

Abstract
The epithelial Na+ channel (ENaC) is composed of three homologous subunits: α, β and γ. We used gene targeting to disrupt the β subunit gene of ENaC in mice. The βENaC-deficient mice showed normal prenatal development but died within 2 days after birth, most likely of hyperkalemia. In the −/− mice, we found an increased urine Na+ concentration despite hyponatremia and a decreased urine K+ concentration despite hyperkalemia. Moreover, serum aldosterone levels were increased. In contrast to αENaC-deficient mice, which die because of defective lung liquid clearance, neonatal βENaC deficient mice did not die of respiratory failure and showed only a small increase in wet lung weight that had little, if any, adverse physiologic consequence. The results indicate that, in vivo, the β subunit is required for ENaC function in the renal collecting duct, but, in contrast to the α subunit, the β subunit is not required for the transition from a liquid-filled to an air-filled lung. The phenotype of the βENaC-deficient mice is similar to that of humans with pseudohypoaldosteronism type 1 and may provide a useful model to study the pathogenesis and treatment of this disorder.
The epithelial Na+ channel (ENaC) plays a key role in regulating salt and water homeostasis (1–3). The channel is expressed in the apical membrane of epithelial cells lining the distal nephron, the colon, and the pulmonary airways. Three homologous subunits, α, β, and γ, form the Na+-selective channel (4–8). Each has cytosolic N and C termini, two transmembrane domains, and a large cysteine-rich extracellular domain (9–11). Heterologous expression in Xenopus laevis oocytes has shown that the α subunit alone is sufficient to generate a small amiloride-sensitive Na+ channel (4, 6, 8). However, when the α subunit is coexpressed with the β and γ subunits, the Na+ currents are much larger (5, 7). In contrast, expression of β alone, γ alone, or β plus γ generates no channel activity (5, 7).
The critical function of ENaC is demonstrated by its disruption in human disease. Lack of Na+ channel activity causes the autosomal recessive disease pseudohypoaldosteronism type 1 (PHA1). In PHA1, the genes encoding one of the ENaC subunits bear mutations that are predicted to disrupt subunit function (12, 13). As a result, newborns with PHA1 develop hyponatremia, hyperkalemia, salt wasting, and elevated aldosterone concentrations. Conversely, gain-of-function mutations cause the autosomal-dominant Liddle’s syndrome, characterized by hypertension (14). Liddle’s mutations disrupt a C-terminal proline-rich protein binding site in the β or γ subunit, which increases the number of Na+ channels in the cell membrane, leading to excess Na+ reabsorption (15, 16).
In an initial attempt to understand the physiology of ENaC, the gene encoding the α subunit was disrupted in mice (17). Homozygous mutant mice died from respiratory failure within 40 hr after birth because they failed to clear liquid from their lungs. In this study, we asked whether deletion of the β subunit also would prevent normal channel function in vivo. Because the α subunit is required to generate channel activity in heterologous expression systems, and because coexpression of the γ subunit with the α subunit generates more channel activity than α alone (5, 7), we hypothesized that a mouse missing only the β subunit might have some Na+ channel activity in the lung; thus, it might display a phenotype different from that of an α null mouse. Moreover, because βENaC loss of function mutations have been described in some PHA1 patients (12), we predicted that such a mouse might provide a model for the study of PHA1.
MATERIALS AND METHODS
Construction of the β Mouse ENaC (βmENaC) Gene Targeting Vector.
A 1,488-bp fragment of βmENaC cDNA was amplified from mouse kidney cDNA (CLONTECH) by using degenerate PCR primers corresponding to the amino acids KKKAMWF (5′ primer) and IEFGEII (3′ primer). This DNA fragment was random prime labeled with [32P]dCTP and was used to screen a D3 embryonic stem (ES) cell genomic DNA library (a gift of J. K. Heath, University of Birmingham, U.K.), following standard procedures (18). A number of positive clones were obtained, and, after preliminary restriction mapping and DNA sequencing, a 13.5-kilobase (kb) βmENaC genomic fragment was chosen to construct the gene targeting vector. This clone contained three exons of the βmENaC gene, the most 5′ of which included the presumed start methionine (by comparison with the rat and human βENaC sequences) (Fig. 1A). We designed a replacement gene targeting vector in which the first exon and part of the first intron of βmENaC were replaced with PGKneo (the phosphoglycerate kinase promoter driving expression of the neomycin-resistance gene), which was in the reverse transcriptional orientation. To prepare the gene targeting vector, a 3.8-kb HindIII-EcoRI fragment downstream of the first exon was blunt-end cloned into the XhoI site of pPNT (a gift of R. Mulligan, Whitehead Institute, Massachusetts Institute of Technology). Subsequently, a 3-kb EcoRI-NotI fragment upstream of the first exon was blunt-end cloned into the EcoRI site. This resulted in a total of 6.8 kb of genomic DNA homologous to βmENaC positioned around the PGKneo cassette. The herpes simplex virus thymidine kinase cassette was located outside of the regions of homology at the βmENaC 5′ end. Thus, a gene-targeted event replaces exon 1, or the first 90 amino acids of βmENaC, with the PGKneo cassette.
Figure 1.

Disruption of the mouse βENaC locus. (A) Gene targeting strategy. (B) Southern blot of BamHI-digested genomic DNA from ES cell clones. Lanes 2 and 8 show targeted ES cell lines. HSVtk, herpes simplex virus thymidine kinase.
Production of βENaC-Deficient Mice.
The βmENaC gene
targeting vector was linearized at the unique NotI site, was
gel-purified, and was electroporated into R1 ES cells (A. Nagy, Mount
Sinai Hospital, Toronto). ES cells were selected in G418 and
gancyclovir, and surviving clones were expanded for screening. Genomic
DNA was isolated from clones of ES cells. The cell pellet was incubated
in lysis buffer (10 mM Tris, pH 7.4/1 mM EDTA/100 mM NaCl/1%
SDS/500 mg/ml proteinase K) for 24–48 hr at 37°C. After standard
extraction with phenol:chloroform:isoamyl alcohol (1:1: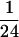 ),
the genomic DNA was precipitated and resuspended in a 10 mM Tris and 1
mM EDTA (pH 8) solution containing 10 μg/ml RNase A. Approximately
25 μg of genomic DNA was digested overnight with BamHI,
was separated on a 1% agarose gel, and was transferred to Hybond N+
(Amersham Pharmacia) overnight in 0.4 M NaOH. A 0.7-kb probe located
downstream from the 3.8-kb βmENaC genomic fragment used for the gene
targeting construct was labeled with [32P]dCTP
and was hybridized to the filters by using standard procedures (18).
The filters were washed twice in 2× standard saline citrate and 0.1%
SDS and once in 0.2× standard saline citrate and 0.1% SDS and was
exposed to X-omat film (Kodak) for 1–5 days. Because a
BamHI site was introduced with the gene targeting construct,
a novel BamHI fragment was observed in targeted ES cell
clones. The probe recognized an ≈18-kb BamHI fragment from
the normal allele whereas an additional BamHI fragment of
≈11 kb was observed in the targeted allele.
),
the genomic DNA was precipitated and resuspended in a 10 mM Tris and 1
mM EDTA (pH 8) solution containing 10 μg/ml RNase A. Approximately
25 μg of genomic DNA was digested overnight with BamHI,
was separated on a 1% agarose gel, and was transferred to Hybond N+
(Amersham Pharmacia) overnight in 0.4 M NaOH. A 0.7-kb probe located
downstream from the 3.8-kb βmENaC genomic fragment used for the gene
targeting construct was labeled with [32P]dCTP
and was hybridized to the filters by using standard procedures (18).
The filters were washed twice in 2× standard saline citrate and 0.1%
SDS and once in 0.2× standard saline citrate and 0.1% SDS and was
exposed to X-omat film (Kodak) for 1–5 days. Because a
BamHI site was introduced with the gene targeting construct,
a novel BamHI fragment was observed in targeted ES cell
clones. The probe recognized an ≈18-kb BamHI fragment from
the normal allele whereas an additional BamHI fragment of
≈11 kb was observed in the targeted allele.
For chimera production, positive ES cell clones either were injected into C57BL/6J blastocysts or were cocultured with eight-cell C57BL/6J embryos. Chimeras then were bred with C57BL/6J females. The genotypes of the pups from heterozygote crosses were determined by PCR by using primer sets to specifically detect normal or targeted alleles. They initially were confirmed by Southern blots. The following primer set was used to amplify across the PGKneo cassette to detect targeted alleles: 5′ primer, 5′-CAACAGACAATCGGCTGCTC-3′; and 3′ primer, 5′-GTCACGACGAGATCCTCGC-3′. This produced a 495-bp fragment. The following primer set was used to detect normal βmENaC alleles: 5′ primer, 5′-GGCAGTGGGGAGTCTTCATC-3′; and 3′ primer, 5′-CTGACCTTGCCTCCATTCCA-3′, which amplified a 339-bp fragment.
Expression Analysis, Histology, and Physiological Measurements.
Reverse transcriptase (RT)–PCR was performed by using a RETROscript kit (Ambion, Austin, TX) according to the manufacturer’s directions. The primers used for the RT-PCR were αENaC forward primer 5′ CTAATGATGCTGGACCACACC 3′ and reverse primer 5′ AAAGCGTCTGCTCCGTGATGC 3′; βENaC forward primers 5′GCCAGTGAAGAAGTACCTGC 3′ and reverse primer 5′CCTGGGTGGCACTGGTGAA 3′; γENaC forward primer 5′AAGAATCTGCCGGTTCGAGGC 3′ and reverse primer 5′TACCACTCCTGGATGGCATTG 3′; and glyceraldehyde-3-phosphate dehydrogenase forward primer 5′CGTCTTCACCACCATGGAGA 3′ and reverse primer 5′ CGGCCATCACGCCACAGTTT 3′. Standard hematoxylin and eosin staining was performed on paraffin-embedded lung and kidney tissues. Serum and urine ion concentrations were measured by a DT60II automatic analyzer (Johnson & Johnson Clinical Diagnostics, Rochester, NY) at the Animal Clinical Laboratory in the University of North Carolina at Chapel Hill, NC. Serum aldosterone levels were measured by using a solid phase radioimmunoassay following the protocols of the manufacturer (Diagnostic Products, Los Angeles). Blood gas analyses were performed by using an AVL 995 (AVL Scientific Corporation, Roswell, GA) on 40 μl of heparinized blood obtained from decapitated newborns. These samples represent mixed arterial and venous blood.
RESULTS
Neonatal Mortality of βmENaC Null Pups.
The scheme for disruption of mouse βENaC by gene targeting is depicted in Fig. 1A. The βmENaC targeting vector was introduced into R1 ES cells by electroporation, and surviving G418 and gancyclovir resistant clones were screened by Southern blotting (Fig. 1B). Six targeted ES cell clones were identified from 196 screened, giving a targeting frequency of ≈3%. Two positive clones were chosen to develop βENaC-deficient mouse lines, and the two lines showed identical phenotypes.
Pups from heterozygous matings were genotyped at three ages (Table 1 and Fig. 2A). When embryos [between embryonic day 12.5 (E12.5) and E19.5] were genotyped, we found that 21% of pups were homozygous-deficient. When the pups were genotyped during the first 8 hr after birth, 18% were homozygous-deficient. However, no homozygous-deficient (−/−) pups were found at the time of weaning (3 weeks), suggesting that loss of βENaC caused a lethal phenotype shortly after birth. Fig. 2B shows a survival curve. After birth, the βENaC −/− pups showed a steady decline in survival, with a maximum survival time of 47 hr. This survival curve is similar to that reported for mice deficient in αENaC, which survived a maximum of 40 hr (17). These results indicate that βENaC null mice survive development and are born but that they die shortly after birth.
Table 1.
Percentage of mice of each genotype determined prenatally, immediately postnatally, and at weaning
| Age | n | Percent of animals
|
||
|---|---|---|---|---|
| +/+ | +/− | −/− | ||
| Prenatal E12.5–E19.5 | 111 | 23 | 56 | 21 |
| Postnatal 0–8 hr | 34 | 32 | 50 | 18 |
| Weaning, 3 weeks | 72 | 35 | 65 | 0 |
Figure 2.

Genotype and survival of mice. (A) Genotype of mice determined by PCR. The 495-bp band represents the targeted βENaC allele, and the 339-bp band represents the normal βENaC allele. (B) Survival curves of wild-type (+/+), heterozygous (+/−), and homozygous-deficient (−/−) pups.
Expression of βmENaC.
Expression of all three ENaC subunits was examined in mice of all genotypes (Fig. 3). By using an RT-PCR assay, no βmENaC specific mRNA was detected in either lung or kidney of βENaC −/− pups. However, βENaC mRNA was detected in wild-type (+/+) and heterozygous (+/−) littermates. In addition, mRNA for both α and γENaC subunits was observed in tissues from pups of each genotype.
Figure 3.

Expression of ENaC subunits determined by RT-PCR. RT-PCR was performed on RNA extracted from kidney (K) and lung (L) from wild-type (+/+), heterozygote (+/−), and homozygous-deficient (−/−) pups. The primers used to specifically detect each subunit are described in Materials and Methods. As a control, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was amplified from each sample.
Disruption of βmENaC Does Not Cause Respiratory Failure.
Pulmonary liquid absorption is increased just before birth, and the activity of ENaC is thought to be critical for the change from a water-filled embryonic lung to an air-filled adult lung (19). These observations were supported by the report that mice deficient in αENaC died of respiratory failure because they failed to clear liquid from their lungs after birth (17). Although βENaC-deficient mice had a similar survival curve, they did not show the respiratory failure of α−/− mice. The β−/− pups appeared normal at birth, moved and suckled normally, had milk in the stomach at death, and could not be distinguished from their littermates. They did not show an abnormal breathing pattern, chest wall retractions, or cyanosis. In fact, they appeared normal even when observed shortly before death.
This behavior suggested that lung liquid clearance is not impaired severely. To test this, we examined the lungs, and on gross inspection found that they were air-filled and not mottled. Lung sections showed no difference in morphology between βENaC +/+ and −/− mice (results not shown). We also measured the wet and dry weights of lungs removed from βENaC −/− mice and compared them to their +/+ and +/− littermates. Although prenatal lung wet:dry weights were not significantly different between the three groups, there was a small but significant increase in wet:dry lung weight of the βENaC −/− animals (Fig. 4). However, the increase in wet:dry lung weight was only half the increase reported for αENaC-deficient mice (17).
Figure 4.

Lung wet/dry weight ratios. Lungs were obtained prenatally (E18.5–E19.5) and at 12–18 hr after birth and were analyzed for water content. Numbers of animals in each group were, for prenatal, 2 +/+, 4 +/−, and 3 −/−; and for postnatal, 8 +/+, 4 +/−, and 7 −/−. Data are mean ± SEM. Asterisk indicates P < 0.05 compared with +/+ animals.
To evaluate lung function further, we performed blood gas analysis of 12- to 18-hr-old pups. There were no significant differences between the groups of mice in CO2 pressure, O2 pressure, O2 saturation, base excess, HCO3 concentration, or pH (Table 2). These data together with those described above indicate that the β−/− mice did not die of respiratory failure. This led us to suspect that there may be other phenotypes associated with loss of βENaC function that are responsible for premature death.
Table 2.
Blood gas analysis
| Genotype | n | pH | CO2 pressure, mmHg | O2 pressure, mmHg | Base excess, mM | HCO3, % | O2 saturation, % |
|---|---|---|---|---|---|---|---|
| +/+ | 7 | 7.31 ± 0.02 | 59.8 ± 11.6 | 80.5 ± 38.1 | 1.6 ± 2.3 | 29.0 ± 3.1 | 89.5 ± 8.6 |
| +/− | 5 | 7.33 ± 0.02 | 60.4 ± 16.0 | 80.7 ± 28.5 | 3.1 ± 1.2 | 30.3 ± 3.8 | 91.9 ± 8.1 |
| −/− | 3 | 7.34 ± 0.02 | 55.1 ± 11.9 | 68.8 ± 33.8 | 1.6 ± 1.0 | 28.2 ± 3.1 | 86.9 ± 11.3 |
Blood taken from 12- to 18-hr-old pups was analyzed for pH, CO2 pressure, O2 pressure, base excess, HCO3 concentration, and O2 saturation. Data are mean ± SD. 1 mmHg = 133 Pa.
Disruption of βmENaC Causes Severe Metabolic Abnormalities.
ENaC function in the kidney is a key component of Na+ absorption and electrolyte homeostasis. Moreover, abnormalities in renal electrolyte transport are responsible for the morbidity and mortality in PHA. Therefore, we evaluated renal histology and function. On gross inspection and on microscopic examination, kidneys of the +/+ and −/− animals showed no difference (data not shown). However, Fig. 5A shows that serum Na+ concentration was decreased in βENaC −/− mice compared with their +/+ and +/− littermates. In addition, the serum K+ concentration was strikingly elevated in the βENaC null mice. The serum Cl− concentration also was elevated slightly in −/− mice. The urine electrolytes (Fig. 5B) of null mice showed a significantly lower K+ concentration and a higher Na+ concentration than controls. The urine Cl− concentration was not different in the three groups. The hyponatremia with high urine Na+ concentrations suggests renal Na+ loss. Consistent with this, the −/− pups weighed less (1.23 ± 0.03 g, n = 3, P = 0.05) than controls (1.40 ± 0.04 g, n = 12) 12 hr after birth. The inability to excrete a urine high in K+, despite the high serum K+ concentration, is consistent with a loss of ENaC in the renal collecting duct.
Figure 5.

Serum and urine electrolyte concentrations. Serum (A) and urine (B) samples were taken from 12- to 18-hr-old pups and were analyzed for Na+, K+, and Cl−. Numbers of animals in each serum group were 15 +/+, 8 +/−, and 6 −/−. Numbers of animals in each urine group were 15 +/+, 8 +/−, and 10 −/−. Data are mean ± SEM. Asterisk indicates P < 0.05 compared with +/+ animals.
Aldosterone Levels Are Increased in βmENaC-Deficient Pups.
Salt balance is regulated, in part, by aldosterone-mediated ENaC subunit transcription and channel activity (reviewed in ref. 20). The defects in electrolyte concentration suggested that there would be compensatory changes in aldosterone levels. Fig. 6 shows that serum aldosterone was higher in the βENaC-deficient mice compared with littermate controls. Increased serum aldosterone combined with renal Na+ loss suggests renal resistance to high levels of aldosterone, as is seen in PHA1.
Figure 6.

Serum aldosterone levels. Serum samples were taken from 12- to 18-hr-old pups and were analyzed for aldosterone. Numbers of animals in each group were 9 +/+, 7 +/−, and 10 −/−. Data are mean ± SEM. Asterisk indicates P < 0.05 compared with +/+ animals.
DISCUSSION
Using gene targeting, we have generated mouse lines that are deficient in βENaC. Prenatally and within a short period after birth, the percentage of −/− mice (21 and 18%, respectively) was close to the predicted 25%, suggesting little, if any, loss during embryogenesis. Moreover, the −/− mice had no detectable internal or external morphologic defects. These results suggest that βENaC is not required for normal embryogenesis or development. This is consistent with the timing of ENaC expression late in development in lung and kidney (21, 22). However, the βENaC null mice survived <2 days; our data suggest they died from hyperkalemia caused by the loss of renal ENaC function. These results reveal that the βENaC subunit is an essential component of the epithelial Na+ channel in vivo.
The βENaC null mice showed several interesting differences from mice deficient in αENaC (17). The αENaC null mice died from respiratory failure with respiratory distress, cyanosis, and lungs that were heavy with fluid and showed alveolar filling on histologic examination. In contrast, the βENaC null mice were not in respiratory distress, their behavior was normal, they had normal lungs on gross inspection, and their pulmonary histology was normal. Although the wet:dry weight ratio was increased, suggesting some alteration of lung liquid clearance, the changes were small and less than those observed in αENaC −/− mice. Moreover, the lack of changes in the blood gases in βENaC −/− mice suggests that there was little respiratory compromise. Thus, αENaC, but not βENaC, appears to be required for lung liquid clearance at the time of birth. The lack of severe pulmonary sequela caused by βENaC disruption could have several explanations. It may be that the remaining α and γ subunits generate some channel activity in the lung, as has been observed with heterologous expression of human and rat α- and γENaC (5, 7). Alternatively, it may be that another, as yet undiscovered subunit can substitute for βENaC in the lung. For example, the human δ subunit can substitute for an α subunit to generate Na+ channels in heterologous expression systems (23). Future studies are required to sort out these and other possibilities.
PHA1 results from a mutation in any one of the ENaC subunits, and the reported phenotypes are similar. It is interesting that humans with PHA1, including those with αENaC mutations, do not show the pulmonary phenotype reported for the αENaC −/− mice (12, 17). The lack of respiratory failure with βENaC disruption in the mouse appears to resemble more closely the situation observed in humans with PHA1 than does the phenotype in mice with a disrupted αENaC gene. This difference suggests that control of liquid balance in the lung is significantly different in mice and humans. This conclusion is also consistent with earlier findings in mice that disruption of the cystic fibrosis transmembrane conductance regulator gene or mutation by deletion of F508 does not reproduce the pulmonary phenotype of patients with cystic fibrosis (24, 25). Thus, even though the βENaC −/− mice may have a pulmonary phenotype more like that of humans with PHA1, caution is warranted in using them as a model to understand human lung disease.
In contrast to the lack of a pulmonary phenotype, the βENaC-deficient mice showed a significant defect in renal function that appears to have caused their death. In the −/− mice, we found an increased urine Na+ concentration despite hyponatremia and a decreased urine K+ concentration despite hyperkalemia. Moreover, serum aldosterone levels were increased. Together, these findings indicate a critical defect in renal collecting duct Na+ channel activity and, secondarily, K+ secretion. In the cortical collecting duct, Na+ absorption through apical ENaC Na+ channels is coupled to K+ secretion into the urine through apical K+ channels (26). The linkage between Na+ absorption and K+ secretion is completed by virtue of the activity of the basolateral membrane Na-K-ATPase, which exchanges Na+ for K+. In general, the rate-determining step for Na+ absorption and K+ secretion is Na+ entry across the apical membrane, and aldosterone stimulates this step by increasing the number and/or activity of apical ENaC Na+ channels. Our data indicate that the β subunit of ENaC is critical to both Na+ absorption and K+ secretion in the distal tubule.
In rodents, the ability of the cortical collecting duct to absorb Na+ and to secrete K+ is not fully developed at birth but requires 1–2 weeks of neonatal life to mature (27, 28). Our finding that lethal hyperkalemia developed within the first 2 days of life strongly suggests that, even though ENaC function may not be fully developed, there is sufficient function to prevent hyperkalemia. Thus, we infer that a functional ENaC is important for the metabolic transition from fetal to neonatal life.
It is difficult to be certain of the requirement for the α subunit in the kidney or to compare the renal phenotype of the αENaC null mice (17) to our βENaC null mice. It is possible that αENaC deficient mice might show a phenotype similar to that which we report, but death from respiratory failure could preclude analysis. Recently, Hummler et al. (29) reported generation of mice with homozygous disruption of αENaC that were also transgenic for the rat αENaC expressed in many tissues, including lung and kidney. Many of the mice survived the neonatal period, and metabolic abnormalities were not obvious at 12 hr. Although serum electrolytes were not measured, at 5–9 days after birth, the mice showed increased urinary Na+ and modest metabolic acidosis. Thus, βENaC null mice have a renal phenotype much more severe than αENaC null mice expressing an αENaC transgene.
The βENaC-deficient mice reproduce much of the pathology observed in humans with PHA1. Patients with PHA1 show hyponatremia, hyperkalemia, dehydration, and elevated aldosterone levels, and, if left untreated, would usually die (12, 13). The βENaC-deficient mice we describe may provide a useful model to understand better human PHA1 and to develop new approaches to therapy.
Acknowledgments
We thank Maggie Price, Paul Reuther, Lisa de Berg, Theresa Mayhew, and our other laboratory colleagues for excellent assistance and discussions. We thank the University of Iowa DNA Core of the Diabetes and Endocrine Research Center (National Institutes of Health Grant DK25295) for help. This work was supported by the Howard Hughes Medical Institute and an O’Brien Kidney Disease Center grant (DK52617). M.J.W. is an Investigator of the Howard Hughes Medical Institute.
ABBREVIATIONS
- ENaC
epithelial Na+ channel
- mENaC
mouse ENac
- PHA1
pseudohypoaldosteronism type 1
- kb
kilobase
- RT
reverse transcriptase
- E12.5
embryonic day 12.5
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Benos D J, Awayda M S, Ismailov I I, Johnson J P. J Membr Biol. 1995;143:1–18. doi: 10.1007/BF00232519. [DOI] [PubMed] [Google Scholar]
- 2.Garty H, Palmer L G. Physiol Rev. 1997;77:359–396. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 3.Fyfe G K, Quinn A, Canessa C M. Semin Nephrol. 1998;18:138–151. [PubMed] [Google Scholar]
- 4.Canessa C M, Horisberger J-D, Rossier B C. Nature (London) 1993;361:467–470. doi: 10.1038/361467a0. [DOI] [PubMed] [Google Scholar]
- 5.Canessa C M, Schild L, Buell G, Thorens B, Gautschi I, Horisberger J D, Rossier B C. Nature (London) 1994;367:463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 6.McDonald F J, Snyder P M, McCray P B, Jr, Welsh M J. Am J Physiol. 1994;266:L728–L734. doi: 10.1152/ajplung.1994.266.6.L728. [DOI] [PubMed] [Google Scholar]
- 7.McDonald F J, Price M P, Snyder P M, Welsh M J. Am J Physiol. 1995;268:C1157–C1163. doi: 10.1152/ajpcell.1995.268.5.C1157. [DOI] [PubMed] [Google Scholar]
- 8.Lingueglia E, Voilley N, Waldmann R, Lazdunski M, Barbry P. FEBS Lett. 1993;318:95–99. doi: 10.1016/0014-5793(93)81336-x. [DOI] [PubMed] [Google Scholar]
- 9.Snyder P M, McDonald F J, Stokes J B, Welsh M J. J Biol Chem. 1994;269:24379–24383. [PubMed] [Google Scholar]
- 10.Renard S, Lingueglia E, Voilley N, Lazdunski M, Barbry P. J Biol Chem. 1994;269:12981–12986. [PubMed] [Google Scholar]
- 11.Canessa C M, Merillat A M, Rossier B C. Am J Physiol. 1994;267:C1682–C1690. doi: 10.1152/ajpcell.1994.267.6.C1682. [DOI] [PubMed] [Google Scholar]
- 12.Chang S S, Grunder S, Hanukoglu A, Rosler A, Mathew P M, Hanukoglu I, Schild L, Lu Y, Shimkets R A, Nelson-Williams C, et al. Nat Genet. 1996;12:248–253. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 13.Strautnieks S S, Thompson R J, Gardiner R M, Chung E. Nat Genet. 1996;13:248–250. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 14.Shimkets R A, Warnock D G, Bositis C M, Nelson-Williams C, Hansson J H, Schambelan M, Gill J R, Jr, Ulick S, Milora R V, Findling J W, et al. Cell. 1994;79:407–414. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 15.Snyder P M, Price M P, McDonald F J, Adams C M, Volk K A, Zeiher B G, Stokes J B, Welsh M J. Cell. 1995;83:969–978. doi: 10.1016/0092-8674(95)90212-0. [DOI] [PubMed] [Google Scholar]
- 16.Schild L, Canessa C M, Shimkets R A, Gautschi I, Lifton R P, Rossier B C. Proc Natl Acad Sci USA. 1995;92:5699–5703. doi: 10.1073/pnas.92.12.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier B C. Nat Genet. 1996;12:325–328. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 18.Ausubel F M, Brent A, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Greene & Wiley; 1992. [Google Scholar]
- 19.Matalon S, Benos D J, Jackson R M. Am J Physiol. 1996;271:L1–L22. doi: 10.1152/ajplung.1996.271.1.L1. [DOI] [PubMed] [Google Scholar]
- 20.Barbry P, Hofman P. Am J Physiol. 1997;273:G571–G585. doi: 10.1152/ajpgi.1997.273.3.G571. [DOI] [PubMed] [Google Scholar]
- 21.Tchepichev S, Ueda J, Canessa C, Rossier B C, O’Brodovich H. Am J Physiol. 1995;269:C805–C812. doi: 10.1152/ajpcell.1995.269.3.C805. [DOI] [PubMed] [Google Scholar]
- 22.Watanabe S, Matsushita K, McCray P B, Jr, Stokes J B. FASEB J. 1998;12:A983. [Google Scholar]
- 23.Waldmann R, Champigny G, Bassilana F, Voilley N, Lazdunski M. J Biol Chem. 1995;270:27411–27414. doi: 10.1074/jbc.270.46.27411. [DOI] [PubMed] [Google Scholar]
- 24.Snouwaert J N, Brigman K K, Latour A M, Malouf N N, Boucher R C, Smithies O, Koller B H. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- 25.Zeiher B G, Eichwald E, Zabner J, Smith J J, Puga A P, McCray P B, Jr, Capecchi M R, Welsh M J, Thomas K R. J Clin Invest. 1995;96:2051–2064. doi: 10.1172/JCI118253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stokes J B. Semin Nephrol. 1993;13:202–212. [PubMed] [Google Scholar]
- 27.Satlin L M. Am J Physiol. 1994;266:F57–F65. doi: 10.1152/ajprenal.1994.266.1.F57. [DOI] [PubMed] [Google Scholar]
- 28.Satlin L M, Palmer L G. Am J Physiol. 1996;270:F391–F397. doi: 10.1152/ajprenal.1996.270.3.F391. [DOI] [PubMed] [Google Scholar]
- 29.Hummler E, Barker P, Talbot C, Wang Q, Verdumo C, Grubb B, Gatzy J, Burnier M, Horisberger J D, Beermann F, et al. Proc Natl Acad Sci USA. 1997;94:11710–11715. doi: 10.1073/pnas.94.21.11710. [DOI] [PMC free article] [PubMed] [Google Scholar]