

Abstract
Sturge-Weber syndrome is a neurocutaneous syndrome that manifests with vascular malformations involving the brain, eye, and skin. We report the case of an elderly patient suffering from Sturge-Weber syndrome who presented with episodic “angry” slapping symptoms to a psychiatric facility. A detailed history, physical and mental-state examination, and elaborate neuropsychological, neuroimaging, and laboratory assessment were undertaken. Clinically, the “angry” slapping episodes were diagnosed as complex partial seizures that improved in frequency with an increase in the dose of the antiepileptic medication. We also have attempted to identify the pathophysiology of such behavioral episodes in Sturge-Weber syndrome. This case report underlines the need for detailed laboratory and neuroimaging work-up in the elderly presenting with atypical symptoms. It also emphasizes the need for identification of and differentiation between similar atypical presentations and appropriate management of resources by the medical staff.
Sturge-Weber syndrome (also called encephalofacial or encephalotrigeminal angiomatosis) is a rare neurocutaneous syndrome that occurs with a frequency of approximately 1 per 50,000 and is characterized by facial port-wine stains in the trigeminal nerve distribution, glaucoma, and vascular lesions in the ipsilateral brain and meninges.1–3 The syndrome occurs almost entirely sporadically and with equal frequency in both sexes.4 Sporadic somatic mutations that embryologically disrupt local angiogenesis have been hypothesized as the etiologic basis of Sturge-Weber syndrome, though a “2-hit hypothesis” (involving sporadic mutations as well as familial occurrence) has also been suggested.5,6
Clinically, patients typically present with cutaneous angiomatosis, glaucoma, and variable neurologic manifestations including seizures, mental retardation, hemianopia, hemiparesis, and learning difficulties.1,5,7 The cutaneous lesions are irregular in shape and are usually located in the dermatome of the ophthalmic division of the trigeminal nerve. Glaucoma is usually open-angle type, almost always ipsilateral to the port-wine stain, and can cause progressive visual loss. Epilepsy, mental retardation, and focal neurologic deficits are commonly associated with Sturge-Weber syndrome. While developmental milestones are usually normal, mild to moderate mental retardation is believed to develop in about half of the patients.1
Patients suffering from Sturge-Weber syndrome most often present to the neurologist for the management of seizures and are very rarely hospitalized in a psychiatric unit. In our case report, we provide the details of such a presentation and highlight the need for identification of periictal behavioral phenomena associated with Sturge-Weber syndrome.
CASE REPORT
An 82-year-old left-handed white man was hospitalized for evaluation, diagnosis, and management of an episode of odd, assaultive behavior and anger spells. The patient had been living in a retirement community facility for 6 years. On the day of hospitalization, he smelled smoke coming into his apartment from the hallway in the morning and called 911. Thereafter, he also allegedly slapped the nurse who came to his apartment for inspection. The police, the firemen, and the staff did not find any evidence of smoke in either the hallway or his apartment. Subsequently, the patient was transferred to the hospital for evaluation and management of his odd and aggressive behavior. During the interview, he was very pleasant and remembered smelling the smoke and slapping the nurse. He, however, had no recollection of events thereafter, including his transfer to the hospital.
There was no associated history of headaches, head trauma, abnormal involuntary movements, geographical disorientation, significant memory lapses, incontinence of urine, or weakness or numbness of limbs. The patient's psychiatric history was negative for depression, anxiety, mood swings, psychosis, or violence, although he described having “anger management issues” in the past. His medical history was significant for seizures, diabetes mellitus, hypertension, glaucoma, and L1 compression. The seizures had an onset at age 22; they were characterized by lip smacking and finger movements but were not associated with generalized tonic-clonic movements. The frequency of seizures had decreased with a combination of lamotrigine and gabapentin, with the last seizure reported 7 months prior to this admission. His other medications included insulin, timolol eye drops, calcium carbonate, and alendronate. There was no significant family psychiatric or medical history, and he had no known allergies. The patient had worked as a florist in the past and had been retired for 20 years.
Physical examination revealed a facial port-wine stain in the left trigeminal nerve distribution. Neurologic examination, including cranial nerves, motor, sensory, and cerebellar examination, was within normal limits and did not reveal any neurologic deficits. On mental-status examination, the patient was calm and cooperative. His mood was euthymic and his affect appropriate. His speech and thought processes were coherent, goal-directed, and logical. No depressive or psychotic features could be elicited, though the patient reported having anger-control problems. He scored 29/30 on the Mini-Mental State Examination (MMSE),8 with no evidence suggestive of delirium. Laboratory results, including complete blood count, complete metabolic profile, thyroid function tests, and B12 and folate levels, were within normal limits.
The differential diagnosis of Sturge-Weber syndrome presenting with complex partial seizures was considered along with cognitive disorder not otherwise specified and impulse-control disorder not otherwise specified. We increased the patient's lamotrigine dose from 300 mg/day to 325 mg/day and added 0.25 mg of risperidone to his treatment regimen for managing a possible episode of aggression. Lamotrigine levels were found to be 8.7 µg/mL, and a repeat MMSE examination showed no change from the baseline. Neuropsychological testing revealed a Wechsler Adult Intelligence Scale-Revised (WAIS-R)9 IQ score of 117, with the vocabulary subscore in the high-average range and the abstract subscore in the superior range. These were essentially unchanged from previous assessments done in 1952 (Wechsler-Bellevue Intelligence Scale) and 1958 (WAIS). He performed very well with items requiring abstract reasoning and new-problem skills. The tests also revealed no problems with spelling, language, grammar, or visuospatial skills. Magnetic resonance imaging of the head revealed abnormal signal intensity in the anterior left temporal lobe, especially the left hippocampus, on both T2-weighted and fluid-attenuated inversion-recovery (FLAIR) imaging with faint contrast enhancement. The gyri were slightly enlarged as compared with the opposite temporal lobe, as well. This constellation of findings was further suggestive of Sturge-Weber syndrome, with complex partial seizures as the primary diagnosis, presenting as repetitive anger spells.
The hospital stay was largely uneventful except for an episode in which the patient was noticed to have a vacant stare, followed by repetitive slapping of his face lasting a few seconds. The patient had no recollection of the episode. While electroencephalogram in the interictal period revealed minimal abnormality, this episode confirmed Sturge-Weber syndrome with complex partial seizures as the primary diagnosis, manifesting as repetitive anger and spells of aggression. The patient was discharged back to the retirement community on a regimen of 0.25 mg/day of risperidone and 325 mg/day of lamotrigine. He has been free of symptoms during 3 months of outpatient follow-up.
DISCUSSION
Sturge-Weber syndrome classically presents with a facial port-wine stain (trigeminal nerve distribution), glaucoma, and vascular malformations in the brain, especially the parieto-occipital region.10–12 Sturge-Weber syndrome occurs with equal frequency in both sexes, with seizures typically developing in the first year of life.4,10 The associated neurologic problems include seizures, migraines, mental retardation, hemiparesis, and learning difficulties.5,10,13,14 Glaucoma, especially open-angle, is believed to occur in 30% of the patients diagnosed with Sturge-Weber syndrome.5,10 The classic vascular malformations possibly result from a failure of the primitive cephalic venous plexus to regress during vascular development.5,15 The association between the facial and brain vascular malformation is believed to result from close proximity of the facial ectoderm with the neural tube region. While the genetic and environmental factors involved in the etiology of Sturge-Weber syndrome are unclear, somatic mutations affecting the fibroblasts have been recently cited as a probable cause.6,16 It has been hypothesized that in Sturge-Weber syndrome, abnormal cortical veins interacting with the overlying angioma produce thrombosis, thus resulting in cortical ischemia, in turn stimulating the angiogenesis seen in the syndrome.5 The associated cortical dysgenesis manifests as seizures that result in tissue hypoxia, further leading to altered vascular function and epileptogenesis (Figure 1). The seizure phenomena have been previously reported to manifest with atypical behavioral presentations including brief ictal and interictal psychosis.17
Figure 1.
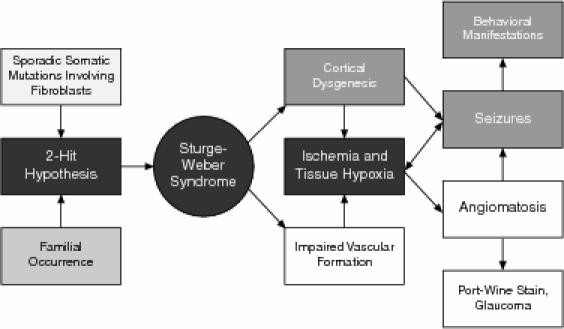
Hypothesized Pathophysiology in Sturge-Weber Syndrome and Correlation With Behavioral Symptoms
In our case, the patient presented with “anger problems” manifesting as slapping the nurse. This episode involving “anger issues” can, however, be explained as a complex partial seizure phenomenon characterized by an olfactory aura (smell of something burning), followed by ictal automatism (slapping) and amnesia for the event. A similar episode was observed during the inpatient stay, when he suddenly stopped eating his meal and slapped himself for a few seconds but had no recollection of this later on. The sudden onset, very short duration of the episode, and favorable response to an increase in lamotrigine dosage further corroborated the diagnosis.
This case underlines the need for a complete laboratory, neuroimaging, and neuropsychological work-up in elderly individuals presenting with atypical behavioral manifestations. It further emphasizes the need for sensitization of the medical and nursing staff to similar clinical presentations and appropriate management of available resources.
CONCLUSIONS
Sturge-Weber syndrome is a neurocutaneous syndrome characterized by vascular and cortical dysgenesis including facial and intracranial angiomas. The epileptic phenomena of Sturge-Weber syndrome can manifest with atypical psychiatric symptoms and need correct identification and appropriate response for optimal management by medical staff.
Drug names: alendronate (Fosamax), gabapentin (Neurontin and others), lamotrigine (Lamictal), risperidone (Risperdal), timolol (Betimol, Istalol, and others).
Footnotes
Dr. Dewan has been a consultant for and has been on the speakers or advisory board of Bristol-Myers Squibb; Drs. Madaan, Ramaswamy, and Sharma report no financial or other relationships relevant to the subject of this article.
REFERENCES
- Thomas-Sohl KA, Vaslow DF, Maria BL. Sturge-Weber syndrome: a review. Pediatr Neurol. 2004;30:303–310. doi: 10.1016/j.pediatrneurol.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Baselga E. Sturge-Weber syndrome. Semin Cutan Med Surg. 2004;23:87–98. doi: 10.1016/j.sder.2004.01.002. [DOI] [PubMed] [Google Scholar]
- Smirniotopoulos JG. Neuroimaging of phakomatoses: Sturge-Weber syndrome, tuberous sclerosis, von Hippel-Lindau syndrome. Neuroimaging Clin North Am. 2004;14:171–183. doi: 10.1016/j.nic.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Bodensteiner JB, Roach ES. Sturge-Weber syndrome: introduction and overview. In: Bodensteiner JB, Roach ES, eds. Sturge-Weber Syndrome. Mt. Freedom, NJ: The Sturge-Weber Foundation. 1999 1–10. [Google Scholar]
- Comi AM. Pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509–516. doi: 10.1177/08830738030180080701. [DOI] [PubMed] [Google Scholar]
- Huq AH, Chugani DC, and Hukku B. et al. Evidence of somatic mosaicism in Sturge-Weber syndrome. Neurology. 2002 59:780–782. [DOI] [PubMed] [Google Scholar]
- Lisotto C, Mainardi F, and Maggioni F. et al. Headache in Sturge-Weber syndrome: a case report and review of the literature. Cephalalgia. 2004 24:1001–1004. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Wechsler D. Wechsler Adult Intelligence Scale-Revised. New York, NY: Psychological Corporation. 1981 [Google Scholar]
- Paller AS. The Sturge-Weber syndrome. Pediatr Dermatol. 1987;4:300–304. doi: 10.1111/j.1525-1470.1987.tb00797.x. [DOI] [PubMed] [Google Scholar]
- Enjolras O, Riche MC, Merland JJ. Facial port-wine stains and Sturge-Weber syndrome. Pediatrics. 1985;76:48–51. [PubMed] [Google Scholar]
- Hussain MS, Emery DJ, and Lewis JR. et al. Sturge-Weber syndrome diagnosed in a 45-year-old man. CMAJ. 2004 170:1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapper J. Headache in Sturge-Weber syndrome. Headache. 1994;34:521–522. doi: 10.1111/j.1526-4610.1994.hed3409521.x. [DOI] [PubMed] [Google Scholar]
- Chapieski L, Friedman A, Lachar D. Psychological functioning in children and adolescents with Sturge-Weber syndrome. J Child Neurol. 2000;15:660–665. doi: 10.1177/088307380001501004. [DOI] [PubMed] [Google Scholar]
- Taly AB, Nagaraja D, and Das S. et al. Sturge-Weber-Dimitri disease without facial nevus. Neurology. 1987 37:1063–1064. [DOI] [PubMed] [Google Scholar]
- Comi AM, Hunt P, and Vawter MP. et al. Increased fibronectin expression in Sturge-Weber syndrome fibroblasts and brain tissue. Pediatr Res. 2003 53:762–769. [DOI] [PubMed] [Google Scholar]
- Sachdev P. Schizophrenia-like psychosis and epilepsy: the status of the association. Am J Psychiatry. 1998;155:325–336. doi: 10.1176/ajp.155.3.325. [DOI] [PubMed] [Google Scholar]