

Abstract
Background: To compare the safety and tolerability of duloxetine with paroxetine and placebo in patients with major depressive disorder (MDD).
Method: Data from four 8-week randomized, double-blind, placebo- and paroxetine-controlled studies of duloxetine for MDD (DSM-IV criteria) were pooled to compare the safety and tolerability of duloxetine 40 to 120 mg/day with paroxetine 20 mg q.d. Two of the 4 trials included a 26-week extension.
Results: The pooled database included 1466 patients (duloxetine, N = 736; paroxetine, N = 359; placebo, N = 371). No deaths occurred in the acute phase trials. Discontinuation rates for adverse events did not differ significantly for duloxetine, 8.0%, and paroxetine, 6.1%. Nausea was the most frequent treatment-emergent adverse event for duloxetine (duloxetine, 14.4%; paroxetine, 12.0%; placebo, 3.8%). Blood pressure and corrected QT (QTc) interval changes were modest and did not differ significantly for the 3 groups. Mean heart rate increased slightly in the duloxetine group, 1.0 beat/minute, and did differ significantly (p < .001) from that in the paroxetine group, but the change is of doubtful importance. Mean changes in laboratory analytes remained within the reference range. Emergent sexual dysfunction was significantly greater among duloxetine- and paroxetine-treated patients than placebo-treated patients (p = .007 vs. duloxetine and p < .001 vs. paroxetine); however, it was significantly lower in duloxetine-treated patients than in paroxetine-treated patients (46.4% vs. 61.4%; p = .015). During the extension phase, weight gain (≥ 7% of initial body weight) was greater in both active-treatment groups than in the placebo group (duloxetine, 10.8%; paroxetine, 13.8%; placebo, 3.1%), but the active-treatment groups did not differ.
Conclusions: Duloxetine is safe and well tolerated in patients with MDD, with safety and tolerability comparable to that of paroxetine.
One of the most important considerations in the choice of an antidepressant is its safety and tolerability. The selective serotonin reuptake inhibitors (SSRIs) replaced the well-established tricyclic antidepressants (TCAs) as agents of first choice in the treatment of depression because of their better safety and tolerability. In recent years, dual-reuptake inhibitors of both serotonin (5-HT) and norepinephrine (NE) have emerged as a new class of antidepressants, referred to as serotonin-norepinephrine reuptake inhibitors (SNRIs). The SNRIs may have a broader spectrum of action than the SSRIs and demonstrate greater efficacy in the treatment of depression and pain associated with depression.1–4 However, currently available antidepressant medications with a dual 5-HT/NE reuptake inhibition mechanism are known to possess safety and tolerability issues, including, but not limited to, cardiovascular and gastrointestinal side effects as well as sexual dysfunction.5–7 These side effects limit the use of SNRIs and may adversely affect long-term treatment adherence. An antidepressant combining the efficacy of a dual-action medication with the safety profile of an SSRI would be desirable.
Duloxetine hydrochloride, also known as (+)-(S)-N-methyl-γ-(1-naphthyloxy)-2-thiophenepropylamine hydrochloride, inhibits the uptake of both 5-HT and NE but lacks significant affinity for muscarinic, histaminergic1, α1-adrenergic, dopaminergic2, 5-HT1A, 5-HT1B, 5-HT1D, 5-HT2A, 5-HT2C, and opioid receptors.8 Compared with venlafaxine, duloxetine's potency for blocking NE reuptake is relatively more equivalent to its potency in blocking 5-HT reuptake (NE/5-HT Ki ratio = 9.4).9 The efficacy of duloxetine in the treatment of major depressive disorder (MDD) has been established in double-blind, placebo-controlled trials.10–13 The present study compared the safety and tolerability of duloxetine over its studied dose range (40–120 mg/day) with paroxetine at 20 mg q.d. in patients with MDD. This report presents the findings from 4 double-blind, placebo-controlled clinical trials that used paroxetine as an active comparator and evaluated the safety and tolerability of oral duloxetine in patients with MDD. These 4 studies include all of the placebo-controlled comparisons of duloxetine and paroxetine performed by Eli Lilly and Company.
METHOD
Study Design
Data from 4 clinical trials were included in the analysis (Table 1). All trials were multisite, randomized, double-blind, placebo-controlled studies with paroxetine as an active comparator.
Table 1.
Basic Study Information
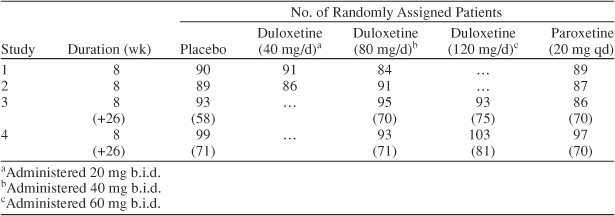
The studies incorporated variable-duration placebo lead-in and lead-out periods in order to blind patients and investigators to the start and end of active therapy. All 4 studies featured an 8-week acute treatment phase in which patients were randomly assigned to receive placebo, paroxetine (20 mg q.d.), or variable doses of duloxetine. In studies 1 and 2, the duloxetine dose was either 40 mg/day (20 mg b.i.d.) or 80 mg/day (40 mg b.i.d.), whereas in studies 3 and 4, the duloxetine dose was either 80 mg/day (40 mg b.i.d.) or 120 mg/day (60 mg b.i.d.). In studies 1 and 2, patients were started on treatment with the fixed dose specified. In studies 3 and 4, patients followed a forced-dose titration schedule. Patients randomly assigned to duloxetine 80 mg/day received duloxetine 20 mg b.i.d. for 3 days, and then the dose was increased to 40 mg b.i.d. Patients randomly assigned to duloxetine 120 mg/day received duloxetine 20 mg b.i.d. for 3 days, then 40 mg b.i.d. for 3 days, and then the dose was increased to 60 mg b.i.d. No dose titration was used for patients assigned to paroxetine 20 mg q.d. Patients in studies 3 and 4 who had a ≥ 30% improvement in the 17-item Hamilton Rating Scale for Depression (HAM-D17)14 total score during acute treatment continued to receive the same treatment for an additional 26 weeks in an extension phase. The extension phase was included in this report in order to determine the cumulative effect of medication on weight and sexual dysfunction.
Study protocols were approved by the ethics committee at each site in accordance with the principles of the Declaration of Helsinki, and all patients provided informed consent before the administration of any study procedures or study drug.
Patients
All study patients were at least 18 years of age and met the criteria for MDD as defined by DSM-IV. In addition, patients had both a Clinical Global Impressions-Severity of Illness scale15 rating ≥ 4 (moderate) and a clinician-rated HAM-D17 total score ≥ 15 at the screening and baseline study visits. Patients were excluded if they had any current primary DSM-IV Axis I diagnosis other than MDD or any anxiety disorder as a primary diagnosis within the year preceding enrollment; any previous diagnosis of bipolar disorder, psychosis, or schizoaffective disorder; a history of substance abuse or dependence within the past year or a positive urine drug screen; a lack of response to at least 2 adequate courses of antidepressant therapy (at least 4 weeks' duration) within the therapeutic dose range during their current MDD episode; serious suicidal risk; a serious medical illness; or a clinically significant laboratory abnormality.
Safety Assessments
Overall discontinuation rate and adverse events
Measures of safety and tolerability included the incidence of serious adverse events (those involving hospitalization, severe or permanent disability, congenital anomaly, or cancer), adverse events associated with discontinuation of the study, patient-reported treatment-emergent adverse events, and overall rates of study discontinuation due to adverse events. Spontaneously reported adverse events were recorded at each visit.
Cardiovascular measures
Weekly blood pressure and heart rate measurements were obtained with the patient in a supine position. A patient was considered hypertensive if supine systolic blood pressure was ≥ 140 mm Hg and a ≥ 10–mm Hg increase from baseline occurred or if supine diastolic blood pressure was ≥ 90 mm Hg and a ≥ 10–mm Hg increase from baseline occurred. Sustained hypertension was defined as meeting the above hypertension criteria for 3 consecutive visits. As a more sensitive index of elevated heart rate, we determined the percentage of patients with a ≥ 10–bpm increase at any time during treatment. Electrocardiogram (ECG) findings (including mean changes from baseline to endpoint in QT, corrected QT [QTc] intervals, and treatment-emergent prolonged QTc intervals) were evaluated. Treatment-emergent QTc prolongation was defined as a ≥ 30–msec change from baseline.
Laboratory analytes
Clinical laboratory tests were performed at screening and at the end of acute treatment.
Weight changes
Weight changes were recorded at each visit. Mean changes in weight were assessed using a likelihood-based repeated-measures approach. Longer-term data were obtained from extension phases in 2 of the trials (studies 3 and 4), in which acute treatment responders received placebo, duloxetine (80–120 mg/day), or paroxetine (20 mg q.d.) for an additional 26 weeks. In addition to mean change in weight, weight gain of at least 7% of initial weight was evaluated. (Weight gain ≥ 7% has been reported as an indicator of clinically important weight gain.16) The incidence of weight changes ≥ 7% at endpoint was compared using Fisher exact test.
Sexual dysfunction
The Arizona Sexual Experience Scale (ASEX) was used in all 4 studies to assess sexual function. The ASEX, developed by McGahuey et al.,17 is a 5-question patient-rated scale investigating interest or drive, psychological arousal, erection/lubrication, ease of achieving orgasm, and satisfaction with orgasm. Responses are measured on a 6-point scale with the total score varying from 5 to 30. Items 3 through 5 are asked only if the patient is sexually active. The ASEX was designed to be bimodal: lower scores indicate increased sexual function, and higher scores indicate decreased sexual function. Total scores near the middle of the range should reflect generally normal sexual function. McGahuey et al.17 defined sexual dysfunction as a total score of ≥ 19, a score of ≥ 5 on any item, or a score of ≥ 4 on any 3 items. The ASEX was administered prior to randomization (baseline), at the end of acute treatment, or at the visit at which a patient discontinued from the trial. In studies 3 and 4, the ASEX was also administered at the end of the extension phase or at the visit at which a patient discontinued from the trial.
Statistical Method
All analyses were conducted on an intent-to-treat basis. All randomly assigned patients were included in the analyses. Data were integrated from the 4 studies with duloxetine dosages ranging from 40 to 120 mg/day pooled as duloxetine in the analyses. Changes from baseline to endpoint (the last nonmissing observation during post-baseline visits) on continuous safety measures, including blood pressure, weight, laboratory analytes, and ECG parameters, were evaluated by an analysis-of-variance (ANOVA) model with the terms of treatment and study. Unless otherwise specified, categorical safety measures (e.g., the incidence of treatment-emergent adverse events) were evaluated using the Fisher exact test.
The primary analytic approach used to assess the incidence of sexual dysfunction, as defined by ASEX criteria, was a generalized linear logistic regression model that included the terms protocol, baseline category, treatment, baseline category-by-treatment interaction, and baseline score (sum of questions 1 and 2). The significance of treatment group differences was assessed with a t test of the logit scale outcomes. The t test requires assumptions regarding normality to be valid. While this would likely not be the case for the observed scale ASEX data (yes/no outcome), this approach was valid because in the generalized linear regression approach, the t test is applied to the “pseudo” variable based on the logit scale data, which does satisfy the normality assumptions.
In analyses of individual ASEX questions, dysfunction was defined as a score ≥ 5. Additionally, at each time point after baseline, patients were categorized as having improved (decrease in score), worsened (increase in score), or remained the same (no change in score) on the total ASEX score and individual items. Differences between treatment groups were then assessed using the Fisher exact test. Treatment effects were tested at a 2-sided significance level of .05, and interaction effects were tested at a significance level of .10.
RESULTS
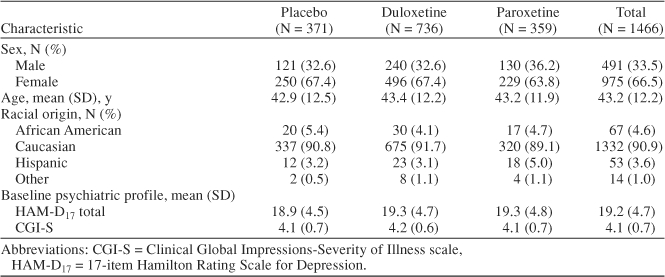
A total of 1466 patients were randomly assigned to placebo (N = 371), duloxetine (N = 736), or paroxetine (N = 359). During the extension phase of studies 3 and 4, 129 patients received placebo; 297, duloxetine; and 140, paroxetine. Basic information for each study is summarized in Table 1. Baseline patient demographics are described in Table 2.
Table 2.
Baseline Patient Demographics
Overall Discontinuation Rate and Adverse Events
There were no deaths during the 8-week acute treatment phase of the studies. Serious adverse events occurred in all groups but were rare. Serious adverse events occurred in 1 placebo patient (0.3%), 2 duloxetine patients (0.3%), and 4 paroxetine patients (1.1%). No statistically significant differences were observed between any of the 3 groups for all reported serious adverse events.
Overall discontinuation rates for any reason did not significantly differ between any of the 3 groups (Figure 1). The incidence of discontinuation due to adverse events was significantly greater for the duloxetine-treated group when compared with the placebo-treated group (8.0% vs. 4.0%, respectively; Fisher exact test, p = .015). However, the rates of discontinuation due to adverse events did not differ significantly between the duloxetine- and paroxetine-treated groups (8.0% vs. 6.1%; Fisher exact test, p = .325) (Figure 1). Rates of discontinuation due to any individual adverse event did not differ significantly between duloxetine- and paroxetine-treated groups. Nausea was the only adverse event for which the discontinuation rate in duloxetine-treated patients was significantly greater than the rate seen for placebo-treated patients (1.2% vs. 0.0%; Fisher exact test, p = .033). Discontinuation because of lack of efficacy was significantly more likely among placebo-treated patients (12.0%) than among duloxetine-treated patients (3.9%; Fisher exact test, p < .001) or among paroxetine-treated patients (5.3%; Fisher exact test, p = .001) (Figure 1).
Figure 1.
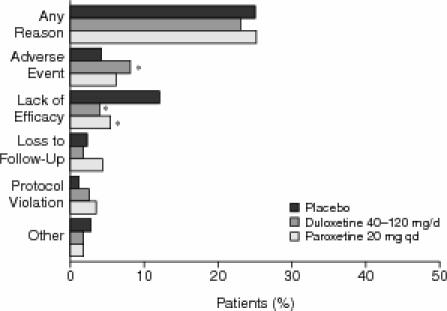
Overall Discontinuation Rates for Any Reason Among Patients Receiving Placebo (N = 371), Duloxetine (40–120 mg/day; N = 736), or Paroxetine (20 mg q.d.; N = 359) During the 8-Week Treatment Phasea
Treatment-emergent adverse events are summarized in Table 3. Among duloxetine-treated patients, the following adverse events had an incidence > 5% and twice the incidence for placebo-treated patients: nausea (14.4%), constipation (10.3%), insomnia (9.0%), dry mouth (8.6%), somnolence (5.8%), increased sweating (5.7%), and fatigue (5.4%). However, none of these adverse-event rates differed significantly between the duloxetine- and paroxetine-treated groups, except for decreased appetite, which occurred in 4.2% of duloxetine-treated patients versus 1.4% of paroxetine-treated patients (Fisher exact test, p = .017).
Table 3.
Treatment-Emergent Adverse Events
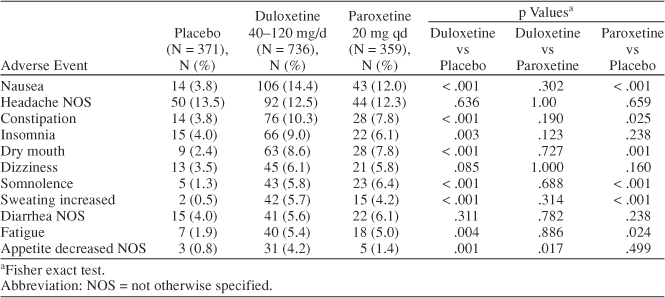
Nausea was the most frequently observed treatment-emergent adverse event among duloxetine-treated patients, occurring at a rate of 14.4% (106/736). In the fixed-dose studies (studies 1 and 2), the rate of emergent nausea increased with dose. At 40 mg/day, the rate was 16.4% (29/177), but at 80 mg/day, the rate was 25.7% (45/175). In studies 3 and 4, in which dose was titrated to a target dose, rates of nausea were lower: 9.6% (18/188) at 80 mg/day and 7.1% (14/196) at 120 mg/day.
Safety
Cardiovascular assessments
Mean baseline-to-endpoint changes in both supine systolic and diastolic blood pressure for duloxetine-treated patients were 0.6 mm Hg and did not increase markedly by dose. Those mean changes did not differ significantly from the corresponding changes in paroxetine-treated patients (Table 4). The rates of treatment-emergent sustained hypertension (defined above) were 1.6% (placebo group), 1.5% (duloxetine group), and 0.28% (paroxetine group). The hypertension rate in the duloxetine-treated patients did not differ significantly from that of the placebo- or paroxetine-treated patients.
Table 4.
Mean Change From Baseline to Endpoint for Vital Signs, Body Weight, and QTc
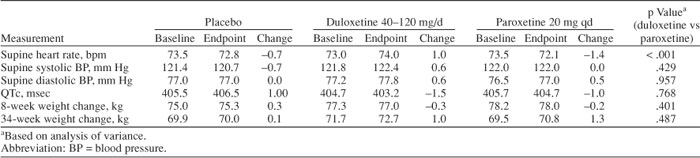
Duloxetine-treated patients exhibited a mean baseline-to-endpoint increase in supine heart rate of 1.0 bpm, compared with mean decreases of 0.7 bpm in placebo-treated patients and 1.4 bpm in paroxetine-treated patients (Table 4). The difference between mean rates for duloxetine and paroxetine was statistically significant (ANOVA, p < .001). The difference in the heart rate in duloxetine-and placebo-treated patients was small and would be of doubtful clinical importance for most patients; however, the mean value may fail to inform about the number of patients with a meaningful increase. To apply a more conservative and sensitive measure, we determined the percentage of patients who experienced a 10-bpm increase at any time during the trial and found that 27.0% of the placebo patients, 32.0% of the duloxetine patients, and 29.0% of the paroxetine patients experienced a 10-bpm increase at any time point. None of the between-group comparisons was statistically significant.
Duloxetine had little effect on QTc intervals or other cardiac intervals. The mean changes in the QTc from baseline to endpoint were −1.5 msec (duloxetine-treated patients), −1.0 msec (paroxetine-treated patients), and +1.0 msec (placebo-treated patients). These changes were not statistically significant or clinically meaningful.
Laboratory values
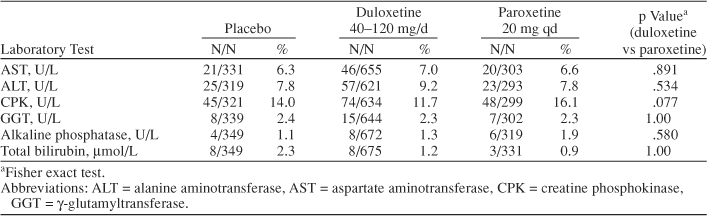
Although statistically significant mean changes in alkaline phosphatase, aspartate amino-transferase (AST), alanine aminotransferase (ALT), and uric acid were observed between duloxetine-treated and placebo-treated patients, these mean changes were within the normal reference range and thus did not appear to be clinically relevant. Rates of abnormal values, present at any time, were also determined (Table 5). No differences in these rates were noted among the treatment groups. We also examined the rates of enzyme elevation > 3 times higher than normal. For ALT, these rates were 0.8% (duloxetine group), 0.3% (placebo group), and 0% (paroxetine group). For AST, the rates were 0.15% (duloxetine group), 0% (placebo group), and 0.3% (paroxetine group). None of the between-group comparisons was statistically significant (Table 5).
Table 5.
Analysis of Laboratory Analytes (treatment-emergent abnormal values at any time)
Sexual functioning
The ASEX ratings were available for the 1466 patients in the 4 studies. Overall rates of sexual dysfunction (based on the main effect of treatment) at the end of acute treatment were 49.3% for placebo-, 55.9% for duloxetine-, and 62.7% for paroxetine-treated patients. The difference in the ASEX total scores between paroxetine- and placebo-treated patients was significant (t = 2.79, df = 1337, p = .005), the difference between duloxetine- and placebo-treated patients was not (t = 1.54, df = 1337, p = .123), and the difference between duloxetine- and paroxetine-treated patients approached significance (t = 1.76, df = 1337, p = .078). However, baseline ratings indicated that a substantial number of patients met the criterion for sexual dysfunction before treatment. Because the presence or absence of sexual dysfunction at baseline might play an important role in understanding the effect of medications, patients with and without dysfunction were examined separately.
A total of 870 patients (59.3%) met criteria for sexual dysfunction at baseline. During acute treatment, sexual dysfunction resolved in 33.3% of these patients, and this rate rose to 42.0% during extended treatment; however, rates of resolution did not differ among the treatment groups during the acute or extended treatment. Approximately 35.0% of the 870 patients experienced worsening of sexual dysfunction during acute treatment, but, again, rates of worsening did not differ significantly by treatment group.
Among the 596 patients without sexual dysfunction at baseline, treatment-emergent sexual dysfunction was more frequent with both duloxetine (46.4%) and paroxetine (61.4%) treatments compared with placebo (28.8%; duloxetine vs. placebo, t = 2.69, df = 1337, p = .007; paroxetine vs. placebo, t = 4.30, df = 1337, p < .001; t tests performed on the logit scale values from the logistic regression). However, patients receiving duloxetine had a significantly lower incidence of treatment-emergent sexual dysfunction compared with paroxetine-treated patients (46.4% vs. 61.4%, t = −2.43, df = 1337, p = .015). Individual ASEX items were also examined to determine the rate of treatment-emergent sexual dysfunction (a rating of ≥ 5). Only ease of orgasm significantly worsened relative to placebo in both drug groups.
During the extended-treatment phase, the incidence of sexual dysfunction did not significantly differ among the treatment groups.
Weight changes
Mean changes of weight during both the 8-week and 34-week studies were minimal, ranging from −0.3 kg to +1.3 kg. No statistically significant differences were observed between the duloxetine- and paroxetine-treated groups for these mean changes (Table 4). Rates of weight gain of at least 7% during 34 weeks of treatment are summarized in Figure 2. Both active-treatment groups (duloxetine 80–120 mg/day and paroxetine 20 mg q.d.) had significantly higher incidence of weight gain (duloxetine group: 10.8%, p = .001; paroxetine group: 13.8%, p < .001; Fisher exact test) when compared with that of placebo (3.1%), but the difference in weight gain between the active-treatment groups was not significant (p = .327, Fisher exact test). The number of patients in the extension phase of studies 3 and 4 was not sufficient to definitively characterize longer-term weight changes associated with duloxetine treatment at different doses.
Figure 2.
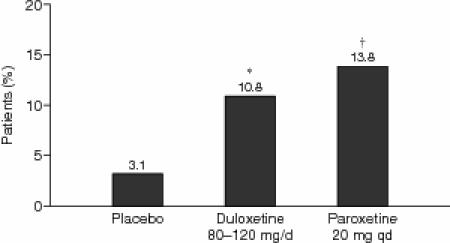
Incidence of ≥ 7% Body Weight Increase From Baseline to Endpoint in Patients Receiving Placebo (N = 192), Duloxetine (80–120 mg/day; N = 381), or Paroxetine (20 mg q.d.; N = 181) During the 34-Week Extended-Treatment Phase
DISCUSSION
Duloxetine appeared to be safe and well tolerated in a dose range from 40 mg/day to 120 mg/day. The overall discontinuation rate due to adverse events for duloxetine-treated patients was only 8.0%, a rate comparable with that for paroxetine-treated patients (6.1%). This result compared favorably with previously reported discontinuation rates for SSRIs (14.9%) and TCAs (19.0%) derived from a meta-analysis18 and also with the discontinuation rate due to adverse events reported for venlafaxine.19 The overall incidence of individual treatment-emergent adverse events associated with duloxetine treatment appeared similar to that for treatment with paroxetine.
Nausea was the most frequently observed treatment-emergent adverse event in the duloxetine group, occurring at an overall rate of 14.4%. This overall rate was comparable with that of paroxetine (12.0%) but varied with the dosing method. The majority of nausea cases occurred early in treatment (within the first 5 days) and were mild to moderate in severity. Only 1.2% of duloxetine-treated patients discontinued study participation due to nausea.
Antidepressants can adversely affect blood pressure. Among the newer antidepressants (SNRIs), venlafaxine has been associated with an increased rate of sustained hypertension.5 In the current study, neither duloxetine nor paroxetine was associated with significant increases in mean blood pressure or sustained hypertension. Duloxetine was associated with a small increase in heart rate, a 1.7-bpm increase compared with placebo. For most patients, this increase in heart rate would not appear to be clinically important. In addition, the percentage of duloxetine-treated patients experiencing a 10-bpm increase in heart rate from baseline did not significantly differ from that for placebo-treated patients. In contrast, TCAs such as nortriptyline, acting primarily on norepinephrine, have been associated with a mean 8-bpm increase in heart rate.20
The incidence of treatment-emergent sexual dysfunction among duloxetine-treated patients compared favorably with that for paroxetine-treated patients. Both drugs were associated with a greater incidence of treatment-emergent sexual dysfunction than placebo; however, patients receiving duloxetine had a significantly lower incidence of sexual dysfunction compared with paroxetine-treated patients (46.4% vs. 61.4%, p = .015). The findings also indicate the value of separate analyses of data for patients with and without sexual dysfunction at baseline. In fact, seldom has sexual dysfunction been assessed in anti-depressant studies before treatment. These data also illustrate that sexual dysfunction may improve in a substantial number of patients during treatment.
Because spontaneous reporting of sexual dysfunction may underestimate the magnitude of this outcome, the ASEX was included in these 4 trials. Although the ASEX has been considered a well-validated scale to assess sexual dysfunction in psychiatric patients,17 other well-established scales such as the Changes in Sexual Functioning Questionnaire (CSFQ)21 are available and have become popular. The CSFQ has more questions and is considered by many to be more inclusive than the ASEX. The studies we report, however, were initiated in 1999 and 2000, when the ASEX was commonly employed. Multiple mechanisms (including serotonergic, dopaminergic, and anticholinergic) have been proposed to account for SSRI-induced sexual dysfunction.22 Serotonergic effects are believed to be the principal cause of treatment-emergent sexual dysfunction during SSRI treatment. The observation that agents that enhance catecholamine function, such as yohimbine and bupropion, appear to have beneficial effects on sexual function suggests that noradrenergic activity may partially mitigate serotonergically induced sexual side effects.23,24 The noradrenergic activity of duloxetine may account for its relatively favorable sexual dysfunction profile compared with paroxetine.
Antidepressants can be associated with weight gain,25 which may in turn lead to patient nonadherence. Both duloxetine and paroxetine were associated with modest weight gain. The number of patients in the extension phase of our studies was not sufficient to definitively characterize longer-term weight changes for duloxetine and paroxetine.
The present studies may have several limitations. First, the ability to generalize the results to typical outpatients is somewhat limited because the study participants had relatively few comorbid medical conditions, few concomitant medications, no current Axis I disorder other than MDD, no current substance abuse, no prior anxiety disorder in the past year, and no prior diagnosis of psychosis. Moreover, no inpatients were included in the present studies. Further studies will be required to address duloxetine's safety in these populations. Second, the present studies employed a forced dose-titration schedule, which is not typical of clinical practice and may have resulted in somewhat different findings than a schedule in which individual dose titrations were permitted. Moreover, analyses of safety measures were likely to be underpowered, and therefore negative findings (i.e., lack of statistical significance) should always be interpreted in light of the magnitude of the difference and its clinical importance.
In conclusion, duloxetine appears to be a safe and well-tolerated SNRI antidepressant in the acute (8 weeks) and longer-term (34 weeks) treatment of MDD at doses from 40 mg/day to 120 mg/day. The safety and tolerability of duloxetine appeared comparable to that for paroxetine.
Drug names: bupropion (Wellbutrin and others), duloxetine (Cymbalta), nortriptyline (Aventyl, Pamelor, and others), paroxetine (Paxil, Pexeva, and others), venlafaxine (Effexor).
Acknowledgments
The authors thank the Du-Flu Product Team, the many patients for their voluntary participation in this clinical trial, and the principal investigators. They also thank Eli Lilly Cymbalta statistical analysts for their programming support and their assistance with statistical analyses, and Svetlana Dominguez, B.A., for editorial assistance.
Footnotes
This work was sponsored by Eli Lilly and Co.
A portion of the data was presented at the 156th annual meeting of the American Psychiatric Association, May 17–22, 2003, San Francisco, Calif.
Dr. Nelson has received honoraria from Abbott, Eli Lilly, Forest, GlaxoSmithKline, Organon, Pfizer, and Wyeth Ayerst; has been a consultant/advisory board member for Abbott, Biovail, Bristol-Myers Squibb, Corcept, Eli Lilly, GlaxoSmithKline, Orexigen, Organon, Pfizer, Sepracor, and Shire; and has received research support from Eli Lilly and Organon. Dr. Pritchett is an employee of Abbott. Dr. Martynov is an employee of Amylin. Drs. Yu, Mallinckrodt, and Detke are employees of Eli Lilly and Co., and Dr. Detke is a major stock shareholder in Eli Lilly.
All authors accept full responsibility for the conduct of this analysis, were given full access to all data from the analysis, and participated in the decision to publish the data.
REFERENCES
- Faravelli C, Cosci F, and Ciampelli M. et al. A self-controlled, naturalistic study of selective serotonin reuptake inhibitors versus tricyclic antidepressants. Psychother Psychosom. 2003 72:95–101. [DOI] [PubMed] [Google Scholar]
- Thase ME, Entsuah AR, Rudolph RL. Remission rates during treatment with venlafaxine or selective serotonin reuptake inhibitors. Br J Psychiatry. 2001;178:234–241. doi: 10.1192/bjp.178.3.234. [DOI] [PubMed] [Google Scholar]
- Steffens DC, Krishnan KR, Helms MJ. Are SSRIs better than TCAs? comparison of SSRIs and TCAs: a meta-analysis. Depress Anxiety. 1997;6:10–18. doi: 10.1002/(sici)1520-6394(1997)6:1<10::aid-da2>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Nelson JC, Mazure CM, and Jatlow PI. et al. Combining norepinephrine and serotonin reuptake inhibition mechanisms for treatment of depression: a double-blind, randomized study. Biol Psychiatry. 2004 55:296–300. [DOI] [PubMed] [Google Scholar]
- Thase ME. Effects of venlafaxine on blood pressure: a meta-analysis of original data from 3744 depressed patients. J Clin Psychiatry. 1998;59:502–508. doi: 10.4088/jcp.v59n1002. [DOI] [PubMed] [Google Scholar]
- Kennedy SH, Dickens SE, and Eisfeld BS. et al. Sexual dysfunction before antidepressant therapy in major depression. J Affect Disord. 1999 56:201–208. [DOI] [PubMed] [Google Scholar]
- Glassman AH. Cardiovascular effects of antidepressant drugs: updated. J Clin Psychiatry. 1998 59suppl 15. 13–18. [PubMed] [Google Scholar]
- Bymaster FP, Dreshfield-Ahmad LJ, and Threlkeld PG. et al. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001 25:871–880. [DOI] [PubMed] [Google Scholar]
- Wong DT, Bymaster FP. Dual serotonin and noradrenaline uptake inhibitor class of anti-depressants: potential for greater efficacy or just hype? Prog Drug Res. 2002;58:169–222. doi: 10.1007/978-3-0348-8183-8_5. [DOI] [PubMed] [Google Scholar]
- Detke MJ, Wiltse CG, and Mallinckrodt CH. et al. Duloxetine in the acute and long-term treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur Neuropsychopharmacol. 2004 14:457–470. [DOI] [PubMed] [Google Scholar]
- Detke MJ, Lu Y, and Goldstein DJ. et al. Duloxetine, 60 mg once daily, for major depressive disorder: a randomized double-blind placebo-controlled trial. J Clin Psychiatry. 2002 63:308–315. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Mallinckrodt C, and Lu Y. et al. Duloxetine in the treatment of major depressive disorder: a double-blind clinical trial. J Clin Psychiatry. 2002 63:225–231. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Lu Y, and Detke M. et al. Duloxetine in the treatment of depression: a double-blind placebo-controlled comparison with paroxetine. J Clin Psychopharmacol. 2004 24:389–399. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy W. ECDEU Assessment Manual for Psychopharmacology. US Dept Health, Education, and Welfare publication (ADM) 76-338. Rockville, Md: National Institute of Mental Health. 1976 218–222. [Google Scholar]
- Kanders BS, Forse RA, and Blackburn GL. Methods in obesity. In: Rakel RE, ed. Conn's Current Therapy, 1992. Philadelphia, Pa: WB Saunders Company. 1991 524–532. [Google Scholar]
- McGahuey CA, Gelenberg AJ, and Laukes CA. et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. J Sex Marital Ther. 2000 26:25–40. [DOI] [PubMed] [Google Scholar]
- Montgomery SA, Henry J, and McDonald G. et al. Selective serotonin reuptake inhibitors: meta-analysis of discontinuation rates. Int Clin Psychopharmacol. 1994 9:47–53.Correction. 1994;9:296. [DOI] [PubMed] [Google Scholar]
- Wellington K, Perry CM. Venlafaxine extended-release: a review of its use in the management of major depression. CNS Drugs. 2001;15:643–669. doi: 10.2165/00023210-200115080-00007. [DOI] [PubMed] [Google Scholar]
- Roose SP, Laghrissi-Thode F, and Kennedy JS. et al. Comparison of paroxe-tine and nortriptyline in depressed patients with ischemic heart disease. JAMA. 1998 279:287–291. [DOI] [PubMed] [Google Scholar]
- Clayton AH, McGarvey EL, Clavet GJ. The Changes in Sexual Functioning Questionnaire (CSFQ): development, reliability, and validity. Psychopharmacol Bull. 1997;33:731–745. [PubMed] [Google Scholar]
- Rosen RC, Lane RM, Menza M. Effects of SSRIs on sexual function: a critical review. J Clin Psychopharmacol. 1999;19:67–85. doi: 10.1097/00004714-199902000-00013. [DOI] [PubMed] [Google Scholar]
- Hollander E, McCarley A. Yohimbine treatment of sexual side effects induced by serotonin reuptake blockers. J Clin Psychiatry. 1992;53:207–209. [PubMed] [Google Scholar]
- Clayton AH, Warnock JK, and Kornstein SG. et al. A placebo-controlled trial of bupropion SR as an antidote for selective serotonin reuptake inhibitor–induced sexual dysfunction. J Clin Psychiatry. 2004 65:62–67. [DOI] [PubMed] [Google Scholar]
- Fava M, Judge R, and Hoog SL. et al. Fluoxetine versus sertraline and paroxetine in major depressive disorder: changes in weight with long-term treatment. J Clin Psychiatry. 2000 61:863–867. [DOI] [PubMed] [Google Scholar]