
As is typical of ciliates, T. thermophila cells exhibit nuclear dimorphism [14]. Each cell has two nuclei, the micronucleus (MIC) and the macronucleus (MAC), containing distinct but closely related genomes. The MIC is diploid and contains five pairs of chromosomes. It is the germline, the store of genetic information for the progeny produced by conjugation in the sexual stage of the T. thermophila life cycle. Conjugation involves meiosis, fusion of haploid MIC gametes to produce a new zygotic MIC, and differentiation of new MACs from mitotic copies of the zygotic MIC (for details, see [15]). After formation of the MAC, cells reproduce asexually until the next sexual conjugation. During this asexual growth, all gene expression occurs in the MAC, which is thus considered the somatic nucleus.
The MAC genome derives from that of the MIC, but the two genomes are quite distinct. During MAC differentiation, several types of developmentally programmed DNA rearrangements occur [16,17] (Figure 2). One such rearrangement is the deletion of segments of the MIC genome known as internally eliminated sequences (IESs). It is estimated that approximately 6,000 IESs are removed, resulting in the MAC genome being an estimated 10% to 20% smaller than that of the MIC [18]. A key aspect of the process is the preferential removal of repetitive DNA, which results in 90% to 100% of MIC repeats being eliminated [19,20]. Thus the process can be considered analogous to and more extreme than other forms of repeat element silencing phenomena such as repeat-induced point mutation (RIP) in Neurospora and heterochromatin formation [21,22]. A second programmed DNA rearrangement is the site-specific fragmentation at each location of the 15–base pair (bp) chromosome breakage sequence (Cbs) [23–25]. During fragmentation, sections of the MIC genome containing each Cbs, as well as up to 30 bp on either side, are deleted [26]. Telomeres are then added to each new end [27], generating some 250 to 300 MAC chromosomes [28,29].
Figure 2. Relationship between MIC and MAC Chromosomes.

The top horizontal bar shows a small portion of one of the five pairs of MIC chromosomes. MAC-destined sequences are shown in alternating shades of gray. MIC-specific IESs (internally eliminated sequences) are shown as blue rectangles, and sites of the 15-bp Cbs are shown as red bars (not to scale). Below the top bar are shown macronuclear chromosomes derived from the above region of the MIC by deletion of IESs, site-specific cleavage at Cbs sites, and amplification. Telomeres are added to the newly generated ends (green bars). Most of the MAC chromosomes are amplified to approximately 45 copies (only three shown). Through the process of phenotypic assortment, initially heterozygous loci generally become homozygous in each lineage within approximately 100 vegetative fissions. Polymorphisms located on the same MAC chromosome tend to co-assort.
Another process that occurs during MAC differentiation is the amplification of the number of copies of the MAC chromosomes. The rDNA chromosome, which encodes the 5.8S, 17S, and 26S rRNAs, is maintained at an average of 9,000 copies per MAC [30]. Six other chromosomes that have been examined are each maintained at an average of 45 copies per MAC [31]. During asexual reproduction, the MAC divides amitotically, with apparently random distribution of chromosome copies that behave as if acentromeric. In contrast, MIC chromosomes are metacentric [32] and are distributed mitotically [33,34]. Parental MAC DNA is not transmitted to sexual progeny, although it does have an epigenetic influence on postzygotic MAC genome rearrangement, mediated by RNA interference [35].
The Tetrahymena research community has coordinated an effort to develop genomic tools for T. thermophila [9,36]. The MAC genome was selected for initial sequencing because it contains all the expressed genes and because the complexity of the assembly process was expected to be reduced due to the lower amounts of repetitive DNA. These advantages, however, are countered by some complexities not seen in other eukaryotic genome projects, including the presence of several hundred medium-sized to small chromosomes, the possibility of unequal copy number of at least some chromosomes, the existence of polymorphisms that are generated during MAC development, and the inability to completely separate the MIC from the MAC prior to DNA isolation.
We report here on the shotgun sequencing, assembly, and analysis of the MAC genome of T. thermophila strain SB210, an inbred strain B derivative that has been extensively used for genetic mapping and for the isolation of mutants. We discuss how the complexities of sequencing the MAC were successfully addressed, as well as the biological and evolutionary implications of our analysis of the genome sequence.
Results/Discussion
Genome Assembly and General Chromosome Structure
Sequencing and assembly.
Using physical isolation methods, MAC were purified from a culture of T. thermophila strain SB210 and used to create multiple differentially sized shotgun sequencing libraries (Table S1). Construction of large (greater than 10 kb) insert libraries was not successful—a common problem in working with AT-rich genomes. Approximately 1.2 million paired end sequences were generated from the libraries and assembled using the Celera Assembler [37]. In an initial assembly, the mitochondrial genome (mtDNA; which was present due to some contamination of the MAC preparation with mitochondria) and the highly amplified rDNA chromosome did not assemble well compared to the published sequences of these molecules [38,39]. This was probably because contigs from these molecules had higher depths of coverage than those from other chromosomes, which caused the Celera Assembler to treat them as repetitive DNA. Thus we divided sequence reads into three bins (mtDNA, rDNA, and bulk MAC DNA) and generated assemblies for each bin separately. This resulted in a moderate improvement, and the three separate assemblies were thus used for all subsequent analyses. Detailed sequence and assembly information is presented in Tables 1 and S2.
Table 1.
Important Genome Statistics

The bulk MAC assembly contains 1,971 scaffolds (contigs that have been linked into larger pieces by mate pair information) with a total estimated span of 104.1 Mb. Perhaps most important, using a combination of computational and experimental identification of telomeres, we have found that many scaffold ends correspond to chromosome ends. One hundred twenty-five scaffolds, encompassing 44% of the assembled genome length, are telomere-capped at both ends and thus likely represent complete MAC chromosomes. One hundred twenty additional scaffolds, encompassing another 31% of the genome, are telomere-capped at one end (Tables 1 and S3).
Assembly accuracy and completeness.
Overall, all analyses indicate that the bulk MAC assemblies are highly accurate. For example, all 75 MAC loci that are in distinct genetic co-assortment groups (and thus should be on different chromosomes [40]) map to different scaffolds, and all pairs of loci that coassort (and thus should be on the same chromosome) either map to the same scaffold or to two non–fully capped scaffolds whose cumulative size is less than that of the corresponding MAC chromosome (Table S4). For the 24 completely assembled chromosome scaffolds for which we know the corresponding chromosome physical size, there is a very strong correlation between physical size and assembly length. In addition, there are no cases where a scaffold is significantly longer than the physical size of the corresponding chromosome (Figure 3A). Finally, all of the 96 MIC sequences known to be adjacent to Cbs sites [24,41,42] that matched to a MAC scaffold did so only at the scaffold's end.
Figure 3. Scaffold Sizes.

(A) Scaffold sizes versus MAC chromosome size. Blue diamonds represent scaffolds capped by telomeres on both ends. Red squares and green triangles represent incomplete scaffolds capped by telomeres at one or neither end, respectively.
(B) Size distribution of scaffolds capped by telomeres on both ends.
The general accuracy of the assemblies indicates that many of the potential difficulties discussed in the Introduction were not significant. For example, we see little evidence for polymorphism among reads, which is likely a reflection of the use of an inbred strain and the process of phenotypic assortment, which leads to whole-genome MAC homozygous lineages [43]. Also, searches for known MIC-specific sequences indicate that the amount of MIC contamination is very low (e.g., Cbs junctions are at 0.044× coverage which is approximately 200-fold less than the bulk MAC chromosomes) and limited to small contigs (most less than 5 kb). The uniform depth of contig coverage and accuracy of assemblies also suggest that the chromosomes are present in roughly similar copy number and that only limited amounts of repetitive DNA are present in the MAC, both of which are discussed further below.
The total scaffold length is much smaller than the predicted genome size of 180 to 200 Mb [14]. Given the accuracy of the assemblies, the large number of chromosomes partially or completely capped, and the fact that all (more than 200) known MAC DNA sequences are found in the assemblies, we conclude that the assemblies represent a very large (more than 95%) fraction of the genome. We conclude therefore that previous genome size estimates were inaccurate (which is not surprising given that they were made almost 30 years ago) and that the genome is close to 105 Mb in size. It is possible, however, that some chromosomes or regions were underrepresented in our libraries due to purification or cloning bias, and thus one cannot infer the absence of any particular gene or feature simply due to its absence from our current assemblies.
Estimating the number of MAC chromosomes.
The total number of MAC chromosomes is unknown. The telomere-capping of scaffolds allows us to place a minimum boundary on this number at 185 (125 plus half of 120). One way of estimating the actual number is through analysis of the non–rDNA telomere-containing reads; 3,328 such reads can be linked to a total of 370 scaffold ends. This corresponds to approximately 9-fold coverage (3,328/370), which is not significantly different from the bulk MAC chromosome coverage of 9.08, indicating that there is no significant underrepresentation of telomere reads (Tables 1 and S3). Thus since there are 4,058 such reads total (the others could not be linked), we estimate that there are approximately 451 telomere ends (4,058/9), and thus that there are approximately 225 chromosomes (451/2). An independent estimate of the actual chromosome number can be made by assuming that the size distribution of fully capped chromosomes (see Figure 3B) is representative of the genome as a whole. Since these 125 capped chromosomes represent 43.5% of the total assembly length, this would predict 287 chromosomes in total (125/0.435). This is likely to be an overestimate, since larger chromosomes are statistically less likely to be in the completely assembled set. Indeed, the average size of completely assembled chromosomes is 359 kb, whereas estimates of the average MAC chromosome size obtained through pulsed-field gel electrophoresis are substantially higher [29,41]. Thus, we conclude that there are between 185 and 287 chromosomes, most likely somewhere near 225.
Absence of many standard global features of eukaryotic chromosomes.
We note that we searched for but could not find many of what are considered standard global features of eukaryotic chromosomes. For example, we could not find sequence or structural features shared across multiple chromosomes that could be considered candidates for centromeric regions. This is consistent with experimental studies [44]. In addition, although in many eukaryotes certain genes and repeat elements cluster near telomeres [45–51], we cannot detect any such clustering here. This is not because there is no variation in these features; for example, GC content (Figure S1) and gene density (Figure S2) do vary greatly. Instead, the absence of similar global structure between MAC chromosomes is likely due to the absence of the processes that help generate the key features of normal eukaryotic chromosomes (e.g., mitosis and meiosis, which in T. thermophila are confined to the MIC).
MAC chromosome copy number is uniform.
The high quality and completeness of the assemblies suggest that copy number variation among at least most MAC chromosomes is relatively small since otherwise the assembler would have treated contigs from overrepresented chromosomes as repetitive DNA. Such uniform copy number is consistent with genetic experimental data for six chromosomes [31], but its generality for all chromosomes has been unknown. We realized that the relative chromosome copy number could be estimated from depth of coverage in our assemblies (assuming that cloning and sequencing success were relatively random). When all scaffolds are examined, the depth of coverage is remarkably uniform (Figure 4). The decrease in uniformity and coverage seen as scaffold size decreases is likely a reflection of both chance low coverage of some regions and some of the small scaffolds being MIC contaminants. When only scaffolds capped by telomeres at both ends are included in the analysis, observed sequence coverage is even more uniform (red diamonds in Figure 4). Although we cannot rule out that some smaller, incompletely assembled chromosomes are maintained at different copy numbers, the observed uniformity indicates that the replication and/or segregation of most or all bulk MAC chromosomes is under coordinated regulation.
Figure 4. Depth of Coverage versus Scaffold Size.

Black diamonds indicate all scaffolds; red diamonds, scaffolds capped with telomeres on both ends.
General Features of Predicted Protein Coding Genes and Noncoding RNAs
Protein coding gene predictions.
We identified 27,424 putative protein-coding genes in the genome (Table 2), a high number for a single-celled species. These gene models were tested by aligning expressed sequence tags (ESTs) to the genome assemblies using PASA [52]. We note that most of these ESTs were generated after the models were built (Table S5). Of the 9,122 EST clusters identified, most have either no conflicts with the gene models (49.5%) or relatively small ones (17.7% have a missed exon and 9.8% suggest the models need to be merged or split). Only 408 (4.4%) clusters are intergenic relative to the gene models. Although these could represent missed genes or gene regions, they could also be noncoding RNAs (ncRNAs) or genomic DNA contamination of cDNA libraries. In addition, the predicted and EST-derived introns are quite similar in size distribution except at the short and long extremes (Figure S3), GC content (16.3% versus 16.7%), and splice sites [only a small number (85) of EST-based introns have exceptions to the 5′-GT…AG-3′ junctions assumed by the model—these could simply be sequencing errors]. These analyses indicate that the gene models are relatively robust and should be more than sufficient for making general predictions about the coding potential of this species.
Table 2.
Characteristics of Ab Initio Predicted Genes

Two other lines of evidence suggest the predicted gene number is not inflated. First, a large number of the predicted genes have matches to known or predicted genes from other species (14,916 have a BLASTP match with an E-value better than 10−10), and second, experimental studies of mRNA complexity predict transcription of at least 25,000 genes of an average size of 1,200 bp [53]. We also note that the sequence of the largest MAC chromosome of another ciliate, Paramecium tetraurelia, indicates a high coding density, and extrapolation to the complete genome predicts at least 30,000 protein-coding genes [54].
ncRNAs and the use of all 64 codons to code for amino acids.
The ncRNAs found in the genome are listed in Table S6. We call attention to a few new findings. Of the 174 putative 5S rRNA genes (Table S6A), 19 do not correspond to any of the four previously reported T. thermophila sequences [55,56]. These 19 differ from one another by single nucleotide substitutions at 34 positions, as well as by various insertions, deletions, and truncations and may represent pseudogenes. In addition, there are two forms of U2 snRNA present (Table S6C), which we have termed U2 (four genes) and U2var (five genes). Functional RNA gene families are expressed ubiquitously during the T. thermophila life cycle and under stress conditions as well (representative data shown in Figure S4). The largest class is tRNAs with 700 identified (Tables S6B and S6D), a number consistent with hybridization-based estimates [57].
One of the more unusual features of T. thermophila and certain other ciliates is the use of an alternative genetic code in which the canonical stop codons UAG and UAA code for glutamine [58]. The importance and age of this alternative code are reflected in the genome by the presence of 39 tRNAs for these codons. Remarkably, analysis of the genome has also revealed the presence of a tRNA that is predicted to decode the remaining stop codon, UGA. Multiple lines of evidence indicate that this is a functioning tRNA for selenocysteine (Sec), the so-called 21st amino acid. In those eukaryotic species that use Sec, most UGA codons still cause translation termination while those mRNAs that encode Sec-containing peptides have a characteristic stem-loop sequence motif in the 3′ UTR region that directs Sec incorporation [59,60]. The putative T. thermophila tRNA-Sec was identified by analysis of the genome sequence and shown to be transcribed and acylated [61], and we have found that it is expressed and charged and that its charging may be under distinct regulatory control from other tRNAs (Figure S4A). In addition, we identified six T. thermophila genes with in-frame UGA codons that align (after editing of the gene models) with known Sec codons of their homologs from other eukaryotic species and that have the stem-loop consensus and thus are likely to encode selenoproteins. Thus we conclude that UGA is almost certainly translated into Sec, which would make T. thermophila the first organism known to use all 64 triplet codons to specify amino acid incorporation.
Genome Evolution
Codon and amino acid usage bias.
Although T. thermophila can use all 64 codons, it does not use all equally. The most significant aspect of the codon usage in this species is that the AT-rich codons tend to be used more frequently than others [62,63]. Thus although the AT bias in the genome is strongest in noncoding regions, where selection is thought to be relaxed, it is seen even in coding regions. In fact, the AT pull is so strong in coding regions that amino-acid composition of proteins is shifted toward those coded by codons with high AT content, as seen in other species with extreme AT bias (e.g., [64]). Although the overall codon usage is biased against GC-rich codons, on a gene-by-gene level there is significant variation in the degree of bias. We have identified two dominant patterns to this gene-by-gene variation. The major pattern is that for most genes, the codons used are simply a reflection of the overall AT content of the gene (Figure 5). The variation among genes is due to genomewide variation in AT content (see Figure 5A), although we have been unable to discern a mechanism underlying this variation (e.g., there is no clustering of high or low AT genes near telomeres). There is, however, a less common pattern in the gene-by-gene variation that is very important. There exists a subset of genes (shown in red) that use a common preferred codon set that is different from that of the average gene, and the codons in this set are not strongly correlated to the genes' AT content. Although the existence of such a preferred codon set for this species has been reported [62,63], analysis of the genome allows the set and the genes that use it to be more precisely defined. In total, using a relatively conservative cutoff (Figure 5B), we have identified 232 such genes.
Figure 5. Codon Usage.

(A) Effective number of codons (ENc; a measure of overall codon bias) for each predicted ORF is plotted versus GC3 (the fraction of codons that are synonymous at the third codon position that have either a guanine or a cytosine at that position). The upper limit of expected bias based on GC3 alone is represented by the black curve; most T. thermophila ORFs cluster below the curve [red dots as in (B)].
(B) Principal component analysis of relative synonymous codon usage in T. thermophila. The 232 genes in the tail of the comma-shaped distribution (those with the most biased codon usage) are colored red.
(C) Principal component analysis of relative synonymous codon usage in P. falciparum.
The use of preferred codons by a gene is thought to allow for more efficient or accurate translation [65]. This appears to be the case here as, of the predicted genes using the preferred subset, many have likely housekeeping functions, and, although they account for only 0.85% of all predicted genes, 12.5% of all ESTs map to them (Table S7). Although some do not have EST matches and theoretically could represent falsely predicted genes, it seems unlikely that spurious genes would use the preferred codon set. Thus we predict that these outlier genes are either highly expressed (in at least some of the conditions normally encountered by the organism) or have some critical function requiring accurate translation.
Codon usage differences between genes are thought to have only small fitness effects. For natural selection to effectively work on codon usage differences and to thus create a preferred subset, factors that enhance genetic drift (e.g., small population sizes, inbreeding) must be weaker than the selective forces [66]. Thus although codon usage is probably under selective pressure in all species, not all are able to evolve preferred codon sets. For example, although it has a similar AT bias to T. thermophila, no preferred set could be detected in the apicomplexan Plasmodium falciparum (Figure 5C), possibly a reflection of its parasitic lifestyle and limited effective population size. The presence of a preferred subset in T. thermophila is likely a reflection of a large effective population size due to its free-living, sexually reproducing lifestyle (see [66,67] for additional discussion on the large population size of this species).
No plastid-derived genes can be identified.
One question of particular interest that the T. thermophila genome might shed light on relates to the timing of the origin of the plastids found in apicomplexans and dinoflagellates, the other members of the alveolates [68,69]. Although the plastids in these lineages differ (e.g., that in apicomplexans, known as an apicoplast, is not even involved in photosynthesis), both are thought to be of red algal origin [70]. This has led to the proposal that the plastids in these lineages are the result of a single endosymbiotic event between an ancestor of apicomplexans and dinoflagellates and a red alga, with the algal nucleus being lost and the algal plastid being kept. A key question is whether this secondary endosymbiosis occurred before or after the ciliates split off from the other two lineages. The possibility that it occurred before the ciliate split is known as the chromalveolate hypothesis [71].
For the chromalveolate hypothesis to be correct, plastid loss would have to have occurred in ciliates, most likely at the base of the ciliate tree since no modern ciliates are known to harbor plastids. If the ancestor of ciliates once had a plastid, it is possible that some plastid-derived genes would have been transferred to the nuclear genome (as has occurred in many lineages including apicomplexans and dinoflagellates [72]), and furthermore that some such genes would still be found in T. thermophila. To test this possibility, we built phylogenetic trees of all genes in the genome and searched for those with a branching pattern consistent with plastid descent (see Materials and Methods). For T. thermophila, we do not see any signal for genes of plastid descent that rises above the noise seen in such automated phylogenetic analyses.
Several lines of evidence suggest that this is not a general flaw in the phylogenetic approach used here. For example, we have used the same approach to identify and catalog the plastid-derived genes in other lineages including the plant Arabidopsis thaliana and the apicomplexan P. falciparum. In addition, such an approach has been used to detect past endosymbioses in other eukaryotic lineages [73]. Finally, using the same approach we identified 91 likely mitochondrion-derived genes (Table S8) in the T. thermophila nuclear genome. This is significant because mitochondrion-derived genes are generally more difficult to identify than plastid-derived genes [74], in part because the plastid symbiosis was more recent [75].
Nevertheless, since it is possible that our phylogenomic screen might have missed some plastid-derived genes, we also did a targeted search for genes that might be expected to be retained, using the apicoplast as a model. Apicoplasts are involved in biosynthesis of fatty acids, isoprenoids, and heme. Fatty acid and isoprenoid biosynthetic pathways are of special interest because the plastid-derived pathways are distinct from analogous pathways in the eukaryotic cytoplasm [76]. In the case of isoprenoid biosynthesis, genes for proteins in the canonical eukaryotic cytosolic mevalonate pathway are present as expected based on experimental studies [77–79], but no enzymes involved in the plastid-derived DOXP pathway were evident. For fatty acid biosynthesis, while T. thermophila does not require an exogenous supply of fatty acids for growth, no evidence for a complete version of a type I (normally cytosolic) pathway could be found. Although at least some genes for a type II pathway are present, these are insufficient for de novo fatty acid synthesis and appear more likely to be derived from the mitochondrion than a plastid.
Based on the general and targeted searches, we conclude that there is presently no evidence for a plastid or ancestrally plastid-derived genes in T. thermophila. This does not preclude the possibility that other ciliates have plastid-derived enzymes or even a plastid, but there is presently no evidence to suggest this despite extensive ultrastructural observations [80,81]. If ciliates do lack all evidence of a plastid, it could either mean that the hypothesized early origin of the chromalveolate plastid is incorrect or that an ancestor of T. thermophila (and perhaps all ciliates) lost its plastid and all detectable plastid-derived genes outright. The latter possibility is not without precedent, as some apicomplexans such as the Cryptosporidia have lost their apicoplasts and have few, if any, plastid-derived genes in their nuclear genomes [82,83]. This loss has been suggested to be the result of metabolic streamlining in response to its parasitic lifestyle. Resolving whether a plastid was present in the ancestor of ciliates will be important to our understanding of the evolution of plastids and their biochemical relationship with eukaryotic hosts.
IES excision targets foreign DNA rather than repetitive DNA per se.
As discussed in the Introduction, there are multiple parallels between the IES excision process and other repeat element silencing phenomena such as RIP and heterochromatin formation. Despite these parallels, the processes differ significantly in their mechanisms of action and therefore likely have different short- and long-term evolutionary consequences. For example, in species with RIP, all repetitive DNA becomes a target for mutational inactivation, which has resulted in a drastic suppression of evolutionary diversification through gene duplication [84,85]. The IES excision process results in the exclusion of certain MIC DNA sequences from the transcriptionally active MAC. Experimental introduction of foreign transgenes into the MIC has shown that as MIC copy number increases, so does the efficiency of transgene excision [86]. One might therefore predict a similar suppression of gene duplication as in RIP. However, rather than targeting repetitive DNA per se, it has been proposed that IES excision specifically targets foreign DNA that has invaded the germline MIC but is not represented in the MAC [35,87,88]. MIC gene duplication and functional diversification should still be possible under this scenario as long as, at each conjugation event, the gene copies have not diverged in sequence enough to be recognized as foreign and excluded from the MAC; since sex is frequent in natural populations of T. thermophila [89], this should be the case. We therefore sought to use the genome sequence data to both test the foreign DNA hypothesis and to examine what the consequences of the IES excision process have been on the evolution of the T. thermophila genome.
Analysis of the genome reveals several lines of evidence that provide strong support for the foreign DNA hypothesis. First, small but nevertheless significant amounts of repetitive DNA are present in the MAC. This is best seen in analysis of the scaffolds that correspond to complete MAC chromosomes which are unlikely to contain MIC IES contamination. These scaffolds contain dispersed repeats that make up 2.3% of the total DNA. This means that some repetitive DNA bypasses the IES excision process. The second line of evidence comes from examining the small contigs and singletons (nonassembled sequences) in the assembly data. Known MIC-specific elements such as the REP and Tlr1 transposons [90,91] are found only in these small contigs, which are thus clearly enriched for MIC-specific DNA (and also for repetitive DNA; see Figure S5). In fact, the small contigs contain homologs of an unusually wide range of transposable element (TE) clades for a single-celled eukaryote [92,93] including many previously unreported in Tetrahymena (Table S9). We do not find any good matches to TEs in any of the large contigs. Thus, transposons in general appear to be filtered out very efficiently by the IES excision process. The tandem and dispersed repeats in the MAC appear to correspond to noninvasive DNA (e.g., the 5S rRNA genes). Taken together, the fact that mobile (and likely invasive) DNA elements are kept out of the MAC, combined with the fact that both tandem and dispersed noninvasive repeats avoid the excision process, indicates strong support for the foreign DNA hypothesis.
In organisms with RIP, since all duplicated DNA is targeted [94], gene diversification by duplication is suppressed. For example, the fraction of all Neurospora crassa genes found in paralogous families is only 19%, a value that falls below the overall correlation line between this fraction and total gene number [84]. In addition, very few gene pairs share greater than 80% amino acid sequence identity [84]. Consistent with the foreign DNA hypothesis, we do not see such signs of suppression of gene family diversification in T. thermophila. Large numbers of paralogous genes are found in the genome (1,970 gene families including 10,851 predicted proteins) (Table 3). The fraction of genes in such families in T. thermophila (39%) is much higher than that seen in N. crassa. Although this fraction is not as high as would be predicted from the observed correlation between total number of genes and the fraction found in paralogous families [84], the fraction of gene pairs sharing greater than 80% amino acid identity is much higher than in N. crassa and similar to that found in other sequenced eukaryotes.
Table 3.
Gene Families

Since it is possible some of the 1,970 gene families could have originated by duplications that occurred prior to the origin of the IES excision process, it is more useful to examine recent duplications. We searched for such duplications in multiple ways, including the identification of genes duplicated in the T. thermophila lineage relative to other lineages for which genomes are available (Table S10) and by searching for pairs of paralogs with very similar sequences. Both of these classes are abundant in T. thermophila, further indicating that the IES excision does not significantly affect expansion of gene families of “native” genes. Thus the ciliate system of targeting invading DNA has significantly different consequences than RIP.
High gene count in T. thermophila.
The expansion of gene families helps explain the high gene count in T. thermophila, which is higher than that of other protists and even surpasses that of some metazoans (Table 4). The duplication events appear to be spread out over evolutionary time with some being ancient and some quite recent. We searched for but did not find evidence for either whole genome or segmental duplications. We do find extensive numbers of tandemly duplicated genes. In total, 1,603 tandem clusters of between two and 15 genes were found, comprising 4,276 total genes; 67% of these clusters are simple gene pairs and 96% contain five or fewer genes. Thus it appears many of the paralogous genes in T. thermophila are the results of separate small duplication events.
Table 4.
Numbers of Protein-Coding Genes in Various Eukaryotes

The high gene count in T. thermophila relative to some other single-celled eukaryotes is not simply a reflection of gene family expansions. For example, when recent gene expansions are collapsed into ortholog sets, we find that humans and T. thermophila share more orthologs with each other (2,280) than are shared between humans and the yeast S. cerevisiae (2,097) or T. thermophila and P. falciparum (1,325) (Figure 6), despite the sister phyla relationships of animals and fungi on the one hand and ciliates and apicomplexans on the other. We note that this does not mean that humans and T. thermophila are overall more similar to each other than either is to species in sister phyla. For example, humans and S. cerevisiae do share some processes that evolved in the common ancestor of fungi and animals. In addition, for orthologs found in all eukaryotes, the human and S. cerevisiae genes are more similar in sequence to each other than either is to genes from T. thermophila. The higher number of orthologs shared between humans and T. thermophila is a reflection of both the loss of genes in other eukaryotic lineages and the retention of a variety of ancestral eukaryotic functions by T. thermophila. Consistent with this conclusion, there are 874 human genes with orthologs in T. thermophila but not S. cerevisiae, 58 of which correspond to loci associated with human diseases (Table S12). Thus genome analysis reveals many cases where T. thermophila can continue to complement experimental studies of yeast as a model system for eukaryotic (and human) cell biology [13].
Figure 6. Orthologs Shared among T. thermophila and Selected Eukaryotic Genomes.

Venn diagram showing orthologs shared among human, the yeast S. cerevisiae, the apicomplexan P. falciparum, and T. thermophila. Lineage-specific gene duplications in each of the organisms were identified and treated as one single gene (or super-ortholog). Pairwise mutual best-hits by BLASTP were then identified as putative orthologs.
Gene Duplication as an Indicator of Important Biological Processes
One motivation for obtaining the genome sequence of an organism is to advance the study of processes already under investigation. Many researchers, including those who have never worked on this species before, have taken advantage of the publicly available data in an effort to achieve this goal (e.g. [24,95–103]). Rather than focus our bioinformatic analysis on these well-studied processes, we decided to search for evidence in the predicted proteome of processes of particular importance to the organism. Our approach was relatively straightforward—we looked for overrepresentations (compared to other eukaryotes) in the lists of paralogous gene families or lineage-specific gene family expansions associated with a variety of processes. This approach was taken for several reasons. First, searches for differences in large gene families are not as biased by annotation errors as searches focused on individual genes. In addition, large gene families clearly contribute to the large number of genes present in T. thermophila compared to other single-celled eukaryotes. We note that many of the available genomes of single-celled eukaryotes are of parasites that were selected for sequencing mostly due to their medical relevance and that these are not representative (e.g., many have quite small genomes). Most important, the presence of large gene families and recent gene duplications are likely indications of functional diversity, recent evolutionary innovations, and selective pressures placed on this organism.
Our analysis of paralogous gene families and in particular the recently duplicated members of such families reveals the importance of processes associated with the sensing of and responding to environmental changes. We highlight five such processes here: signal transduction, membrane transport, proteolytic digestion, construction and manipulation of cell shape and movement, and membrane trafficking. These processes are all critical to the free-living heterotrophic lifestyle of this organism. In the following sections, we discuss what the analysis of the genome reveals about these processes in T. thermophila with a particular focus on expansions of genes associated with these functions relative to other species.
Signal transduction and the expansions of kinase families.
A variety of genes with putative roles in signal transduction were identified in our screens of paralogous genes. Of these, we chose to perform an in depth analysis of the kinases because they are such a diverse family of proteins and because they have been found to have critical roles in sensory and regulatory processes across the tree of life. In total, 1,069 predicted protein kinases (Tables 5 and S11A) were identified in the genome. This corresponds to approximately 3.8% of the predicted proteome, a fraction significantly larger than the approximately 2.3% in fungi, Drosophila, and vertebrates [104]. Among these, representatives were found of 54 of the known kinase families and subfamilies [105]. Some families found in a wide diversity of eukaryotes [106] were not detected. This includes the checkpoint kinase CHK1/RAD53, the PI3 kinase–related kinase TRRAP, two cyclin-dependent kinases (CDK7 and CDK8, which may be functionally replaced by the related expanded CDC2 family), and two poorly conserved classes (Bub1 and Haspin) that may have been missed by sequence homology searches. Despite the reported presence of phosphotyrosine in T. thermophila [107], no clear members of the tyrosine kinase group could be identified. However, the genome encodes some proteins that might be alternative tyrosine kinases including multiple dual-specificity kinases (e.g., Wee1, Ste7, TTK, and Dyrk) as well as five members of the related TKL group, which may mediate tyrosine phosphorylation in the slime mold Dictyostelium discoideum [106]. Twelve kinase classes are found in T. thermophila and humans but not yeast, and thus are apparent examples of the retention of ancestral eukaryotic functions discussed above. Several of the genes in these classes have been implicated in the etiology of human disease (Dyrk1A, DNAPK, SGK1, RSK2, Wnk1, and Wnk4) [108].
Table 5.
Distribution of Selected Protein Kinase Classes in T. thermophila and Other Classified Kinomes

A key feature of the T. thermophila kinome is the expansion of several kinase classes relative to other sequenced organisms (Table 5). The implications of some of these expansions can be predicted based on the known functions of family members. For example, the mitotic kinase families Aurora, CDC2, and PLK are all substantially expanded, perhaps reflecting the additional signaling complexity required by two nuclei that simultaneously engage in very different processes within the same cell cytoplasm. Also expanded are multiple kinases that interact with the microtubule network [109,110] [e.g., Nima-related kinases (NRKs) and the ULK family], possibly reflecting diversification of cytoskeletal systems (discussed more below). Of the kinase families with known functions, the most striking expansion is the presence of 83 histidine protein kinases (HPKs), which are generally involved in transducing signals from the external environment [111]. HPKs are found predominantly in two-component regulatory systems of bacteria, archaea, protists, and plants and are absent from metazoans. Most of the T. thermophila HPKs have substrate receiver domains, and many are predicted to be transmembrane receptors.
The full meaning of the kinome diversity in T. thermophila is hard to predict as a great deal of the diversification has occurred in classes for which the functions are poorly understood. For example, in many of the known kinase families, the T. thermophila proteins are highly diverse in sequence, both relative to those in other species as well as to each other (e.g., see Figure S6). The scope of the diversification in T. thermophila is perhaps best seen in the fact that 630 (approximately 60%) of the kinases could not be assigned to any known family or subfamily [105]. Overall, 37 novel classes of kinases and hundreds of unique proteins were identified in this genome. The presence of so many novel kinases and expansions in many known classes of kinases is both an indication of the versatility of the eukaryotic protein kinase domain seen in other lineages [112] and suggestive of a great elaboration of ciliate-specific functions.
Diversification of membrane transport systems.
Many of the most greatly expanded T. thermophila gene families encode proteins predicted to be involved in membrane transport. Membrane transporters play critical roles in responding to variations in the environment and making use of available resources. We therefore conducted a more thorough analysis of the predicted transporters in this species. Overall, T. thermophila possesses a robust and diverse collection of predicted membrane transport systems (Tables 6 and S11B). Comparison to other eukaryotes [113] reveals some interesting differences in terms of both classes of transporters and predicted substrates being moved. For example, T. thermophila has more representatives in each of the four major families than do humans. In addition, it encodes a much higher number of transporters in the ABC superfamily, voltage-gated ion channels (VICs), and P-type ATPases than any other sequenced eukaryotic species (Table 6) including the other free-living protists, the diatom Thalassiosira pseudonana, and the slime mold D. discoideum. Regarding substrates, an extremely extensive set of transporters likely specific for inorganic cations has been identified (Table 6). Most of these are channel-type transporters and cation-transporting P-type ATPases. Interestingly, despite the apparent massive amplification of cation transporters, T. thermophila has a very limited repertoire of transporters for inorganic anions: only one member each for sulfate, phosphate, arsenite, and chromate ion were identified, and there are no predicted anion channels. The reason for the difference in the amplification of cation versus anion transporters is unclear.
Table 6.
Comparison of the Numbers of Membrane Transporters in T. thermophila and Other Eukaryotes by Family and Predicted Substrate

As with kinases, some of the most interesting properties are revealed by examination of the lineage-specific duplications of transporters. The recent clusters include K+ channel proteins (285 members), ABC transporters (152 members), cation-transporting ATPases (59 members), K+ channel beta subunit proteins (22 members), oxalate:formate antiporters (24 members), sugar transporters (22 members), and phospholipid-transporting ATPases (20 members). The expansion of the K+ channel proteins, which are VIC-type transporters, was particularly large and was pursued further.
In total, 308 VIC-type K+-selective channels have been predicted, many more than in any other sequenced species and over three times as many as identified in humans (89). A multigene family of potassium ion channels has also been identified in P. tetraurelia [114] and thus may be a general characteristic of some ciliates. Some lines of evidence suggest that this expansion in ciliates could be adaptive. First, K+ channels control the passive permeation of K+ across the membrane, which is essential for ciliary motility [115]. Second, a novel adenylyl cyclase with a putative N-terminal K+ ion channel regulates the formation of the universal second messenger cAMP in ciliates and apicomplexans [116,117], which could assist in responding to sudden changes of the ionic environment. T. thermophila encodes six homologs of this adenylate cyclase/K+ transporter, whereas the parasitic apicomplexans P. falciparum and Cryptosporidium parvum encode only one each.
The robust transporter systems present are likely a reflection of T. thermophila's behavioral and physiological versatility as a free-living single-celled organism and its exposure to a wide range of different substrates in its natural environment. Examination of the specific types of expansions suggests that functions associated with transport of K+ and other cations have been greatly diversified. Thus such functions may play a role in many of the unique aspects of the biology of this species and ciliates in general.
Proteolytic processing.
T. thermophila is a voracious predator and thus might be expected to have a wide diversity of proteolytic enzymes. Analysis of the predicted proteins in T. thermophila reveals some conflicting results relating to this idea. On the one hand, many of the largest clusters of lineage-specific duplications are of proteases (e.g., papain, leishmanolysin). On the other hand, the total number of proteases identified (480) is relatively low in terms of the fraction of the proteome (1.7%) compared to other model organisms that have been sequenced and annotated [118–120]. The conflict is most likely a reflection of the diversity of physiological processes in which proteases function [121]. Thus we examined the subclassification of types of proteases present in more detail.
Using the Merops protease nomenclature, which is based on intrinsic evolutionary and structural relationships [119] the T. thermophila proteases were divided into five catalytic classes and 40 families. These are: 43 aspartic proteases belonging to two families, 211 cysteine proteases belonging to 11 families, 139 metalloproteases belonging to 14 families, 73 serine proteases belonging to 12 families, and 14 threonine proteases belonging to the T1 family (Tables 7 and S11C). Some unique features of T. thermophila can be seen by comparison to P. falciparum which is the most closely related sequenced species to have a detailed analysis of its proteases published [122]. Twenty-one protease families are present in both genomes. For example, the highly conserved threonine proteases and the ubiquitin carboxyl-terminal hydrolase families (C12 and C19) reflect the crucial role of the ATP-dependent ubiquitin-proteasome system, which has been implicated in cell-cycle control and stress response [123]. Nineteen protease families are present in T. thermophila but not P. falciparum. One of these includes leishmanolysin (M8), originally identified in the kinetoplastid parasite Leishmania major and thought to be involved in processing surface proteins [124–126]. This family is greatly expanded (to 48 members, including 15 in a tandem array) in T. thermophila and suggests that surface protein processes may be important here, although the functions of leishmanolysin-related proteases in nonkinetoplastid eukaryotes remain unclear. The carboxypeptidase A (M14) and carboxypeptidase Y (S10) families are expanded to 28 and 25 members, respectively, in T. thermophila, which may reflect numerous and diverse functions. Only four protease families present in P. falciparum are not found in T. thermophila. Among these are metacaspase (C14), an ancestral type of caspase that is characteristic of apoptosis or apoptosis-like signal transduction pathways [127].
Table 7.
Protease Complements in T. thermophila and Other Model Organisms

The largest clusters of expanded proteases in T. thermophila are all cysteine proteases, which comprise 44% of the total protease complement. The two most prominent families from this class are the papain family (C1), which is the most abundant and complex family, with 114 members, and the ubiquitin carboxyl-terminal hydrolase 2 family (UCH2, C19) with 47 members. It is possible that the biochemical activity among the paralogs within these families is conserved but that they are used in different parts of the cell (or outside the cell) or in different developmental stages in T. thermophila.
Cytoskeletal components and regulators.
Ciliates have highly complex cytoskeletal architecture [128] with highly polarized cell types which assemble 18 types of microtubular organelles in specific locations along the anteroposterior and dorsoventral axis. We therefore sought to determine whether this diversity was reflected in the genome. As with the protease analysis described above, initial comparisons of the number of particular types of cytoskeletal and microtubule-associated proteins was somewhat ambiguous (the numbers for humans and T. thermophila are shown in Tables 8 and S11D). For example, although kinesin and dynein motors as well as kinases associated with microtubules appear to be expanded, structural components of the cilia and participants in the intraflagellar transport pathway are not. In addition, some cytoskeletal protein types are apparently absent from T. thermophila; these include intermediate filament proteins (including nuclear lamins) as already suggested by biochemical studies [129], some microtubule-associated proteins (MAP2, MAP4, and Tau, for which no nonanimal eukaryotic homologs have been found) and some actin-binding proteins (e.g., α-actinin). To better understand what role genes involved in microtubule and cytoskeletal functions might have played in the diversification of this species, we focused analysis on some of the genes with apparent expansions: tubulins, dyneins, and regulatory proteins.
Table 8.
Numbers of Loci Encoding Selected Types of Cytoskeletal Genes in T. thermophila and H. sapiens
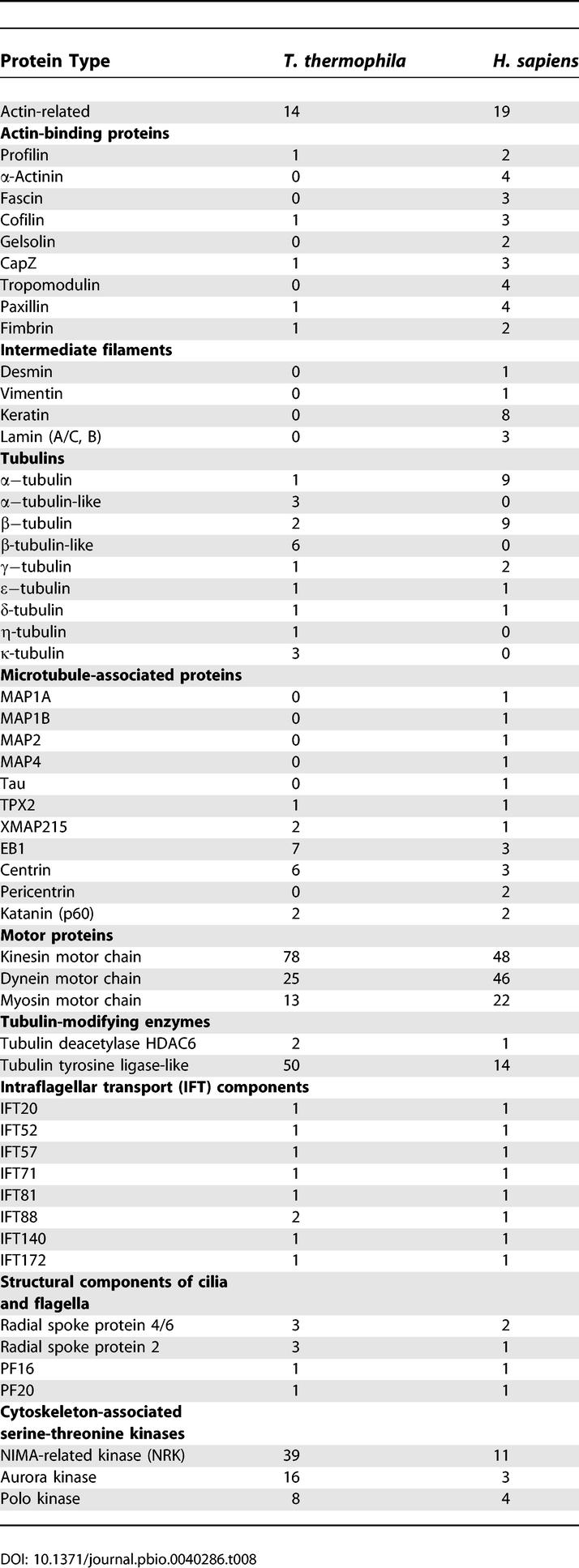
Tubulins.
Tubulins are the key structural components of microtubules and they come in many forms in eukaryotes [130]. In the T. thermophila genome, phylogenetic analysis of tubulin homologs (Figure 7) reveals the presence of one or two genes, each within the essential alpha (α), beta (β), and gamma (γ) subfamilies (as reported previously [131–133]) and one in each of the delta (δ), epsilon (ɛ), and eta (η), which are found in organisms that possess centrioles/basal bodies [134–136]. In addition, T. thermophila encodes noncanonical tubulin homologs that can be divided into two categories. In the first category are genes that are most similar to the canonical α- or β-tubulins. These nine genes (three α-like and six β-like) lack characteristic motifs for the tail domain post-translational modifications (polyglutamylation and polyglycylation) that are essential to the function of their canonical counterparts [137–139]. Three of the β-like genes (BLT1/TTHERM_01104960, TTHERM_01104970, and TTHERM_01104980) form a tandem cluster with intergenic intervals of less than 2 kb. We hypothesize that these genes function, perhaps redundantly, in formation or function of some of the many highly specialized microtubule systems of T. thermophila cells. Experimental analysis of BLT1, a β-like tubulin, indicated that its product localizes to a small subset of microtubules and is not incorporated into growing ciliary axonemes (K. Clark and M. Gorovsky, unpublished data). Genetic deletion of this gene or of the α-like gene TTHERM_00647130 did not yield an obvious phenotype (R. Xie and M. A. Gorovsky, unpublished data).
Figure 7. Tubulin Gene Diversity in T. thermophila .

The figure shows a neighbor-joining tree built from a clustalX alignment. Species abbreviations: Hs, H. sapiens; Dm, D. melaogaster; Sc, S. cerevisiae; Tt, T. thermophila; Pt, P. tetraurelia; Cr, C. reinhardtii; Tb, T. brucei; Ec, E. coli; Xl, Xenopus laevis. A prokaryotic tubulin ortholog, Escherichia coli FtsZ, was used as the outgroup.
The second category of noncanonical tubulin homologs consists of three novel proteins (TTHERM_00550910, TTHERM_01001250, and TTHERM_01001260) that fall into a clade with P. tetraurelia iota tubulin. Two of these (TTHERM_01001250 and TTHERM_01001260) are closely related to each other (Figure 7) and closely linked in the genome and thus likely arose by a recent tandem duplication. The functions of these genes are unknown, but because they are, so far, unique to ciliates, they might be responsible for microtubule functions specific to this phylum.
Dyneins.
Dyneins, which were first discovered in Tetrahymena [140], are molecular motors that translocate along microtubule tracks, a process critical to many activities in T. thermophila including ciliary beating, karyokinesis, MAC division, cortical organization, and phagocytosis. Many of these activities are critical for sensing and responding to changes in the environment. Each dynein complex consists of one, two, or three heavy chains (containing the motor activity) and specific combinations of smaller subunits, including intermediate, light-intermediate, and light chains, which regulate motor activity and the tethering of dynein to its molecular cargo [141–143]. In organisms with cilia or flagella, there are multiple isoforms of dyneins, including the axonemal outer arm dyneins, the axonemal inner arm dyneins, and nonaxonemal or “cytoplasmic” dyneins. Each is specialized in its intracellular location and the cellular task it performs [144].
In total we identified 21 light chains, five intermediate chains, two light-intermediate chains, and 25 heavy chains (Table S13). The expression of each gene, as well as the exon/intron structures of most, was confirmed by RT-PCR and, if necessary, sequencing of the RT-PCR product. For the most part, the families of T. thermophila dynein subunits appear to be similar to those of other model organisms; however, there are some interesting differences. T. thermophila light chains LC3A and 3B are most similar to the green alga Chlamydomonas reinhardtii's LC3 and LC5 [145]. These proteins belong to the larger family of thioredoxin-related proteins, and, without biochemical evidence identifying one or both of the proteins as part of a dynein complex, it may be premature to label these as dynein components. Light chain LC4 belongs to the calmodulin-related family of proteins and may regulate calcium-dependent ciliary reversal. T. thermophila expresses two LC4 genes, perhaps providing alternative or additional ways to control ciliary motility compared to species that express only one. In other systems, LC8 is associated with several different dynein and nondynein complexes, and T. thermophila expresses one canonical LC8 as well as five divergent LC8-like genes, with unknown functions.
Perhaps the most interesting revelation is that T. thermophila expresses 25 dynein heavy chains. These include the 14 DYH genes previously described [146,147] and 11 new ones, all of which appear to be axonemal. The complexity of the DYH family may represent a mechanism by which the organism can fine-tune ciliary activity, produce specialized cilia (e.g., oral and posterior cilia), and/or generate large numbers of new cilia quickly. Along these lines, there has also been an expansion in other motor proteins. For example, there are 78 kinesins, more than in any other sequenced organism ([101] and Table 8). In addition, although there are fewer myosins than in humans (13 versus 22), 12 of 13 of the T. thermophila genes comprise a single novel myosin class not found in other organisms [102,148].
Regulation of microtubules and microtubule-associated processes.
Among the expanded genes in T. thermophila are a variety implicated in the regulation of microtubules or microtubule-associated processes. One example is the tubulin tyrosine ligase-like domain proteins of which multiple members have been identified as enzymes responsible for polyglutamylation of either α- or β-tubulin [149]. T. thermophila encodes 50 tubulin tyrosine ligase-like proteins compared with 14 in human. Another example is the NRK family of protein kinases which, as mentioned above, has undergone a large expansion in T. thermophila. NRKs are often found associated with microtubular organelles [150] such as centrioles, basal bodies, and flagella and play multiple roles, including the regulation of centrosome maturation [151] and flagellar excision [152]. We identified 39 NRKs in T. thermophila, roughly three times the number of such loci in humans. Phylogenetic and functional analyses have suggested that this diversification has adapted the members of this family for distinct subcellular localizations and cytoskeletal roles [103]. Thus, such gene expansions could allow differentially targeted protein isoforms to regulate the function of the same organelle type in different locations or generate different properties of the same structural building materials (e.g., microtubules), which are used as frameworks to build different types of organelles.
Secretory pathways and membrane trafficking.
Besides the conventional organelles, T. thermophila maintains several more specialized membrane-bound compartments, including alveoli (shared with other alveolates), a contractile vacuole (found in many protists), and separate, functionally distinct macronuclei and micronuclei [128]. It also has multiple pathways for plasma membrane internalization, as well as both constitutive and regulated exocytosis [128,153]. The sorting and trafficking of membrane components are critical functions for all these activities. Analysis of the genome reveals homologs of many of the key proteins known from other eukaryotes to be involved in vesicle formation and fusion, including all major classes of coat proteins (Table S14). One interesting finding that came from genome analysis is that T. thermophila encodes eight dynamin-related proteins, more than most other sequenced unicellular eukaryotes, and two of them, Drp1p and Drp2p, have evolved a new function in endocytosis [96] (A. Rahaman and A. P. Turkewitz, unpublished data). Furthermore, phylogenetic analysis indicated that the recruitment of dynamin to a role in endocytosis occurred independently by convergent evolution in the animal and ciliate lineages [96].
The diversification of membrane trafficking is more apparent in regard to Rab proteins, which are small monomeric GTPases that regulate membrane fusion and fission events. T. thermophila, with 69 Rabs (Table S15), has a number more along the lines of humans (which have 60) than many single-celled species, such as Saccharomyces cerevisiae, which has 11 [154] and Trypanosoma brucei, which has 16 [155]. Based on localization and functional studies, including comparisons between yeast and humans [156], Rabs have been divided into eight groups [157]. Phylogenetic analysis (Figure S7) indicates that T. thermophila encodes representatives of all but groups IV and VII, which are involved in late endocytosis and Golgi transport, respectively. For group VII this appears to reflect a lineage-specific loss, since the genomes of both T. brucei and Entamoeba histolytica have several homologs in this group. Two T. thermophila Rabs appear homologous to Rab28 and Rab32, which have not been assigned to any of these groups; Rab32 was previously thought to be restricted to mammalian lineages. Rab groups II and V, involved in endocytosis, are especially large in T. thermophila and include several Rab2, Rab4, and Rab11 homologs in group II. This may reflect the intricacy of maintaining at least two major pathways of membrane internalization. Additionally, 29 Rabs in T. thermophila fail to cluster with any of the Rab groups found more widely among eukaryotes. Within this group, 20 cluster into three clades, designated Tetrahymena clades I, II, and III in Figure S7, which may represent ciliate-specific radiations. The remaining nine are very divergent and may represent very ancient duplication events and/or changes related to recruitment for novel function. Because unambiguous alignment among such divergent Rabs is difficult, their relationships will become clearer as additional related genomes are sequenced.
Recently, large numbers of Rabs have been found in a variety of amoeboid protists including D. discoideum, E. histolytica [158], and the parabasalid Trichomonas vaginalis [159]. The diversification in these species was proposed to relate to their amoeboid lifestyle [159]. However, the presence of significant diversification in T. thermophila suggests that different protist lifestyles may be accompanied by their own brand of significant Rab diversification.
Tetrahymena Genome Database
An integral part of the effort to make the genomic resources and analyses described above widely available to researchers working with T. thermophila and other organisms has been the creation of the Tetrahymena Genome Database (TGD; http://www.ciliate.org), a Web-accessible resource on the genetics and genomics of T. thermophila. TGD provides information about the T. thermophila MAC genome, its genes and gene products, facts about the ciliate scientific community, and tools for querying the genome and collected scientific literature. TGD was created using the database environment developed for the Saccharomyces Genome Database and software tools contributed to the Generic Model Organism Database (GMOD) project.
Information from the published literature on T. thermophila is distilled in multiple ways. Results from published studies of T. thermophila genes are curated and provided, including community-approved gene names, other nonstandard aliases, nucleotide and amino acid sequences, and literature citations. In addition, free-text descriptions are associated with predicted gene models, and full-text searching is provided using Textpresso [160]. To enable intra- and cross-species comparisons, when information on characterized genes is curated, TGD staff members capture aspects of a gene product's biology (i.e., molecular function, biological role, and cellular localization) using terms from the Gene Ontology (http://www.geneontology.org). This is complemented by automated functional annotation of all predicted genes. Other resources include tools for searching the annotation by keywords, similarity searching using BLAST and BLAT, Gbrowse-based genome visualization [161], information about Tetrahymena research laboratories, links to other ciliate-related resources, and various tutorials. The TGD staff is always available to help individual researchers by answering questions, finding information, and generating datasets specific to their needs.
Conclusions and Future Plans
In sequencing and assembling the T. thermophila MAC genome, there were many anticipated major challenges not commonly seen in eukaryotic genome projects. Overall, however, the assemblies are remarkably accurate and represent excellent coverage of the genome. This is likely in large part due to low levels of repetitive DNA, one of the features of the MAC genome that initially led us to select it for sequencing. The sequence data in our current assemblies are certainly complete enough for detailed analyses of the predicted biology of this species as we have reported here and others have shown. In addition, the genome sequence is already being used in many functional genomic studies taking advantage of the powerful experimental tools available. Along these lines, it will be of great value to do comparative analyses with the genome sequences of other ciliates such as P. tetraurelia and Oxytricha trifallax, which are in progress.
One of our main goals is to obtain a complete sequence of the MAC genome, and there are still some challenges left to its achievement. Since we were unable to obtain quality sequence data from large insert clones, any region of the MAC genome containing significant amounts of repetitive DNA would not have assembled well. To overcome this pitfall we are now using HAPPY mapping [162] as an alternative approach to obtaining such linking information. Also, it is known that at least the ends of at least two MAC chromosomes present immediately following conjugation disappear during subsequent vegetative growth, perhaps an indication that these chromosomes are incapable of long-term maintenance [41]. As expected, we do not find sequences corresponding to these ends in our database. Thus alternative methods will be required to obtain the sequences of these regions and any others lost during early vegetative growth. Despite these challenges, all the evidence suggests that it will be possible to close the entire MAC genome.
Of course, the entire MAC genome alone does not provide us with a complete picture of the T. thermophila genome. Sequencing the MIC genome will be more challenging due to the greater abundance of repetitive DNA. However, we will be able to use the MAC genome as a scaffold and thus in a way MIC sequencing will be equivalent to genome closure rather than an independent project. We have already begun in this area by determining the sequence adjacent to MIC Cbs junctions and mapping these to MAC assemblies as well as the reverse—using MAC telomere-adjacent sequences to pull out MIC Cbs-flanking regions [24,41].
Having a MIC sequence and mapping the MIC to the MAC will be useful in understanding many aspects of T. thermophila biology that we cannot study through the MAC. These include centromere function, MIC telomere features, and the extent to which the MAC and MIC in T. thermophila and other ciliates are the equivalent of somatic and germ cells. Perhaps most important, having both genomes will allow detailed analyses of the genome-wide DNA rearrangement process. It is only by having both genome sequences that we can fully understand the biology of this fascinating species.
Materials and Methods
Cell growth, DNA isolation, and library construction.
T. thermophila cell lines currently in laboratory use were first isolated from the wild in the 1950s [163] and were maintained by serial passage and inbreeding for over 16 y before viable freezing methods were developed. Strain SB210 [164] is the end result of about 25 sexual reorganizations in laboratory culture, including a series of sexual inbreedings by the equivalent of brother-sister matings giving rise to the inbred strain B genetic background [165]. Following the final conjugation, a thoroughly assorted cell line was isolated after at least three serial single-cell isolations (SCIs). The last SCI was approximately 150 fissions after conjugation. These serial SCIs provided abundant opportunity to isolate a cell line that had become pure for most of the MAC developmental diversity but not necessarily all because assortment brings about a stochastic, exponential decay in diversity. The chosen cell line was then subjected to a genomic exclusion cross [166], which generates a whole-genome homozygous MIC but does not generate a new MAC. At least one additional SCI occurred at this step, after which this cell line was frozen. As needed, frozen stocks were replenished following a minimal number of vegetative fissions. The strain has been deposited in the Tetrahymena Stock Center at Cornell University as suggested [167].
A culture was started from a fresh thaw of strain SB210. Purified macronuclei were prepared by differential sedimentation, and DNA was extracted from the purified macronuclei as described [168]. The preparation was checked by Southern blot hybridization to verify that the level of contamination with MIC DNA was low. Genomic libraries were prepared as described [169]. DNA was randomly sheared, end-polished with consecutive polynucleotide kinase and T4 DNA polymerase treatments, and size-selected by electrophoresis in 1% low-melting-point agarose. After ligation to BstXI adapters (Invitrogen, Carlsbad, California, United States; catalog No. N408–18), DNA was purified by three rounds of gel electrophoresis to remove excess adapters, and the fragments, now with 3′-CACA overhangs, were inserted into BstXI-linearized plasmid vector (pHOS2, a medium-copy pBR322 derivative) with 3′-TGTG overhangs. Libraries with average sizes of inserts were constructed: 1.8, 2.5, 3.5, 5.0, and 8.5 kb (Table S1). Libraries with larger insert sizes were unstable, presumably due to the high AT content in the genomic DNA.
Sequencing was done from paired-ends primarily at the J. Craig Venter Science Foundation Joint Technology Center. Possible contaminating sequences from other projects have been filtered out using BLASTN searches against all other genome projects conducted at the same time at TIGR and the Joint Technology Center. Whole genome assemblies were performed using the Celera Assembler [37] with modifications implemented by researchers at the J. Craig Venter Science Foundation and TIGR. Sequence reads corresponding to the mitochondrial and rDNA chromosomes were identified using the latest version of the MUMmer program [170] and comparison to the published sequences.
Linking open ends of assembled scaffolds to telomeres.
The initial assembly contained 85 telomere-capped scaffold ends. However, these ends correspond to a minority of the total number of non–rDNA telomere–containing sequence reads, which we estimate to be 4,058. Computational and experimental methods were used to identify and confirm scaffold ends that were very close to a telomere, marking the end of a chromosome.
One method matched read-mates of telomere-containing reads (Tel-reads) that the assembly program failed to incorporate into scaffolds. These were identified by searching the sequence read database for exact matches to a 12-mer encompassing two telomeric repeats (GGGGTTGGGGTT). Read-mates were identified for 95% of the Tel-reads. Two internal 40-nt tags were extracted from each Tel-read mate and tested for at least one exact match with the terminal 5 kb of every scaffold (or the entire scaffold if less than 10 kb long). After clustering the matches, a nonredundant list of Tel-linked scaffold ends was generated.
A second method matched previously identified MIC DNA sequences flanking cloned Cbs junctions to scaffold ends (see Figure 2). Telomeres are added within 30 bp of the Cbs element. Thus, if Cbs-adjacent sequence from MIC DNA can be aligned with a MAC scaffold end, the end can be inferred to be telomere-linked. BLASTN searches were carried out with the “no filter” option because very AT-rich sequence was being compared.
A third method involved PCR walking from scaffold open ends to telomeres. Primers designed from scaffold ends were used in combination with the generic 14-nt telomere primer, 5′-CCCCAACCCCAACC-3′. The authenticity of each PCR product was confirmed by sequencing.
Cloning and sequencing RAPDs and sizing their associated MAC chromosomes.
Conditions and reagents for RAPD PCR were as in [171]. The 10-mer primers were from Operon Technologies. The polymorphic RAPD PCR products were size-fractionated by electrophoresis in a 1.5% agarose gel. Polymorphic bands were excised and the DNA was extracted with a QIAquick gel extraction kit (Qiagen, Chatsworth, California, United States). The DNA was reamplified using the same PCR conditions and primer combination initially used to detect the polymorphism. Amplified fragments were cloned into the pCR2.1-TOPO vector (Invitrogen) according to the manufacturer's directions. Insert-containing clones, identified as white colonies, were screened for insert size by colony PCR as in [172]. The authenticity of each correctly sized insert was confirmed by hybridization to a Southern blot of RAPD products from a panel of ten Tetrahymena strains in which the alleles of the RAPD locus were meiotically segregating [40].
Plasmid DNA was isolated using a QIAprep Miniprep kit (Qiagen, Valencia, California, United States), and inserts were sequenced using the Big Dye Terminator Cycle-Sequencing-Ready Reaction kit (PE Applied Biosystems, Foster City, California, United States). Nucleotide sequences were determined using an ABI 310 Genetic Analyzer. Insert sequences were then searched against the assemblies using BLASTN.
High-molecular-weight DNA was prepared by embedding live cells from strain SB210 in agarose plugs and lysing them using a modification of Birren and Lai [173]. The DNA plugs were inserted into the wells of a 1% Pulsed Field Certified Agarose gel (Bio-Rad, Hercules, California, United States) in 1× TAE buffer. Preliminary sizing of MAC chromosomes was obtained from gels run using the following conditions: 30 h at 6 V/cm with a 60- to 120-s switch time ramp at an included angle of 120°, 1× TAE recirculated at 10 °C. Running conditions were varied when the above conditions did not provide adequate resolution in the size range of a particular MAC chromosome (E. P. Hamilton, unpublished data). The DNA in the gel was acid-depurinated, neutralized, and transferred to a positively charged nylon membrane by downward alkaline transfer (CHEF-DR III Instruction Manual and Applications Guide; Bio-Rad). After blotting, the DNA was crosslinked to the membrane using a Bio-Rad GS Gene Linker. 32P-labeled probes were made from the PCR products obtained from each RAPD clone. Methods for making probes, Southern hybridization, and autoradiography were as in [40].
cDNA library construction and sequencing.
cDNA libraries were generated from cells in either the conjugative or vegetative stages of the life cycle. For the conjugative library, cells from a mating between strains CU428 and B2086 were harvested at 3, 6, and 10 h after mixing, and RNA was purified using TRIzol. PolyA+ RNA was isolated and cDNA was generated by Amplicon Express (Pullman, Washington, United States). Inserts were cloned into EcoRI and XhoI sites in pBluescript IISK+ (Stratagene, La Jolla, California, United States) and had an average size of 1.4 kb. Clones were picked at random and sequenced from the 5′ end of the transcript using the T3 primer. For the vegetative library, which was made by DNA Technologies (Gaithersburg, Maryland, United States), CU428 cells were harvested in exponential growth and RNA was purified using TRIzol. PolyA+ mRNA was isolated using oligo(dT) cellulose, cDNA was generated, and inserts were cloned into the EcoRV and NotI sites of the pcDNA3.1(+) vector (Invitrogen). Clones were picked at random and sequenced from the 5′ end using the custom pcDNA(−48) primer. All sequences were submitted to the dbEST division of GenBank, to the Taxonomically Broad EST Database (TBestDB) at http://tbestdb.bcm.umontreal.ca/searches/login.php, and to TIGR's Tetrahymena Gene Index at http://www.tigr.org/tigr-scripts/tgi/T_index.cgi?species=t_thermophila. Subsequent analyses used comparisons of the conjugative sequences with all vegetative sequences including those in GenBank not generated at TIGR.
Functional ncRNA analysis.
Most ncRNA annotations (Table S6) were generated using covariance model (CM) scans [174]. Transfer RNA annotations are those provided by the CM-based tRNAscanSE program [175] run with default parameters. Most other scans were based on CMs defined by the Rfam database [176,177] (release 7.0, March 2005; 503 families). With a few exceptions, we used rigorous filters [178] built from the Rfam models to identify exactly those sequences that match the Rfam models with scores at or above Rfam's family-specific “gathering” cutoff. One exception was RF00005 (tRNA), as mentioned above. Another exception was RF00012, the U3 small nucleolar RNA, for which the Rfam model found no hits. Instead, we manually added one known Tetrahymena U3 sequence [179] to the Rfam seed alignment, built a CM from it, and rescanned the genome, finding the four U3 sequences reported in Table S6C. The third class of exceptions consisted of the 44 Rfam families using the “local alignment” feature of CMs. These families were scanned using ML-heuristic filters [180], with a scan threshold chosen for each such family such that approximately 1% of the genome was scored by the CM. This setting generally shows good sensitivity but is not guaranteed to find all sequences that match the Rfam model, unlike the rigorous scans above. Hits against the Rfam T_box (RF00230), group I self-splicing introns (RF00028), and ctRNA_pND324 (RF00238) involved in bacterial plasmid copy control all appear implausible and are also unexpected by phylogenetic criteria. Hits against Rfam small nucleolar RNAs (RF00086, RF00133, RF00309) also appeared to be false positives, as were most hits to the iron response element (RF00037) and selenocysteine insertion sequence (RF00031) families. Other families not discussed here or in Table S6 yielded no hits above threshold. See http://www.cs.washington.edu/homes/ruzzo/papers/Tthermophila for full details about the ncRNA scans. It should be noted that our annotation approach may be prone to reporting ncRNA pseudogenes and that its accuracy may be affected by the high AT content of the genome.
Protein-coding gene finding and coding region analysis.
The gene finder TIGRscan ([181], since renamed GeneZilla) was trained for T. thermophila using a two-phase bootstrapping process [182], due to the dearth of curated training data available at the time. In the first round of training (termed “long-ORFs”), all parameters were estimated from a set of 193 full-length cDNAs from the apicomplexan P. falciparum (including surrounding regions from the genomic sequence; 1.6 Mb total) except for the exon state, which was trained on 2,130 nonoverlapping, long ORFs (each at least 3,000 bp in length). The default polyadenylation signal state and TATA-box state for this gene finder utilize human TRANSFAC weight matrices [183]; these were not modified. The gene finder was then used to predict genes in the raw T. thermophila genomic sequence, and the predictions were used to bootstrap the parameter estimation during the second round of training (termed “hybrid”). Sixty curated T. thermophila genes which became available during the second round of training were analyzed and their coding statistics were used to improve the exon state by averaging with the original long-ORF statistics, appropriately weighted to eliminate length bias. Exon length distributions were estimated from the 60-gene set, with appropriate smoothing. Interpolated and noninterpolated Markov chains [184] were utilized by the content states, with the order of dependency (3rd for exons and introns, 0th for intergenic, and 1st for UTR) selected so as to optimize prediction accuracy on the 60-gene set. Splice site and start/stop codon states were re-trained from pooled data consisting of the 60 curated genes and the original P. falciparum training data, using an 80%:20% T. thermophila/P. falciparum weighting to mitigate the effects of overtraining due to small sample sizes in the sixty gene set. Weight matrices utilized by the latter states were reduced to approximately 22 bp when it was noticed that longer matrices interfered with the prediction of short introns. The “hybrid” and “long-ORFs” parameterizations were tested on a set of 300 partial genes inferred from ESTs that were assembled against the chromosomes using the PASA program [52]. The “hybrid” parameterization was chosen because it was about three times more accurate at the exon level than “long-ORFs” (see Table S16).
Multivariate analysis of codon usage was performed with the codonW package (http://codonw.sourceforge.net). Correspondence analysis of relative synonymous codon usage values was carried out to examine the major source of codon usage variation. Amino acid composition of the predicted aggregate proteome was compared with the corresponding data downloaded from dictyBase for the slime mold D. discoideum and from Ensembl for Homo sapiens.
To find candidate tandem gene duplicates, we analyzed pairwise alignments between neighboring genes using BLASTP. An all-versus-all BLASTP search was performed using all Tetrahymena gene-encoded proteins, requiring a maximum E-value of 1e−20, and reporting the best 20 matches. Matching genes found at adjacent genome locations were chained together and reported as candidate tandem gene arrays, allowing only a total of two nonmatching genes to intervene matching genes in a single array.
A Lek clustering algorithm [169] was applied for paralogous gene family classification of the predicted proteins in the T. thermophila genome. All predicted proteins were searched with BLASTP against each other. Links were established between genes at an E-value cutoff of 1 × 10−20. Lek similarity scores, which were defined as the number of BLASTP hits shared by any pair of proteins divided by the combined number of hits for either of the two genes, were calculated for all pairs of proteins. The links for which the Lek similarity scores were above a cutoff of 0.66 were used to build gene family clusters by a single-linkage clustering algorithm. Biological function roles were assigned to the gene families based on the top BLASTP hits for individual genes in each family against a nonredundant protein database.
Organelle-derived genes and APIS.
Searches for plastid and mitochondrial related genes were performed using the APIS program. APIS (J. H. Badger, unpublished data) is a system that automatically generates and summarizes phylogenetic trees for each gene in a genome. It is implemented as a series of Ruby scripts, and the results are viewable on an internal Web server which allows the user to explore the data and results in an interactive manner. APIS obtains homologs by comparing each query protein against a database of proteins from complete genomes, and extracting the full length sequences of homologs with E-values less than 1e−10. The homologs are then aligned by MUSCLE [185] and bootstrapped neighbor-joining trees are produced using QuickTree [186]. As QuickTree (unlike most programs) produces bootstrapped trees with meaningful branch lengths, the trees are then midpoint rooted. Then a taxonomic analysis is performed of the proteins that are neighbors in the tree with the query protein. This analysis makes use of the NCBI taxonomy assigned to the other proteins in the tree. For each taxonomic level (e.g., kingdom, phylum, class, etc.), the query protein is assigned to a bin. If in the tree the query protein is within a clade of sequences that are all from group X (for the taxonomic level being examined) then the query protein is placed in a bin labeled “contained within group X.” If the query protein branches next to (but not within) a clade of sequences from the same group, it is placed in a bin labeled “outgroup of X.” If the neighbors of the query sequence are in multiple groups, no binning is done for that taxonomic level.
Candidates for mitochondrially derived genes were separately identified by BLASTP searches using known mitochondrial proteins as queries [187,188]. Phylogenetic trees were then constructed for individual candidates in the context of all completely sequenced genomes and representatives of mitochondria. Genes whose closest neighbors were exclusively α-proteobacteria and/or mitochondria were classified as possibly mitochondrion derived.
Analysis of repetitive DNA and TEs.
The location and characterization of tandem minisatellite and microsatellite repeats were done using Tandem Repeats Finder [189], using the default parameter values. The location, length, period size, %GC, and consensus sequence of each repeat were extracted for all scaffolds and listed with the scaffold number and size. Vmatch (http://www.vmatch.de) was used to search for repeats that are at least 50 bp long and 100% identical (Table S17). We note that repeats that are larger than the average insert size of our libraries would not be able to be uniquely placed into any assembly by the Celera Assembler and thus do not show up in our analysis.
The T. thermophila genome was searched against two sets of TEs using BLASTN and/or TBLASTN [190], with default parameters and E-value cutoff at 1 × 10−5. One of the TE sets consisted of 12 complete or partial ciliate TEs, namely Tec1, Tec2, and Tec3 from Moneuplotes crassus, TBE1 from O. fallax, and REP1, REP2.2, REP3, REP6, TIE1, TIE2, TIE3, and Tlr from T. thermophila [90,91,191,192]. The other TE set consisted of 44 representative elements of the transposon superfamily mariner/Tc1/IS630 [192], including members of the mariner, Tc1, DD39D (plant), DD37D (nematodes and insects), and DD37D (mosquitoes), Ant1/Tec, and Pogo families. In addition, the genome was scanned for homology to TE-encoded ORFs using PSI-TBLASTN [190]. Briefly, a reference ORF from each major family of autonomous transposons and retrotransposons was searched against the nonredundant protein database using BLAST-PGP with two iterations, generating a TE ORF family-specific profile. Each reference TE ORF and corresponding family profile were searched against the genomic sequence using PSI-TBLASTN, and all matches with E-value at most 1e−5 were captured for subsequent analysis. Finally, a few scaffolds with putatively complete transposases belonging to the mariner/Tc1/IS630 superfamily were further investigated for the presence of the inverted terminal repeats (ITRs) that typically flank these elements. Identification of paired ITRs was done using Owen [193] and searches were done against known consensus ITR sequences of mariner and Tc1 elements to find individual ITRs.
Analysis of functional categories with gene family expansions.
Protein kinase genes were identified by comparison of peptide predictions to a set of protein kinase profile hidden Markov models [104] and by BLAST against divergent kinase sequences. A small number of gene predictions were split or fused to adjacent predictions based on presence of split or multiple kinase domains. Kinases were classified by comparison of kinase domain sequences to a set of group-, family-, and subfamily-specific hidden Markov models as well as by BLAST-based clustering of T. thermophila and previously classified kinases.
Predicted protein sequences were searched against a curated database of membrane transport proteins [113] for similarity to known or putative transport proteins using BLASTP. All proteins with significant hits (E-value less than 0.001) were collected and searched against the NCBI nonredundant protein and Pfam databases [194]. Transmembrane protein topology was predicted by TMHMM [195]. A Web-based interface was implemented to facilitate the annotation processes, which incorporates number of hits to the transporter database; BLAST and hidden Markov model search E-value and score; number of predicted transmembrane segments; and the description of top hits to the nonredundant protein database (http://www.membranetransport.org) [113,196].
A total of over 30,000 sequences of characterized and predicted proteases were obtained from the Merops database (http://www.merops.ac.uk release 7.00) [119]. These sequences were searched against the T. thermophila predicted protein sequences using BLASTP with default settings and an E-value cutoff of less than 10−10 for defining protease homologs. Partial sequences (less than 80% of full-length) and redundant sequences were excluded. The domain/motif organization of predicted T. thermophila proteases was revealed by an InterPro search. For each putative protease, the known protease sequence or domain with the highest similarity was used as a reference for annotation; the catalytic type and protease family were predicted in accordance with the classification in Merops, and the enzyme was named in accordance with SWISS-PROT enzyme nomenclature (http://www.expasy.ch/cgi-bin/lists?peptidas.txt) and literature.
Tubulin superfamily genes were identified by a BLASTP search using T. thermophila α-tubulin Atu1p as the query. Twenty-one candidate predicted ORFs were identified, but two showed only moderate sequence similarity to either the amino- (TTHERM_00834920) or the carboxyl- (TTHERM_00896110) terminal halves of α- or β- tubulin and were not considered further. The 19 remaining were aligned with representative tubulins from other organisms and a neighbor-joining tree constructed using default settings of ClustalX (version 1.81) with 1,000 bootstrap runs. A prokaryotic tubulin ortholog, Escherichia coli FtsZ, was used as the outgroup (see Figure 7).
Using dynein subunit sequences obtained in the green alga C. reinhardtii or in other species when appropriate, we searched the T. thermophila MAC genome for orthologous sequence with TBLASTN. Candidate sequences were aligned with the sequences available in the databases of dynein subunits characterized in other experimental systems. Exon-intron borders were first approximated using the characteristics of the 64 introns previously experimentally determined in three dynein heavy chains, DYH1, DYH2, and DYH4. The 64 T. thermophila introns are AT rich (average 88%), are bounded by 5′-GT and AG-3′ and are relatively short (average 80 nucleotides; range, 50 to 332). The exon-intron borders and the expression of each gene were confirmed by RNA-directed PCR and, if necessary, sequencing of the amplified RT-PCR product. The verification of the exon-intron organizations of most of the heavy chains has not been completed.
Peptide sequence of Rab1A from H. sapiens was used to query T. thermophila gene predictions using BLASTP. Candidate Rab homologs were screened to include predicted proteins with complete Rab domains. These sequences were individually used in BLASTP searches of GenBank to confirm that Rab proteins from another species were the closest match. The minimum E score cut-off was 5e−13, but the majority of homologs scored better than 1e−30. The top scoring Rab1 homolog from T. thermophila (TTHERM_00316280) was used in an additional BLASTP search of the T. thermophila genome to confirm that all Rab homologs were identified by the initial query. Homologs of other GTPases in the Rabl, Ral, Rap, Ras, Rho, and Arf families began to appear along with the lower scoring Rab homologs and were discarded from the set. Rab protein sequences from H. sapiens (Ensembl database), Drosophila melanogaster (Flybase), and S. cerevisiae (Saccharomyces Genome Database), along with those identified as described above from T. thermophila, were aligned using ClustalX. The alignment was refined by eye and gaps removed. The tree in Figure S7 was generated using the neighbor-joining module in Phylip 3.6. Trees constructed using maximum-likelihood and parsimony methods largely corroborated this topology. T. thermophila Rab homologs associated with clades of previously identified Rabs were given putative names where consistent BLASTP results were evident and are arranged in Table S15 according to functional groups. Preliminary annotations from the TGD were queried to identify predicted coat protein homologs. Others were identified in queries with peptide sequence from D. melanogaster homologs. T. thermophila homologs were used in BLASTP queries of GenBank to confirm annotations. Further analysis of AP subunits, clathrin, and dynamin-related proteins is found in [96].
Sequence availability.
All of the sequences, assemblies, and gene predictions can be downloaded from the TIGR ftp site (ftp://ftp.tigr.org/pub/data/Eukaryotic_Projects/t_thermophila). The sequence reads and traces can be downloaded from the NCBI trace archive at ftp://ftp.ncbi.nih.gov/pub/TraceDB/tetrahymena_thermophila. Assemblies, sequence reads, and gene predictions can be searched using multiple similarity search methods at the TIGR, TGD, and NCBI Web sites. Sequences are also available in Genbank (see below).
Supporting Information
(A) Scaffolds larger than 1 Mb were sorted by size and concatenated to make a pseudo molecule. Statistics of nucleotide composition were calculated for 2,000 bp sliding windows with a shift length of 1,000 bp. Yellow, GC skew; blue, GC%; purple, χ2 score. The green lines delimit the scaffolds (long) or contigs within each scaffold (short).
(B) Analysis of three T. thermophila scaffolds of diverse size. Red boxes, genes on forward strand; green boxes, genes on reverse strand; blue, χ2 score; orange, GC%; brown, GC skew; salmon, AT skew. The vertical light gray lines delimit contigs within each scaffold. Scaffold sizes: 8254645, 1,076 kb; 8254654, 510 kb; 8254072, 37.3 kb.
(246 KB PDF)
Using scaffolds larger than 100 kb, the percentage of predicted gene coding sequence was calculated within 10-kb windows. For the overall gene density (black bars), a sliding 10-kb window was applied at 2-kb intervals. Gray bars represent gene density in the 10-kb adjacent to each telomere.
(92 KB PDF)
Comparison of the percentage of introns in various size classes for both ab initio predicted genes (gray bars) and introns confirmed by EST sequencing (black bars).
(17 KB PDF)
(A) tRNA charging and expression. Total RNA was harvested from T. thermophila in log-phase growth (lanes 1 and 2) or after resuspension in 10 mM Tris starvation buffer for the times indicated. Total RNA samples were resolved by acid/urea acrylamide gel electrophoresis and transferred to nylon membrane; the same total RNA sample either untreated or deacylated at alkaline pH was used for lanes 1 and 2. Probing was performed using end-radiolabeled oligonucleotides specific for the tRNA of interest.
(B) Expression levels of ncRNAs under various conditions. Total RNA was harvested from T. thermophila under the growth or development conditions indicated, resolved, transferred, and probed as in (A). As an internal control for even loading, the same blot was hybridized to detect tRNA-Sec and SRP RNA (RNA PolIII transcripts found predominantly in the cytoplasm and involved in translation) and also to U1 and U2 snRNAs (RNA PolII transcripts found predominantly in the nucleus and involved in mRNA splicing).
(420 KB PDF)
Orange points represent scaffolds that have been capped with telomeres at both ends.
(30 KB PDF)
Neighbor-joining tree built from ClustalW alignment of polo kinase domains. Species abbreviations: Hs, H. sapiens; Dm, D. melanogaster; Ce, Caenorhabditis elegans; Sc, S. cerevisiae; Dd, D. discoideum; Tt, T. thermophila. Note that T. thermophila has multiple members of both the polo and sak subfamilies, and that even within the T. thermophila–specific cluster, sequences are as divergent as orthologs from vertebrates and lower metazoans. The bar indicates scale of average substitutions per site.
(71 KB PDF)
Unrooted neighbor-joining tree for Rab GTPases. Bootstrap values over 40% (from 100 replicates) are indicated near corresponding branches. Predicted T. thermophila genes are in bold. Other Rabs are from H. sapiens (Hs), D. melanogaster (Dm), and S. cerevisiae (Sc). Proposed Rab families [157] are shown in colored blocks. Asterisks indicate Rabs for which there is functional evidence (**) or at least localization data (*) consistent with their groupings. T. thermophila genes cluster with the members of each Rab family except VII and IV (not shown in a box). There are three clades comprised exclusively of T. thermophila gene predictions (clades I, II, and III) shown in dark gray boxes.
(39 KB PDF)
(28 KB DOC)
(52 KB DOC)
(352 KB DOC)
(167 KB DOC)
(30 KB DOC)
(A) 5S.
(B) tRNA.
(C) Other ncRNAs.
(D) tRNA gene IDs.
(1.0 MB DOC)
(388 KB DOC)
(114 KB DOC)
(73 KB DOC)
(1.9 MB DOC)
(A) Kinases.
(B) Membrane transporters.
(C) Proteases.
(D) Cytoskeletal related.
(3.6 MB DOC)
(90 KB DOC)
(134 KB DOC)
(59 KB DOC)
(159 KB DOC)
(25 KB DOC)
(93 KB DOC)
Accession Numbers
The GenBank (http://www.ncbi.nlm.nih.gov/Genbank) accession numbers for the T. thermophila genes are TTHERM_00047660, 00141160, 00279820, 00486500, 00522580, and 00823430 and for three dynein heavy chains, DYH1, DYH2, and DYH4, are AF346733, AY770505, and AF072878, respectively. The sequence contigs (AAGF01000001 to AAGF01002955), the scaffold assemblies (CH445395 to CH445797 and CH670346 to CH671913), and the gene predictions (EAR80512 to EAS07932) are available from GenBank. The Gene Identification numbers in Figure 7 obtained from JGI Chlamy v2.0 (http://genome.jgi-psf.org/chlre2/chlre2.home.html) are Ec_FtsZ, 16128088; Dm_alpha-1, 135396; Hs_alpha-1, 5174477; Cr_alpha-1, 135394; Tb_alpha,135440; Sc_alpha, 1729835; Pt_alpha, 1460090; Dm_beta-1, 158739; Hs_beta-1, 135448; Cr_beta, 8928401; Tb_beta, 135500; Pt_beta-1, 417854; Sc_beta, 1174608; Dm_gamma-1, 45644955; Hs_gamma-1, 31543831; Sc_gamma, 1729859; Cr_gamma, 8928436; Pt_delta, 10637981; Hs_delta, 50592998; Cr_delta, 75277286; Tb_delta, 13508430; Hs_epsilon, 7705915; Pt_epsilon, 18477270; Tb_epsilon, 259797; Xl_eta, 4266842; Pt_eta, 9501681; Tb_zeta, 7341314; Pt_iota, 18478276; Pt_theta, 18478274; Pt_kappa, 32812838; and Cr_epsilon (C_460065). The Ensembl Gene ID (http://www.ensembl.org) for Rab1A from H. sapiens is ENSG00000138069.
Acknowledgments
We would like to acknowledge the Tetrahymena research community and the members of our Tetrahymena Scientific Advisory Board for advice, support, encouragement, and assistance. In addition, we would like to specifically acknowledge many people for assistance: John Gill (sample tracking); Hean Koo (contaminant identification and trace archive and EST submission); Shannon Smith, Susan van Aken, and William Nierman (library construction); Sam Angiuoli (Web and BLAST page maintenance); Jeff Shao (database construction); Jessica Vamathevan (initial work on genome closure); Tamara Feldblyum, Terry Utterback, and the staff at the J. Craig Venter Institute's Joint Technology Center (sequencing); Lauren Smith and Jyoti Shetty (fosmid construction); Malcolm Gardner (advice); Martin Shumway (general software engineering support); Owen White (general informatics support); Leslie Bisignano and Lynn McKenna (grants support); Aimee Turner (financial operations); Tinu Akinyemi (administrative support); and Claire Fraser (for supporting the scientific research within TIGR).
Abbreviations
- bp
base pairs
- Cbs
chromosome breakage sequence
- CM
covariance model
- EST
expressed sequence tag
- IES
internal eliminated sequence
- ITR
inverted terminal repeat
- MAC
macronucleus/macronuclear
- MIC
micronucleus/micronuclear
- ncRNA
noncoding RNA
- RIP
repeat induced point mutation
- SCI
single-cell isolation
- Sec
selenocysteine
- TE
transposable element
- TGD
Tetrahymena Genome Database
- TIGR
The Institute for Genomic Research
- VIC
voltage-gated ion channel
Footnotes
¤a Current address: University of California Davis Genome Center, Section of Evolution and Ecology, School of Biological Sciences and Department of Medical Microbiology and Immunology, School of Medicine, University of California Davis, Davis, California, United States of America
¤b Current address: Center for Bioinformatics and Computational Biology, University of Maryland, College Park, Maryland, United States of America
¤c Current address: Duke Institute for Genome Sciences and Policy, Duke University, Durham, North Carolina, United States of America
¤d Current address: Agilent Technologies, Inc., Santa Clara, California, United States of America
¤e Current address: Department of Medical Parasitology, New York University School of Medicine, New York, New York, United States of America
¤f Current address: Dupont Agriculture and Nutrition, Wilmington, Delaware, United States of America
¤g Current address: Fred Hutchinson Cancer Research Center, Seattle, Washington, United States of America
¤h Current address: Department of Molecular, Cellular and Developmental Biology, Yale University, New Haven, Connecticut, United States of America
¤j Current address: School of Botany, The University of Melbourne, Melbourne, Australia
¤k Current address: Meharry Medical College, Nashville, Tennessee, United States of America
¤l Current address: Dartmouth-Hitchcock Medical Center, Lebanon, New Hampshire, United States of America
¤m Current address: Lung Biology Center, University of California San Francisco, San Francisco, California, United States of America
Author contributions. JAE coordinated the project. JAE, RSC, EPH, and EO wrote and edited the majority of the manuscript. JAE, RSC, MW, DW, JHB, and MT performed multiple bioinformatics analyses. MT, JRW, PA, MF, RKS, and BJH coordinated the annotation. KMJ and LJT carried out genome closure. ALD and SLS generated and analyzed genome assemblies. JCS, KMK, and LS analyzed mobile DNA elements. WHM generated gene models. QR conducted analyses of membrane transporters. JMC, JG, and REP generated and analyzed ESTs. GM analyzed protein kinases. NCE and APT analyzed membrane trafficking. DJA and DEW analyzed dyneins. YW and HC analyzed proteases. KC, BAS, SRL, WLR, KW, and ZW analyzed ncRNA. DW, JG, MAG, JF, and CCT analyzed cytoskeletal associated proteins. PJK, RFW, NJP, and JHB searched for plastid-derived genes. JMC, NAS, and CJK built TGD. CdT, HFR, SCW, and RAB performed the RAPD analyses. EPH, EO, SLS, JAE, and MW examined genome structure.
Funding. This project was supported by grants to JAE from the National Science Foundation Microbial Genome Sequencing Program (EF-0240361) and the National Institutes of Health–National Institute of General Medical Sciences (R01 GM067012–03). We also acknowledge Genome Canada for support of EST library construction and sequencing through the Protist EST Project and grant RR-009231 to EO from the National Institutes of Health (the National Center for Research Resources) which supported the RAPD and Cbs work and an EO subcontract to NSF grant MCB-0132675 which supported sequence analyses related to number of chromosomes and their copy number.
Competing interests. The authors have declared that no competing interests exist.
References
- Collins K, Gorovsky MA. Tetrahymena thermophila . Curr Biol. 2005;15:R317–R318. doi: 10.1016/j.cub.2005.04.039. [DOI] [PubMed] [Google Scholar]
- Nanney DL, Simon EM. Laboratory and evolutionary history of Tetrahymena thermophila . Methods Cell Biol. 2000;62:3–25. doi: 10.1016/s0091-679x(08)61527-7. [DOI] [PubMed] [Google Scholar]
- Zaug AJ, Cech TR. The intervening sequence RNA of Tetrahymena is an enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]
- Blackburn EH, Gall JG. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena . J Mol Biol. 1978;120:33–53. doi: 10.1016/0022-2836(78)90294-2. [DOI] [PubMed] [Google Scholar]
- Yao MC, Yao CH. Repeated hexanucleotide C-C-C-C-A-A is present near free ends of macronuclear DNA of Tetrahymena . Proc Natl Acad Sci U S A. 1981;78:7436–7439. doi: 10.1073/pnas.78.12.7436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greider CW, Blackburn EH. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell. 1985;43:405–413. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- Brownell JE, Zhou J, Ranalli T, Kobayashi R, Edmondson DG, et al. Tetrahymena histone acetyltransferase A: A homolog to yeast Gcn5p linking histone acetylation to gene activation. Cell. 1996;84:843–851. doi: 10.1016/s0092-8674(00)81063-6. [DOI] [PubMed] [Google Scholar]
- Asai DJ, Forney JD, editors. Tetrahymena thermophila. San Diego: Academic Press; 2000. 580. p. [Google Scholar]
- Turkewitz AP, Orias E, Kapler G. Functional genomics: The coming of age for Tetrahymena thermophila . Trends Genet. 2002;18:35–40. doi: 10.1016/s0168-9525(01)02560-4. [DOI] [PubMed] [Google Scholar]
- Kim K, Weiss LM. Toxoplasma gondii: The model apicomplexan. Int J Parasitol. 2004;34:423–432. doi: 10.1016/j.ijpara.2003.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donald RG, Roos DS. Gene knock-outs and allelic replacements in Toxoplasma gondii: HXGPRT as a selectable marker for hit-and-run mutagenesis. Mol Biochem Parasitol. 1998;91:295–305. doi: 10.1016/s0166-6851(97)00210-7. [DOI] [PubMed] [Google Scholar]
- Radke JR, Behnke MS, Mackey AJ, Radke JB, Roos DS, et al. The transcriptome of Toxoplasma gondii . BMC Biol. 2005;3:26. doi: 10.1186/1741-7007-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DS, Gao Y, Asokan K, Gaertig J. The circumsporozoite protein of Plasmodium falciparum is expressed and localized to the cell surface in the free-living ciliate Tetrahymena thermophila . Mol Biochem Parasitol. 2002;122:119–126. doi: 10.1016/s0166-6851(02)00079-8. [DOI] [PubMed] [Google Scholar]
- Prescott DM. The DNA of ciliated protozoa. Microbiol Rev. 1994;58:233–267. doi: 10.1128/mr.58.2.233-267.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martindale DW, Allis CD, Bruns PJ. Conjugation in Tetrahymena thermophila. A temporal analysis of cytological stages. Exp Cell Res. 1982;140:227–236. doi: 10.1016/0014-4827(82)90172-0. [DOI] [PubMed] [Google Scholar]
- Yao MC, Chao JL. RNA-guided DNA deletion in Tetrahymena: An RNAi-based mechanism for programmed genome rearrangements. Annu Rev Genet. 2005;39:537–559. doi: 10.1146/annurev.genet.39.073003.095906. [DOI] [PubMed] [Google Scholar]
- Yao MC, Duharcourt S, Chalker DL. Genome-wide rearrangements of DNA in ciliates. In: Craig N, Craigie R, Gellert M, Lambowitz A, editors. Mobile DNA II. Herndon (Virginia): ASM Press; 2002. pp. 730–758. [Google Scholar]
- Yao MC, Choi J, Yokoyama S, Austerberry CF, Yao CH. DNA elimination in Tetrahymena: A developmental process involving extensive breakage and rejoining of DNA at defined sites. Cell. 1984;36:433–440. doi: 10.1016/0092-8674(84)90236-8. [DOI] [PubMed] [Google Scholar]
- Yao MC, Gorovsky MA. Comparison of the sequences of macro- and micronuclear DNA of Tetrahymena pyriformis. Chromosoma. 1974;48:1–18. doi: 10.1007/BF00284863. [DOI] [PubMed] [Google Scholar]
- Iwamura Y, Sakai M, Muramatsu M. Rearrangement of repeated DNA sequences during development of macronucleus in Tetrahymena thermophila . Nucleic Acids Res. 1982;10:4279–4291. doi: 10.1093/nar/10.14.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T. Molecular biology. An RNA-guided pathway for the epigenome. Science. 2002;297:2215–2218. doi: 10.1126/science.1077903. [DOI] [PubMed] [Google Scholar]
- Selker EU. Molecular biology. A self-help guide for a trim genome. Science. 2003;300:1517–1518. doi: 10.1126/science.1086053. [DOI] [PubMed] [Google Scholar]
- Fan Q, Yao MC. A long stringent sequence signal for programmed chromosome breakage in Tetrahymena thermophila . Nucleic Acids Res. 2000;28:895–900. doi: 10.1093/nar/28.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton EP, Williamson S, Dunn S, Merriam V, Lin C, et al. The highly conserved family of Tetrahymena thermophila chromosome breakage elements contains an invariant 10-base-pair core. Eukaryot Cell. 2006;5:771–780. doi: 10.1128/EC.5.4.771-780.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Yao CH, Monks B. The controlling sequence for site-specific chromosome breakage in Tetrahymena . Cell. 1990;63:763–772. doi: 10.1016/0092-8674(90)90142-2. [DOI] [PubMed] [Google Scholar]
- Fan Q, Yao M. New telomere formation coupled with site-specific chromosome breakage in Tetrahymena thermophila . Mol Cell Biol. 1996;16:1267–1274. doi: 10.1128/mcb.16.3.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu GL, Blackburn EH. Developmentally programmed healing of chromosomes by telomerase in Tetrahymena . Cell. 1991;67:823–832. doi: 10.1016/0092-8674(91)90077-c. [DOI] [PubMed] [Google Scholar]
- Altschuler MI, Yao MC. Macronuclear DNA of Tetrahymena thermophila exists as defined subchromosomal-sized molecules. Nucleic Acids Res. 1985;13:5817–5831. doi: 10.1093/nar/13.16.5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover RK, Brunk CF. Macronuclear DNA molecules of Tetrahymena thermophila . Mol Cell Biol. 1986;6:900–905. doi: 10.1128/mcb.6.3.900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapler GM. Developmentally regulated processing and replication of the Tetrahymena rDNA minichromosome. Curr Opin Genet Dev. 1993;3:730–735. doi: 10.1016/s0959-437x(05)80091-7. [DOI] [PubMed] [Google Scholar]
- Doerder FP, Deak JC, Lief JH. Rate of phenotypic assortment in Tetrahymena thermophila . Dev Genet. 1992;13:126–132. doi: 10.1002/dvg.1020130206. [DOI] [PubMed] [Google Scholar]
- Ray C., Jr Preparation of chromosomes of Tetrahymena pyriformis for photomicrography. Stain Technol. 1956;31:271–274. doi: 10.3109/10520295609113817. [DOI] [PubMed] [Google Scholar]
- LaFountain JR, Jr, Davidson LA. An analysis of spindle ultrastructure during prometaphase and metaphase of micronuclear division in Tetrahymena . Chromosoma. 1979;75:293–308. doi: 10.1007/BF00293473. [DOI] [PubMed] [Google Scholar]
- LaFountain JR, Jr, Davidson LA. An analysis of spindle ultrastructure during anaphase of micronuclear division in Tetrahymena . Cell Motil. 1980;1:41–61. doi: 10.1002/cm.970010105. [DOI] [PubMed] [Google Scholar]
- Mochizuki K, Gorovsky MA. Small RNAs in genome rearrangement in Tetrahymena . Curr Opin Genet Dev. 2004;14:181–187. doi: 10.1016/j.gde.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Orias E. Toward sequencing the Tetrahymena genome: Exploiting the gift of nuclear dimorphism. J Eukaryot Microbiol. 2000;47:328–333. doi: 10.1111/j.1550-7408.2000.tb00057.x. [DOI] [PubMed] [Google Scholar]
- Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, et al. A whole-genome assembly of Drosophila . Science. 2000;287:2196–2204. doi: 10.1126/science.287.5461.2196. [DOI] [PubMed] [Google Scholar]
- Brunk CF, Lee LC, Tran AB, Li J. Complete sequence of the mitochondrial genome of Tetrahymena thermophila and comparative methods for identifying highly divergent genes. Nucleic Acids Res. 2003;31:1673–1682. doi: 10.1093/nar/gkg270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engberg J, Nielsen H. Complete sequence of the extrachromosomal rDNA molecule from the ciliate Tetrahymena thermophila strain B1868VII. Nucleic Acids Res. 1990;18:6915–6919. doi: 10.1093/nar/18.23.6915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong L, Klionsky L, Wickert S, Merriam V, Orias E, et al. Autonomously replicating macronuclear DNA pieces are the physical basis of genetic coassortment groups in Tetrahymena thermophila . Genetics. 2000;155:1119–1125. doi: 10.1093/genetics/155.3.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy-Hanley D, Bisharyan Y, Fridman V, Gerber J, Lin C, et al. Genome-wide characterization of Tetrahymena thermophila chromosome breakage sites. II. Physical and genetic mapping. Genetics. 2005;170:1623–1631. doi: 10.1534/genetics.104.031435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao MC, Zheng K, Yao CH. A conserved nucleotide sequence at the sites of developmentally regulated chromosomal breakage in Tetrahymena . Cell. 1987;48:779–788. doi: 10.1016/0092-8674(87)90075-4. [DOI] [PubMed] [Google Scholar]
- Karrer KM. Tetrahymena genetics: Two nuclei are better than one. Methods Cell Biol. 2000;62:127–186. doi: 10.1016/s0091-679x(08)61529-0. [DOI] [PubMed] [Google Scholar]
- Cervantes MD, Xi X, Vermaak D, Yao MC, Malik HS. The CNA1 histone of the ciliate Tetrahymena thermophila is essential for chromosome segregation in the germline micronucleus. Mol Biol Cell. 2006;17:485–497. doi: 10.1091/mbc.E05-07-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryde FE, Gorham HC, Louis EJ. Chromosome ends: All the same under their caps. Curr Opin Genet Dev. 1997;7:822–828. doi: 10.1016/s0959-437x(97)80046-9. [DOI] [PubMed] [Google Scholar]
- Wellinger RJ, Sen D. The DNA structures at the ends of eukaryotic chromosomes. Eur J Cancer. 1997;33:735–749. doi: 10.1016/S0959-8049(97)00067-1. [DOI] [PubMed] [Google Scholar]
- Barry JD, Ginger ML, Burton P, McCulloch R. Why are parasite contingency genes often associated with telomeres? Int J Parasitol. 2003;33:29–45. doi: 10.1016/s0020-7519(02)00247-3. [DOI] [PubMed] [Google Scholar]
- Gao W, Khang CH, Park SY, Lee YH, Kang S. Evolution and organization of a highly dynamic, subtelomeric helicase gene family in the rice blast fungus Magnaporthe grisea . Genetics. 2002;162:103–112. doi: 10.1093/genetics/162.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Trask BJ. The complex structure and dynamic evolution of human subtelomeres. Nat Rev Genet. 2002;3:91–102. doi: 10.1038/nrg727. [DOI] [PubMed] [Google Scholar]
- Teunissen AW, Steensma HY. Review: The dominant flocculation genes of Saccharomyces cerevisiae constitute a new subtelomeric gene family. Yeast. 1995;11:1001–1013. doi: 10.1002/yea.320111102. [DOI] [PubMed] [Google Scholar]
- Louis EJ. The chromosome ends of Saccharomyces cerevisiae . Yeast. 1995;11:1553–1573. doi: 10.1002/yea.320111604. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Delcher AL, Mount SM, Wortman JR, Smith RK, Jr, et al. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003;31:5654–5666. doi: 10.1093/nar/gkg770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzone FJ, Stathopoulos VA, Grass D, Gorovsky MA, Angerer RC. Regulation of protein synthesis in Tetrahymena. RNA sequence sets of growing and starved cells. J Biol Chem. 1983;258:6899–6905. [PubMed] [Google Scholar]
- Zagulski M, Nowak JK, Le Mouel A, Nowacki M, Migdalski A, et al. High coding density on the largest Paramecium tetraurelia somatic chromosome. Curr Biol. 2004;14:1397–1404. doi: 10.1016/j.cub.2004.07.029. [DOI] [PubMed] [Google Scholar]
- Erdmann VA, Wolters J, Huysmans E, Vandenberghe A, De Wachter R. Collection of published 5S and 5.8S ribosomal RNA sequences. Nucleic Acids Res. 1984;12(Suppl):r133–r166. doi: 10.1093/nar/12.suppl.r133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luehrsen KR, Fox GE, Woese CR. The sequence of Tetrahymena thermophila 5S ribosomal ribonucleic acid. Curr Microbiol. 1980;4:123–126. [Google Scholar]
- Kimmel AR, Gorovsky MA. Numbers of 5S and tRNA genes in macro- and micronuclei of Tetrahymena pyriformis . Chromosoma. 1976;54:327–337. doi: 10.1007/BF00292813. [DOI] [PubMed] [Google Scholar]
- Horowitz S, Gorovsky MA. An unusual genetic code in nuclear genes of Tetrahymena . Proc Natl Acad Sci U S A. 1985;82:2452–2455. doi: 10.1073/pnas.82.8.2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DM, Copeland PR. Mechanism and regulation of selenoprotein synthesis. Annu Rev Nutr. 2003;23:17–40. doi: 10.1146/annurev.nutr.23.011702.073318. [DOI] [PubMed] [Google Scholar]
- Hatfield DL, Gladyshev VN. How selenium has altered our understanding of the genetic code. Mol Cell Biol. 2002;22:3565–3576. doi: 10.1128/MCB.22.11.3565-3576.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrimali RK, Lobanov AV, Xu XM, Rao M, Carlson BA, et al. Selenocysteine tRNA identification in the model organisms Dictyostelium discoideum and Tetrahymena thermophila . Biochem Biophys Res Commun. 2005;329:147–151. doi: 10.1016/j.bbrc.2005.01.120. [DOI] [PubMed] [Google Scholar]
- Wuitschick JD, Karrer KM. Analysis of genomic G + C content, codon usage, initiator codon context and translation termination sites in Tetrahymena thermophila . J Eukaryot Microbiol. 1999;46:239–247. doi: 10.1111/j.1550-7408.1999.tb05120.x. [DOI] [PubMed] [Google Scholar]
- Wuitschick JD, Karrer KM. Codon usage in Tetrahymena thermophila . Methods Cell Biol. 2000;62:565–568. doi: 10.1016/s0091-679x(08)61558-7. [DOI] [PubMed] [Google Scholar]
- Eichinger L, Pachebat JA, Glockner G, Rajandream MA, Sucgang R, et al. The genome of the social amoeba Dictyostelium discoideum . Nature. 2005;435:43–57. doi: 10.1038/nature03481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kinouchi M, Kudo Y, Ikemura T. Codon usage and tRNA genes in eukaryotes: Correlation of codon usage diversity with translation efficiency and with CG-dinucleotide usage as assessed by multivariate analysis. J Mol Evol. 2001;53:290–298. doi: 10.1007/s002390010219. [DOI] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003;302:1401–1404. doi: 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Katz LA, Snoeyenbos-West O, Doerder FP. Patterns of protein evolution in Tetrahymena thermophila: Implications for estimates of effective population size. Mol Biol Evol. 2006;23:608–614. doi: 10.1093/molbev/msj067. [DOI] [PubMed] [Google Scholar]
- Fast NM, Xue L, Bingham S, Keeling PJ. Re-examining alveolate evolution using multiple protein molecular phylogenies. J Eukaryot Microbiol. 2002;49:30–37. doi: 10.1111/j.1550-7408.2002.tb00336.x. [DOI] [PubMed] [Google Scholar]
- Gajadhar AA, Marquardt WC, Hall R, Gunderson J, Ariztia-Carmona EV, et al. Ribosomal RNA sequences of Sarcocystis muris, Theileria annulata and Crypthecodinium cohnii reveal evolutionary relationships among apicomplexans, dinoflagellates, and ciliates. Mol Biochem Parasitol. 1991;45:147–154. doi: 10.1016/0166-6851(91)90036-6. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Williamson DH, Wilson RJ. A circular DNA in malaria parasites encodes an RNA polymerase like that of prokaryotes and chloroplasts. Mol Biochem Parasitol. 1991;44:115–123. doi: 10.1016/0166-6851(91)90227-w. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. Principles of protein and lipid targeting in secondary symbiogenesis: Euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J Eukaryot Microbiol. 1999;46:347–366. doi: 10.1111/j.1550-7408.1999.tb04614.x. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. Genome sequence of the human malaria parasite Plasmodium falciparum . Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoes A, Zourmpanou D, Leon-Avila G, van der Giezen M, Tovar J, et al. Protein import, replication, and inheritance of a vestigial mitochondrion. J Biol Chem. 2005;280:30557–30563. doi: 10.1074/jbc.M500787200. [DOI] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana . Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Dyall SD, Brown MT, Johnson PJ. Ancient invasions: From endosymbionts to organelles. Science. 2004;304:253–257. doi: 10.1126/science.1094884. [DOI] [PubMed] [Google Scholar]
- Ralph SA, van Dooren GG, Waller RF, Crawford MJ, Fraunholz MJ, et al. Tropical infectious diseases: Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol. 2004;2:203–216. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- Erwin JA, Beach D, Holz GG., Jr Effect of dietary cholesterol on unsaturated fatty acid biosynthesis in a ciliated protozoan. Biochim Biophys Acta. 1966;125:614–616. doi: 10.1016/0005-2760(66)90053-1. [DOI] [PubMed] [Google Scholar]
- Holz GG, Jr, Erwin J, Rosenbaum N, Aaronson S. Triparanol inhibition of Tetrahymena, and its prevention by lipids. Arch Biochem Biophys. 1962;98:312–322. doi: 10.1016/0003-9861(62)90189-3. [DOI] [PubMed] [Google Scholar]
- Holz GG, Jr, Wagner B, Erwin J, Britt JJ, Bloch K. Sterol requirements of a ciliate Tetrahymena corlissi Th-X. I. A nutritional analysis of the sterol requirements of T. corlissi Th-X. II. Metabolism of tritiated lopohenol in T. corlissi Th-X. Comp Biochem Physiol. 1961;2:202–217. doi: 10.1016/0010-406x(61)90062-7. [DOI] [PubMed] [Google Scholar]
- Corliss JO. The impact of electron microscopy on ciliate systematics. Am Zool. 1979;19:573–587. [Google Scholar]
- Lynn DH. The organization and evolution of microtubular organelles in ciliated protozoa. Biol Rev. 1981;56:243–292. [Google Scholar]
- Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum . Science. 2004;304:441–445. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- Huang J, Mullapudi N, Lancto CA, Scott M, Abrahamsen MS, et al. Phylogenomic evidence supports past endosymbiosis, intracellular and horizontal gene transfer in Cryptosporidium parvum . Genome Biol. 2004;5:R88. doi: 10.1186/gb-2004-5-11-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, et al. The genome sequence of the filamentous fungus Neurospora crassa . Nature. 2003;422:859–868. doi: 10.1038/nature01554. [DOI] [PubMed] [Google Scholar]
- Galagan JE, Selker EU. RIP: The evolutionary cost of genome defense. Trends Genet. 2004;20:417–423. doi: 10.1016/j.tig.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Liu Y, Song X, Gorovsky MA, Karrer KM. Elimination of foreign DNA during somatic differentiation in Tetrahymena thermophila shows position effect and is dosage dependent. Eukaryot Cell. 2005;4:421–431. doi: 10.1128/EC.4.2.421-431.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki K, Fine NA, Fujisawa T, Gorovsky MA. Analysis of a piwi-related gene implicates small RNAs in genome rearrangement in tetrahymena. Cell. 2002;110:689–699. doi: 10.1016/s0092-8674(02)00909-1. [DOI] [PubMed] [Google Scholar]
- Yao MC, Fuller P, Xi X. Programmed DNA deletion as an RNA-guided system of genome defense. Science. 2003;300:1581–1584. doi: 10.1126/science.1084737. [DOI] [PubMed] [Google Scholar]
- Doerder FP, Gates MA, Eberhardt FP, Arslanyolu M. High frequency of sex and equal frequencies of mating types in natural populations of the ciliate Tetrahymena thermophila . Proc Natl Acad Sci U S A. 1995;92:8715–8718. doi: 10.1073/pnas.92.19.8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillingham JS, Thing TA, Vythilingum N, Keuroghlian A, Bruno D, et al. A nonlong terminal repeat retrotransposon family is restricted to the germ line micronucleus of the ciliated protozoan Tetrahymena thermophila . Eukaryot Cell. 2004;3:157–169. doi: 10.1128/EC.3.1.157-169.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuitschick JD, Gershan JA, Lochowicz AJ, Li S, Karrer KM. A novel family of mobile genetic elements is limited to the germline genome in Tetrahymena thermophila . Nucleic Acids Res. 2002;30:2524–2537. doi: 10.1093/nar/30.11.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritham EJ, Feschotte C, Wessler SR. Unexpected diversity and differential success of DNA transposons in four species of entamoeba protozoans. Mol Biol Evol. 2005;22:1751–1763. doi: 10.1093/molbev/msi169. [DOI] [PubMed] [Google Scholar]
- Silva JC, Bastida F, Bidwell SL, Johnson PJ, Carlton JM. A potentially functional mariner transposable element in the protist Trichomonas vaginalis . Mol Biol Evol. 2005;22:126–134. doi: 10.1093/molbev/msh260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Garrett PW, Kinsey JA, Selker EU. Specificity of repeat-induced point mutation (RIP) in Neurospora: Sensitivity of nonNeurospora sequences, a natural diverged tandem duplication, and unique DNA adjacent to a duplicated region. Genetics. 1991;127:711–717. doi: 10.1093/genetics/127.4.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GR, Smith DG, Michael Siu KW, Pearlman RE, Turkewitz AP. Genomic and proteomic evidence for a second family of dense core granule cargo proteins in Tetrahymena thermophila . J Eukaryot Microbiol. 2005;52:291–297. doi: 10.1111/j.1550-7408.2005.00045.x. [DOI] [PubMed] [Google Scholar]
- Elde NC, Morgan G, Winey M, Sperling L, Turkewitz AP. Elucidation of clathrin-mediated endocytosis in Tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genetics. 2005;1:e52. doi: 10.1371/journal.pgen.0010052. DOI: 10.1371/journal.pgen.0010052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann L, Erkelenz M, Aldag I, Tiedtke A, Hartmann MW. Biochemical and molecular characterisation of Tetrahymena thermophila extracellular cysteine proteases. BMC Microbiol. 2006;6:19. doi: 10.1186/1471-2180-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuribara S, Kato M, Kato-Minoura T, Numata O. Identification of a novel actin-related protein in Tetrahymena cilia. Cell Motil Cytoskeleton. 2006;63:437–446. doi: 10.1002/cm.20136. [DOI] [PubMed] [Google Scholar]
- Lee SR, Collins K. Two classes of endogenous small RNAs in Tetrahymena thermophila . Genes Dev. 2006;20:28–33. doi: 10.1101/gad.1377006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemm-Wolf AJ, Morgan G, Giddings TH, Jr, White EA, Marchione R, et al. Basal body duplication and maintenance require one member of the Tetrahymena thermophila centrin gene family. Mol Biol Cell. 2005;16:3606–3619. doi: 10.1091/mbc.E04-10-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickstead B, Gull K. A “holistic” kinesin phylogeny reveals new kinesin families and predicts protein functions. Mol Biol Cell. 2006;17:1734–1743. doi: 10.1091/mbc.E05-11-1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Gavin RH. Myosin genes in Tetrahymena . Cell Motil Cytoskeleton. 2005;61:237–243. doi: 10.1002/cm.20078. [DOI] [PubMed] [Google Scholar]
- Wloga D, Camba A, Rogowski K, Manning G, Jerka-Dziadosz M, et al. Members of the Nima-related kinase family promote disassembly of cilia by multiple mechanisms. Mol Biol Cell. 2006;17:2799–2810. doi: 10.1091/mbc.E05-05-0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global analysis of protein kinase genes in sequenced genomes. Available: http://kinase.com. Accessed 15 July 2006.
- Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- Goldberg JM, Manning G, Liu A, Fey P, Pilcher KE, et al. The dictyostelium kinome—Analysis of the protein kinases from a simple model organism. PLoS Genet. 2006;2:e38. doi: 10.1371/journal.pgen.0020038. DOI: 10.1371/journal.pgen.0020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen ST, Guerra CF, Awan A, Wheatley DN, Satir P. Insulin receptor-like proteins in Tetrahymena thermophila ciliary membranes. Curr Biol. 2003;13:R50–R52. doi: 10.1016/s0960-9822(02)01425-2. [DOI] [PubMed] [Google Scholar]
- Manning G, Caenepeel S. Protein kinases in human disease. 2005–06 Catalog and technical reference. Beverly (Massachusetts): Cell Signaling Technologies; 2005. pp. 402–409. [Google Scholar]
- O'Connell MJ, Krien MJ, Hunter T. Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 2003;13:221–228. doi: 10.1016/s0962-8924(03)00056-4. [DOI] [PubMed] [Google Scholar]
- Okazaki N, Yan J, Yuasa S, Ueno T, Kominami E, et al. Interaction of the Unc-51-like kinase and microtubule-associated protein light chain 3 related proteins in the brain: Possible role of vesicular transport in axonal elongation. Brain Res Mol Brain Res. 2000;85:1–12. doi: 10.1016/s0169-328x(00)00218-7. [DOI] [PubMed] [Google Scholar]
- Wolanin PM, Thomason PA, Stock JB. Histidine protein kinases: Key signal transducers outside the animal kingdom. Genome Biol. 2002;3:reviews3013. doi: 10.1186/gb-2002-3-10-reviews3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK. Genomic analysis of the eukaryotic protein kinase superfamily: A perspective. Genome Biol. 2003;4:111. doi: 10.1186/gb-2003-4-5-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Q, Kang KH, Paulsen IT. TransportDB: A relational database of cellular membrane transport systems. Nucleic Acids Res. 2004;32:D284–D288. doi: 10.1093/nar/gkh016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes WJ, Ling KY, Saimi Y, Kung C. PAK paradox: Paramecium appears to have more K(+)-channel genes than humans. Eukaryot Cell. 2003;2:737–745. doi: 10.1128/EC.2.4.737-745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung C, Saimi Y. The physiological basis of taxes in Paramecium . Annu Rev Physiol. 1982;44:519–534. doi: 10.1146/annurev.ph.44.030182.002511. [DOI] [PubMed] [Google Scholar]
- Hennessey T, Machemer H, Nelson DL. Injected cyclic AMP increases ciliary beat frequency in conjunction with membrane hyperpolarization. Eur J Cell Biol. 1985;36:153–156. [PubMed] [Google Scholar]
- Weber JH, Vishnyakov A, Hambach K, Schultz A, Schultz JE, et al. Adenylyl cyclases from Plasmodium, Paramecium and Tetrahymena are novel ion channel/enzyme fusion proteins. Cell Signal. 2004;16:115–125. doi: 10.1016/s0898-6568(03)00129-3. [DOI] [PubMed] [Google Scholar]
- Puente XS, Sanchez LM, Overall CM, Lopez-Otin C. Human and mouse proteases: A comparative genomic approach. Nat Rev Genet. 2003;4:544–558. doi: 10.1038/nrg1111. [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ. MEROPS: The peptidase database. Nucleic Acids Res. 2004;32:D160–D164. doi: 10.1093/nar/gkh071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C. A genomic perspective on human proteases. FEBS Lett. 2001;498:214–218. doi: 10.1016/s0014-5793(01)02490-5. [DOI] [PubMed] [Google Scholar]
- Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of proteolytic enzymes. San Diego: Academic Press; 1998. 1666. p. [Google Scholar]
- Wu Y, Wang X, Liu X, Wang Y. Data-mining approaches reveal hidden families of proteases in the genome of malaria parasite. Genome Res. 2003;13:601–616. doi: 10.1101/gr.913403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochtler M, Ditzel L, Groll M, Hartmann C, Huber R. The proteasome. Annu Rev Biophys Biomol Struct. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- Gruszynski AE, DeMaster A, Hooper NM, Bangs JD. Surface coat remodeling during differentiation of Trypanosoma brucei . J Biol Chem. 2003;278:24665–24672. doi: 10.1074/jbc.M301497200. [DOI] [PubMed] [Google Scholar]
- LaCount DJ, Gruszynski AE, Grandgenett PM, Bangs JD, Donelson JE. Expression and function of the Trypanosoma brucei major surface protease (GP63) genes. J Biol Chem. 2003;278:24658–24664. doi: 10.1074/jbc.M301451200. [DOI] [PubMed] [Google Scholar]
- Yao C, Donelson JE, Wilson ME. The major surface protease (MSP or GP63) of Leishmania sp. Biosynthesis, regulation of expression, and function. Mol Biochem Parasitol. 2003;132:1–16. doi: 10.1016/s0166-6851(03)00211-1. [DOI] [PubMed] [Google Scholar]
- Madeo F, Herker E, Maldener C, Wissing S, Lachelt S, et al. A caspase-related protease regulates apoptosis in yeast. Mol Cell. 2002;9:911–917. doi: 10.1016/s1097-2765(02)00501-4. [DOI] [PubMed] [Google Scholar]
- Frankel J. Cell biology of Tetrahymena thermophila . Methods Cell Biol. 2000;62:27–125. doi: 10.1016/s0091-679x(08)61528-9. [DOI] [PubMed] [Google Scholar]
- Williams NE. Preparation of cytoskeletal fractions from Tetrahymena thermophila . Methods Cell Biol. 2000;62:441–447. doi: 10.1016/s0091-679x(08)61548-4. [DOI] [PubMed] [Google Scholar]
- Dutcher SK. Long-lost relatives reappear: Identification of new members of the tubulin superfamily. Curr Opin Microbiol. 2003;6:634–640. doi: 10.1016/j.mib.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Gaertig J, Thatcher TH, McGrath KE, Callahan RC, Gorovsky MA. Perspectives on tubulin isotype function and evolution based on the observation that Tetrahymena thermophila microtubules contain a single alpha- and beta-tubulin. Cell Motil Cytoskeleton. 1993;25:243–253. doi: 10.1002/cm.970250305. [DOI] [PubMed] [Google Scholar]
- McGrath KE, Yu SM, Heruth DP, Kelly AA, Gorovsky MA. Regulation and evolution of the single alpha-tubulin gene of the ciliate Tetrahymena thermophila . Cell Motil Cytoskeleton. 1994;27:272–283. doi: 10.1002/cm.970270308. [DOI] [PubMed] [Google Scholar]
- Shang Y, Li B, Gorovsky MA. Tetrahymena thermophila contains a conventional gamma-tubulin that is differentially required for the maintenance of different microtubule-organizing centers. J Cell Biol. 2002;158:1195–1206. doi: 10.1083/jcb.200205101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis-Williams P, Fleury-Aubusson A, de Loubresse NG, Geoffroy H, Vayssie L, et al. Functional role of epsilon-tubulin in the assembly of the centriolar microtubule scaffold. J Cell Biol. 2002;158:1183–1193. doi: 10.1083/jcb.200205028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz F, Dupuis-Williams P, Klotz C, Forquignon F, Bergdoll M, et al. Genetic evidence for interaction between eta- and beta-tubulins. Eukaryot Cell. 2004;3:212–220. doi: 10.1128/EC.3.1.212-220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz F, Krzywicka A, Klotz C, Keller A, Cohen J, et al. The SM19 gene, required for duplication of basal bodies in Paramecium, encodes a novel tubulin, eta-tubulin. Curr Biol. 2000;10:1451–1454. doi: 10.1016/s0960-9822(00)00804-6. [DOI] [PubMed] [Google Scholar]
- Duan J, Gorovsky MA. Both carboxy-terminal tails of alpha- and beta-tubulin are essential, but either one will suffice. Curr Biol. 2002;12:313–316. doi: 10.1016/s0960-9822(02)00651-6. [DOI] [PubMed] [Google Scholar]
- Thazhath R, Liu C, Gaertig J. Polyglycylation domain of beta-tubulin maintains axonemal architecture and affects cytokinesis in Tetrahymena . Nat Cell Biol. 2002;4:256–259. doi: 10.1038/ncb764. [DOI] [PubMed] [Google Scholar]
- Xia L, Hai B, Gao Y, Burnette D, Thazhath R, et al. Polyglycylation of tubulin is essential and affects cell motility and division in Tetrahymena thermophila . J Cell Biol. 2000;149:1097–1106. doi: 10.1083/jcb.149.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons IR, Rowe AJ. Dynein: A protein with adenosine triphosphatase activity from cilia. Science. 1965;149:424–426. doi: 10.1126/science.149.3682.424. [DOI] [PubMed] [Google Scholar]
- Gibbons IR, Lee-Eiford A, Mocz G, Phillipson CA, Tang WJ, et al. Photosensitized cleavage of dynein heavy chains. Cleavage at the “V1 site” by irradiation at 365 nm in the presence of ATP and vanadate. J Biol Chem. 1987;262:2780–2786. [PubMed] [Google Scholar]
- King SM. The dynein microtubule motor. Biochim Biophys Acta. 2000;1496:60–75. doi: 10.1016/s0167-4889(00)00009-4. [DOI] [PubMed] [Google Scholar]
- Sakato M, King SM. Design and regulation of the AAA+ microtubule motor dynein. J Struct Biol. 2004;146:58–71. doi: 10.1016/j.jsb.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Asai DJ, Koonce MP. The dynein heavy chain: Structure, mechanics and evolution. Trends Cell Biol. 2001;11:196–202. doi: 10.1016/s0962-8924(01)01970-5. [DOI] [PubMed] [Google Scholar]
- Asai DJ, Wilkes DE. The dynein heavy chain family. J Eukaryot Microbiol. 2004;51:23–29. doi: 10.1111/j.1550-7408.2004.tb00157.x. [DOI] [PubMed] [Google Scholar]
- Sailaja G, Lincoln LM, Chen J, Asai DJ. Evaluating the dynein heavy chain gene family in Tetrahymena . Methods Mol Biol. 2001;161:17–27. doi: 10.1385/1-59259-051-9:017. [DOI] [PubMed] [Google Scholar]
- Xu W, Royalty MP, Zimmerman JR, Angus SP, Pennock DG. The dynein heavy chain gene family in Tetrahymena thermophila . J Eukaryot Microbiol. 1999;46:606–611. doi: 10.1111/j.1550-7408.1999.tb05136.x. [DOI] [PubMed] [Google Scholar]
- Foth BJ, Goedecke MC, Soldati D. New insights into myosin evolution and classification. Proc Natl Acad Sci U S A. 2006;103:3681–3686. doi: 10.1073/pnas.0506307103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Rogowski K, Wloga D, Regnard C, Kajava AV, et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science. 2005;308:1758–1762. doi: 10.1126/science.1113010. [DOI] [PubMed] [Google Scholar]
- Osmani SA, Engle DB, Doonan JH, Morris NR. Spindle formation and chromatin condensation in cells blocked at interphase by mutation of a negative cell cycle control gene. Cell. 1988;52:241–251. doi: 10.1016/0092-8674(88)90513-2. [DOI] [PubMed] [Google Scholar]
- Fry AM, Meraldi P, Nigg EA. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998;17:470–481. doi: 10.1093/emboj/17.2.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahjoub MR, Montpetit B, Zhao L, Finst RJ, Goh B, et al. The FA2 gene of Chlamydomonas encodes a NIMA family kinase with roles in cell cycle progression and microtubule severing during deflagellation. J Cell Sci. 2002;115:1759–1768. doi: 10.1242/jcs.115.8.1759. [DOI] [PubMed] [Google Scholar]
- Turkewitz AP. Out with a bang! Tetrahymena as a model system to study secretory granule biogenesis. Traffic. 2004;5:63–68. doi: 10.1046/j.1600-0854.2003.00155.x. [DOI] [PubMed] [Google Scholar]
- Bock JB, Matern HT, Peden AA, Scheller RH. A genomic perspective on membrane compartment organization. Nature. 2001;409:839–841. doi: 10.1038/35057024. [DOI] [PubMed] [Google Scholar]
- Ackers JP, Dhir V, Field MC. A bioinformatic analysis of the RAB genes of Trypanosoma brucei . Mol Biochem Parasitol. 2005;141:89–97. doi: 10.1016/j.molbiopara.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Stenmark H, Olkkonen VM. The Rab GTPase family. Genome Biol. 2001;2:reviews3007. doi: 10.1186/gb-2001-2-5-reviews3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira-Leal JB, Seabra MC. Evolution of the Rab family of small GTP-binding proteins. J Mol Biol. 2001;313:889–901. doi: 10.1006/jmbi.2001.5072. [DOI] [PubMed] [Google Scholar]
- Saito-Nakano Y, Loftus BJ, Hall N, Nozaki T. The diversity of Rab GTPases in Entamoeba histolytica . Exp Parasitol. 2005;110:244–252. doi: 10.1016/j.exppara.2005.02.021. [DOI] [PubMed] [Google Scholar]
- Lal K, Field MC, Carlton JM, Warwicker J, Hirt RP. Identification of a very large Rab GTPase family in the parasitic protozoan Trichomonas vaginalis . Mol Biochem Parasitol. 2005;143:226–235. doi: 10.1016/j.molbiopara.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Muller HM, Kenny EE, Sternberg PW. Textpresso: An ontology-based information retrieval and extraction system for biological literature. PLoS Biol. 2004;2:e309. doi: 10.1371/journal.pbio.0020309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Mungall C, Shu S, Caudy M, Mangone M, et al. The generic genome browser: A building block for a model organism system database. Genome Res. 2002;12:1599–1610. doi: 10.1101/gr.403602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dear PH, Cook PR. Happy mapping: Linkage mapping using a physical analogue of meiosis. Nucleic Acids Res. 1993;21:13–20. doi: 10.1093/nar/21.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott AM, Gruchy DF. The occurence of mating types in Tetrahymena . Biol Bull (Woods Hole, MA) 1952;105:301. [Google Scholar]
- Mayo KA, Orias E. Further evidence for lack of gene expression in the Tetrahymena micronucleus. Genetics. 1981;98:747–762. doi: 10.1093/genetics/98.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SL, Gibson I. Genetics of Tetrahymena . In: Elliott AM, editor. Biology of Tetrahymena. Stroudsburg (Pennsylvania): Dowden, Hutchinson and Ross; 1973. pp. 307–373. [Google Scholar]
- Allen SL. Genomic exclusion: A rapid means for inducing homozygous diploid lines in Tetrahymena pyriformis, syngen 1. Science. 1967;155:575–577. doi: 10.1126/science.155.3762.575. [DOI] [PubMed] [Google Scholar]
- Ward N, Eisen J, Fraser C, Stackebrandt E. Sequenced strains must be saved from extinction. Nature. 2001;414:148. doi: 10.1038/35102737. [DOI] [PubMed] [Google Scholar]
- Gorovsky MA, Yao MC, Keevert JB, Pleger GL. Isolation of micro- and macronuclei of Tetrahymena pyriformis. Methods Cell Biol. 1975;9:311–327. doi: 10.1016/s0091-679x(08)60080-1. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, et al. The sequence of the human genome. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Brickner J, Nakano KJ, Orias E. Genetic map of randomly amplified DNA polymorphisms closely linked to the mating type locus of Tetrahymena thermophila . Genetics. 1995;141:1315–1325. doi: 10.1093/genetics/141.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton E, Bruns P, Lin C, Merriam V, Orias E, et al. Genome-wide characterization of Tetrahymena thermophila chromosome breakage sites. I. Cloning and identification of functional sites. Genetics. 2005;170:1611–1621. doi: 10.1534/genetics.104.031401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birren B, Lai E. Pulsed field gel electrophoresis—A practical guide. New York: Academic Press; 1993. [Google Scholar]
- Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res. 1994;22:2079–2088. doi: 10.1093/nar/22.11.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: An RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, et al. Rfam: Annotating noncoding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–D124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Ruzzo WL. Exploiting conserved structure for faster annotation of noncoding RNAs without loss of accuracy. Bioinformatics. 2004;20((Suppl 1)):I334–I341. doi: 10.1093/bioinformatics/bth925. [DOI] [PubMed] [Google Scholar]
- Orum H, Nielsen H, Engberg J. Sequence and proposed secondary structure of the Tetrahymena thermophila U3-snRNA. Nucleic Acids Res. 1993;21:2511. doi: 10.1093/nar/21.10.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg Z, Ruzzo WL. Sequence-based heuristics for faster annotation of noncoding RNA families. Bioinformatics. 2006;22:35–39. doi: 10.1093/bioinformatics/bti743. [DOI] [PubMed] [Google Scholar]
- Majoros WH, Pertea M, Salzberg SL. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics. 2004;20:2878–2879. doi: 10.1093/bioinformatics/bth315. [DOI] [PubMed] [Google Scholar]
- Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Fricke E, Geffers R, Gossling E, Haubrock M, et al. TRANSFAC: Transcriptional regulation, from patterns to profiles. Nucleic Acids Res. 2003;31:374–378. doi: 10.1093/nar/gkg108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL, Pertea M, Delcher AL, Gardner MJ, Tettelin H. Interpolated Markov models for eukaryotic gene finding. Genomics. 1999;59:24–31. doi: 10.1006/geno.1999.5854. [DOI] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Bateman A, Durbin R. QuickTree: Building huge Neighbour-Joining trees of protein sequences. Bioinformatics. 2002;18:1546–1547. doi: 10.1093/bioinformatics/18.11.1546. [DOI] [PubMed] [Google Scholar]
- Scharfe C, Zaccaria P, Hoertnagel K, Jaksch M, Klopstock T, et al. MITOP, the mitochondrial proteome database: 2000 Update. Nucleic Acids Res. 2000;28:155–158. doi: 10.1093/nar/28.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfe C, Zaccaria P, Hoertnagel K, Jaksch M, Klopstock T, et al. MITOP: Database for mitochondria-related proteins, genes and diseases. Nucleic Acids Res. 1999;27:153–155. doi: 10.1093/nar/27.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershan JA, Karrer KM. A family of developmentally excised DNA elements in Tetrahymena is under selective pressure to maintain an open reading frame encoding an integrase-like protein. Nucleic Acids Res. 2000;28:4105–4112. doi: 10.1093/nar/28.21.4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao H, Tu Z. Expanding the diversity of the IS630-Tc1-mariner superfamily: discovery of a unique DD37E transposon and reclassification of the DD37D and DD39D transposons. Genetics. 2001;159:1103–1115. doi: 10.1093/genetics/159.3.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogurtsov AY, Roytberg MA, Shabalina SA, Kondrashov AS. OWEN: Aligning long collinear regions of genomes. Bioinformatics. 2002;18:1703–1704. doi: 10.1093/bioinformatics/18.12.1703. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Eddy SR, Birney E, Bateman A, Durbin R. Pfam: Multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 1998;26:320–322. doi: 10.1093/nar/26.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Ren Q, Paulsen IT. Comparative analyses of fundamental differences in membrane transport capabilities in prokaryotes and eukaryotes. PLoS Comput Biol. 2005;1:e27. doi: 10.1371/journal.pcbi.0010027. DOI: 10.1371/journal.pcbi.0010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldauf SL. The deep roots of eukaryotes. Science. 2003;300:1703–1706. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Scaffolds larger than 1 Mb were sorted by size and concatenated to make a pseudo molecule. Statistics of nucleotide composition were calculated for 2,000 bp sliding windows with a shift length of 1,000 bp. Yellow, GC skew; blue, GC%; purple, χ2 score. The green lines delimit the scaffolds (long) or contigs within each scaffold (short).
(B) Analysis of three T. thermophila scaffolds of diverse size. Red boxes, genes on forward strand; green boxes, genes on reverse strand; blue, χ2 score; orange, GC%; brown, GC skew; salmon, AT skew. The vertical light gray lines delimit contigs within each scaffold. Scaffold sizes: 8254645, 1,076 kb; 8254654, 510 kb; 8254072, 37.3 kb.
(246 KB PDF)
Using scaffolds larger than 100 kb, the percentage of predicted gene coding sequence was calculated within 10-kb windows. For the overall gene density (black bars), a sliding 10-kb window was applied at 2-kb intervals. Gray bars represent gene density in the 10-kb adjacent to each telomere.
(92 KB PDF)
Comparison of the percentage of introns in various size classes for both ab initio predicted genes (gray bars) and introns confirmed by EST sequencing (black bars).
(17 KB PDF)
(A) tRNA charging and expression. Total RNA was harvested from T. thermophila in log-phase growth (lanes 1 and 2) or after resuspension in 10 mM Tris starvation buffer for the times indicated. Total RNA samples were resolved by acid/urea acrylamide gel electrophoresis and transferred to nylon membrane; the same total RNA sample either untreated or deacylated at alkaline pH was used for lanes 1 and 2. Probing was performed using end-radiolabeled oligonucleotides specific for the tRNA of interest.
(B) Expression levels of ncRNAs under various conditions. Total RNA was harvested from T. thermophila under the growth or development conditions indicated, resolved, transferred, and probed as in (A). As an internal control for even loading, the same blot was hybridized to detect tRNA-Sec and SRP RNA (RNA PolIII transcripts found predominantly in the cytoplasm and involved in translation) and also to U1 and U2 snRNAs (RNA PolII transcripts found predominantly in the nucleus and involved in mRNA splicing).
(420 KB PDF)
Orange points represent scaffolds that have been capped with telomeres at both ends.
(30 KB PDF)
Neighbor-joining tree built from ClustalW alignment of polo kinase domains. Species abbreviations: Hs, H. sapiens; Dm, D. melanogaster; Ce, Caenorhabditis elegans; Sc, S. cerevisiae; Dd, D. discoideum; Tt, T. thermophila. Note that T. thermophila has multiple members of both the polo and sak subfamilies, and that even within the T. thermophila–specific cluster, sequences are as divergent as orthologs from vertebrates and lower metazoans. The bar indicates scale of average substitutions per site.
(71 KB PDF)
Unrooted neighbor-joining tree for Rab GTPases. Bootstrap values over 40% (from 100 replicates) are indicated near corresponding branches. Predicted T. thermophila genes are in bold. Other Rabs are from H. sapiens (Hs), D. melanogaster (Dm), and S. cerevisiae (Sc). Proposed Rab families [157] are shown in colored blocks. Asterisks indicate Rabs for which there is functional evidence (**) or at least localization data (*) consistent with their groupings. T. thermophila genes cluster with the members of each Rab family except VII and IV (not shown in a box). There are three clades comprised exclusively of T. thermophila gene predictions (clades I, II, and III) shown in dark gray boxes.
(39 KB PDF)
(28 KB DOC)
(52 KB DOC)
(352 KB DOC)
(167 KB DOC)
(30 KB DOC)
(A) 5S.
(B) tRNA.
(C) Other ncRNAs.
(D) tRNA gene IDs.
(1.0 MB DOC)
(388 KB DOC)
(114 KB DOC)
(73 KB DOC)
(1.9 MB DOC)
(A) Kinases.
(B) Membrane transporters.
(C) Proteases.
(D) Cytoskeletal related.
(3.6 MB DOC)
(90 KB DOC)
(134 KB DOC)
(59 KB DOC)
(159 KB DOC)
(25 KB DOC)
(93 KB DOC)