

Abstract
The effect of chromatin organization on EGFP-tagged histone protein dynamics within the cell nucleus has been probed using fluorescence correlation and recovery measurements on single living HeLa cells. Our studies reveal that free fraction of core-particle histones exist as multimers within the cell nucleus whereas the linker histones exist in monomeric forms. The multimeric state of core histones is found to be invariant across mammalian and polytene chromosomes and this is ATP dependent. In contrast, the dynamics of the linker histones exhibits two distinct diffusion timescales corresponding to its transient binding and unbinding to chromatin governed by the tail domain residues. Under conditions of chromatin condensation induced by apoptosis, the free multimeric fraction of core histones is found to become immobile, while the monomeric linker histone mobility is partially reduced. In addition, we observe differences in nuclear colocalization of linker and core particle histones. These results are validated through Brownian dynamics simulation of core and linker histone mobility. Our findings provide a framework to understand the coupling between the state of chromatin assembly and histone protein dynamics that is central to accessing regulatory sites on the genome.
INTRODUCTION
Core and linker histone proteins form the major class of nuclear proteins that condense the genome into a highly organized chromatin assembly within living cells (1). DNA wraps around core histones that exist in an octameric structure, with two dimers of H2A-H2B and a tetramer of H3-H4, called the nucleosome (2). The linker histones then clamp the entry and exit sites of DNA around the core histone octamer (3). The array of nucleosomes is repeated, forming the chromatin structure. The DNA-histone interactions are remodeled during cellular processes such as genome duplication, gene expression, and DNA repair (4). For this histone proteins undergo a variety of epigenetic modifications on their unstructured tail residues leading to the tightening, loosening, or removal of nucleosomes by chromatin remodeling machines (5). The dynamic removal and reassembly of histones and their variants are considered essential in the maintenance of epigenetic memory (6). The functional roles of core and linker histones are found to be distinct arising due to their differential nature of DNA association. Although the core histones with their epigenetic modifications play a central role in regulating access to DNA (7), the linker histones are thought to participate in maintaining the dynamic higher order chromatin structure (8). Although some of the linker histone variants play a role in somatic development (9), the implicit roles of linker histones remain unclear (10). Because histones are found to be regulators of cellular processes, understanding their in vivo dynamics in the context of chromatin organization and nuclear architecture has therefore become important.
Recent progress in fluorescence-based live-cell monitoring techniques (11) and modifying histone proteins with GFP fusion proteins (12) have provided new means to study their mobility within the nucleus. These studies have revealed that the core and linker histones that package the genome are highly dynamic within living cells. Core histones are found to exchange with a τ1/2 ∼ 130 min (13) whereas the linker histones have a τ1/2 ∼ minutes (14,15). Most core histones are found to be associated with the chromatin and are maintained in position through mitosis. Among the free fraction of core histones, the H2A-H2B dimer exchanges more rapidly than the H3-H4 tetramer (13), suggesting that the bound H3-H4 may provide stable epigenetic markers (16). It is also found that replication independent dynamic exchange of the core histones is deposited to transcriptionally active sites (17). The exchange of H2A and H2B is found to be mediated by processes that are ATP dependent (18). In contrast, the mobility of linker histones and their subtypes is dependent on their tail residues (19) and affected by the phosphorylation of these residues (20). Despite the existing information on histone mobility within the nucleus, the underlying mechanisms of their diffusion remain unclear.
In this article, we explore quantitatively the nature of core and linker histone protein diffusion and its coupling to local chromatin assembly within living cells. The hindrance to histone protein mobility offered by the heterogeneous chromatin assembly is first established. We then show that the core histones exist in multimeric states and that their mobility is invariant across organisms. In contrast the linker histone mobility shows two distinct timescales, one arising due to normal diffusion and the other due to its interactions with chromatin. Colocalization experiments of histones reveal spatial heterogeneity in their enrichment within living cells. Functional perturbations to chromatin, such as ATP depletion and cell death, suggest differential roles for core and linker histone dynamics. The results are then validated through detailed numerical simulation to deduce the underlying diffusive mechanisms of core and linker histone protein dynamics.
MATERIALS AND METHODS
Plasmid construction and cell culture
The H2B-EGFP expressing pBOS plasmid was gifted to us by Kanda et al. (12). Human H4-EGFP plasmid was made in two steps. The gene was first amplified from HeLa genomic DNA using primers (X60483, H4-fwd, 5′GGGGTACCATGTCTGGCC GCGG 3′ and H4-rev, 5′ CGGGATCCCCACCGAAACCGTAGAG 3′), was digested and ligated into pBOS-H2B-EGFP using KpnI and BamHI restriction sites. Then the BamHI-EGFP-NotI fragment from the resulting plasmid was replaced with a BglII-EGFP-NotI fragment from pEGFP N1 (Clontech, Palo Alto, CA) plasmid, to generate a 23 amino acid residue linker between H4 and EGFP, driven by an elongation factor (EF) 1α promoter. Human H1.1 gene, driven by CMV promoter, was amplified from HeLa genomic DNA using primers (X57130; H1.1-fwd, 5′ GGGGGATCCATGTCTGAAACAGTGCCTC 3′ and H1.1-rev, 5′ AAAACCGGTTTCTTGGGTGCCGCTTTC 3′) and cloned into pEGFP N1 vector using BamHI and AgeI restriction sites. Human H1.2 and H1.4 genes were amplified from HeLa genomic DNA using primers (X57129; H1.2-fwd, 5′ GGGGTACCATGTCC GAGACTGCTCCTG 3′; H1.2-rev, 5′CGGGATCCCGTTTCTTCTTGGGCG3′ and M60748; H1.4-fwd, 5′GGGGTACCATGTCCGAGACTGCGCC3′; H1.4-rev, 5′CGGGATCCTTTTTCTTGGCTGCCGC 3′) and cloned into pEGFP N1 vector using KpnI and BamHI restriction sites. Human H1.5 gene was also amplified from HeLa genomic DNA using primers (X83509; H1.5-fwd, 5′GGGGTACCATGTCGGAAACCGC3′; H1.5-rev, 5′CGGGATCCT TCTTTTTGGCAGCC3′) and ligated into pEGFP N1 vector at BamHI site on the 3′ end and a blunt end on the 5′ end. The tailless H1.1 was constructed by screening for the core domains of the proteins using the Conserved Domain Search database (http://www.ncbi.nlm.nih.gov/structure/cdd/wrpsb.cgi) and then amplifying the core domains alone. The tailless H1.1 gene was amplified from pH1.1-EGFP N1 by using primers (tailless H1.1-fwd, 5′GGGGGATCCATGTCCGTGTCAGAGC3′; tailless H1.1-rev, 5′AAAACCGGTTTGGTTTCCACGGAGG 3′) and cloned into pEGFP N1 using AgeI and BamHI restriction sites.
HeLa cells were cultured in Dubelco's minimum eagle medium (DMEM) (Gibco, Paisley, UK) supplemented with 10% fetal bovine serum (FBS) (Gibco) and penicillin-streptomycin (Gibco), in a 5% CO2 incubator. HeLa wild-type cells were transfected with 200 ng of DNA using lipofectamine 2000 (Invitrogen, Carlsbad, CA). Stable cell lines expressing H2B-EGFP were generated using selection by blasticidin. ATP depletion was realized by incubating cells for 1 h with 6 mM 2-deoxy-D-glucose and 10 mM sodium azide in Medium1 buffer at 37°C.
To flow sort the chromatin samples, HeLa cells were collected in M1 buffer (Tris-HCl 50 mM, MgCl2 100 mM, NH4Cl 100 mM, 4% PEG 3350, pH 7.5) lysed by mechanical shearing with a 23-gauge needle and sonication. Then the fluorescently labeled chromatin was sorted out using a Becton-Dickinson (Franklin Lakes, NJ) FACSvantage flow sorter. The sorted chromatin was allowed to settle on poly-d-lysine (Sigma, St. Louis, MO) coated coverslips, and the tetramethylrhodamine (TMR)-dextran was externally added to the sample for fluorescence correlation spectroscopy (FCS) experiments.
Salivary gland cells (with polytene chromosomes), derived from the Drosophila larvae (transgenic flies bearing histone C-terminal H2B fused to EGFP), were used. The salivary glands, from the third instar larvae, were dissected in Ringers medium (or medium containing 600 mM NaCl for control experiments) using standard protocols. In brief, under an inspection microscope, the middle portion of the larvae is held using one micro-needle. Another pair of fine-tipped forceps is used to pull out the mouth region of the larvae. The salivary glands are dissected from this open preparation. The glands are then transferred onto a clean microscope No. 1 coverslip in the Ringer's medium and sealed using another coverslip. Care is taken to ensure that the cells are intact during the sample preparation procedure and the samples are stable for microscopic observations.
Fluorescence microscopy and FRAP experiments
All imaging experiments were done in Medium1 buffer (150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 20 mM HEPES, pH 7.3), supplemented with 1% glucose, except in ATP depletion experiments. A Zeiss Confocor (model-LSM510-Meta/Confocor2) fluorescence microscope equipped with fluorescence correlation spectroscopy was used in our experiments. For imaging, fluorescence recovery after photobleaching (FRAP) and FCS experiments, we used a C-Apochromat 40×/ 1.2 N.A water corrected objective. Confocal images (512 × 512 pixels, 12 bit images, pinhole aperture size ∼1 airy units) were acquired. EGFP and its fusion proteins were excited with the 488-nm line of an argon-ion laser (Lasos, Jena, Germany) and the emission collected with a 500–530 nm bandpass filter. Dextrans conjugated to tetramethyl rhodamine (TMR-dextrans (Molecular Probes, Eugene, OR)) was excited with a 543-nm helium-neon laser line, and imaged with a long pass 560-nm emission filter, whereas a 560–615 nm bandpass filter was used in FCS studies.
Fluorescence correlation spectroscopy
In FCS experiments, the correlation of the time course of intensity fluctuations of the fluorescence signal I(t) was measured. From the intensity time series the autocorrelation function, 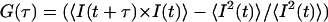 was calculated, where τ is the correlation time. The data was collected for a period of 10-s intervals and averaged over 10 runs to get the autocorrelation function and the corresponding fits. All FCS data is an average of more than 20 sets. The pinhole size was kept at 70 μm for 488-nm laser line (confocal diameter of ∼300 nm) and 78 μm for 543-nm laser line (confocal diameter of ∼360 nm). In our experiments, appropriate laser power of 488-nm line (argon-ion laser, Lasos) was used to avoid artefacts arising due to photobleaching and to ensure high counts per particle.
was calculated, where τ is the correlation time. The data was collected for a period of 10-s intervals and averaged over 10 runs to get the autocorrelation function and the corresponding fits. All FCS data is an average of more than 20 sets. The pinhole size was kept at 70 μm for 488-nm laser line (confocal diameter of ∼300 nm) and 78 μm for 543-nm laser line (confocal diameter of ∼360 nm). In our experiments, appropriate laser power of 488-nm line (argon-ion laser, Lasos) was used to avoid artefacts arising due to photobleaching and to ensure high counts per particle.
The following functions were used to fit the experimentally obtained autocorrelation curves. For unhindered three-dimensional (3D) diffusion:
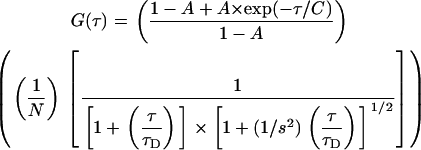 |
(1) |
Here N is the number concentration of the fluorescent species in the confocal volume, τD is the diffusion timescale, such that diffusion constant D = ω2/4τD, where ω is the XY spread of the confocal spot, and s is the structure parameter; A and C are the triplet fraction and triplet timescales, respectively. However, for molecules like TMR-dextrans, activated notch, or the histone core particles, diffusion inside the cell nucleus cannot be adequately described by this model. To understand these results, we used the modified autocorrelation function with an anomalous subdiffusion term β, which is a useful parameter to describe the underlying heterogeneity of the matrix (26,28,29).
 |
(2) |
The data obtained for the linker histone dynamics was fitted to a sum of two diffusing species. For this, we used the maximum entropy method, MEMFCS (35), used in FCS experiments, to fit our data to independently verify our assumption of using sum of two diffusing species to describe the dynamics of linker histones.
 |
(3) |
where, N1 is the inverse of the total number of bright molecules (EGFP tagged linker histones) in the confocal volume and N2 corresponds to the fraction of the species having correlation timescale τD1.
Numerical simulation of Brownian diffusion
Labview software from National Instruments (Austin, TX) is used to simulate Brownian diffusion of particles. Here the particles undergo 3D random walk with equal probability in each direction. A particle crossing the confocal volume, located around (0,0,0) position with a Gaussian profile, 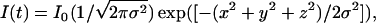 contribute to the intensity (fluorescence) signal, depending upon its coordinate in the profile. We then calculate the total intensity in each time trace for N such particles. From the intensity signal, we calculate the autocorrelation function
contribute to the intensity (fluorescence) signal, depending upon its coordinate in the profile. We then calculate the total intensity in each time trace for N such particles. From the intensity signal, we calculate the autocorrelation function 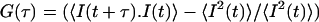 and averaged over a number of traces.
and averaged over a number of traces.
RESULTS
Heterogeneity in chromatin organization is reflected in translational diffusion of molecules within the cell nucleus
Heterogeneity in chromatin assembly is the prime determinant of diffusion inside the cell nucleus (21). To correlate the size and function of the molecules with the chromatin architecture we have monitored, using FCS, the diffusion of differently sized noninteracting particles (EGFP and TMR-dextrans) inside the cell nucleus. Fig. 1 shows fluorescence images of HeLa cells expressing EGFP and HeLa cells incorporated with TMR-dextrans. Typical FCS curves associated with differently sized molecules are shown in Fig. 1. A small (RH ∼ 1.5 nm), noninteracting particle such as EGFP is insensitive to the architecture and a 3D unhindered diffusion (Eq. 1) is sufficient to explain its diffusion behavior with D = 26.4 ± 2.7 μm2/s. Notably, EGFP diffusion either in the nucleus or in the cytoplasm show similar viscosity, in line with previous findings (22). Next, 10 kDa TMR-dextran (RH ∼ 3 nm) was incorporated into HeLa cells by a hypotonic shock to the cells (23). However, the diffusion data for a large noninteracting molecule like TMR-dextran, could not be adequately described by simple 3D unhindered diffusion. The data was fitted to Eq. 2 and the fit parameter β (mean value of the anomalous diffusion factor) was fixed at ∼0.67, which reflects the average chromatin mesh size. Here the diffusion timescale was D = 19.8 ± 4.9 μm2/s.
FIGURE 1.

Heterogeneity in chromatin organization and its reflection on the translation diffusion of molecules within the cell nucleus. The fluorescence images of (i) EGFP-transfected and (ii) TMR-D incorporated HeLa cell nucleus. (Scale bar, 5 μm) and their associated autocorrelation function curves for EGFP (⋆), and TMR-D (•) in the HeLa cell nucleus. (Inset) Probability histogram of β-factor for TMR-D inside the cell nucleus (⋄), in the buffer (○), and in the sorted chromosome (★).
To determine if the chromatin assembly primarily defines the anomalous factor β, we studied TMR-dextrans diffusion through sorted chromosomes from HeLa cells stably transfected with H2B-EGFP. When data were fitted with β as a free parameter, the histograms of the anomalous diffusion factor inside the cell nucleus and in the sorted chromosomes practically overlap; see Fig. 1 (inset), indicating that diffusion inside cells is primarily governed by the local chromatin architecture. For a small molecule like EGFP, when β is maintained as a free-fitting parameter, it attains a value close to one reflecting unhindered 3D diffusion.
Core histone dynamics
Core histone mobility is sensitive to chromatin assembly and exists in a multimeric state
Histone proteins condense the DNA into a highly organized chromatin assembly. We explored the diffusion of the free fraction of core histone proteins (H2B) to assess their dynamic interaction with the chromatin assembly as well as other core histones. The mobility of core histone H2B tagged with EGFP (H2B-EGFP (12)) within the cell nucleus was measured using FCS and the associated correlation curves are shown in Fig. 2 a. The typical correlation timescale is τD = 830.5 ± 232 μs (the corresponding diffusion constant D = 7.3 ± 1.9 μm2/s) under normal physiological conditions. The standard deviation in the timescales reflects the heterogeneity in the chromatin assembly. Interestingly, the mean correlation timescale for H2B-EGFP is much higher than what is expected from its molecular size (∼41 kDa), suggesting that it exists in a multimeric state. To test this, we measured the correlation timescale of H2B-EGFP in the cytoplasm, in transiently transfected HeLa cells overexpressing H2B-EGFP. The correlation timescale of H2B-EGFP in the cytoplasm τD = 268 ± 65.2 μs (with β = 0.67) and the corresponding diffusion constant D = 22.2 ± 5.4 μm2/s were indicative of a monomeric state. It is likely that after the core histones translocate to the nucleus they multimerize. Similar results were obtained for another core histone H4-EGFP (Supplementary Material).
FIGURE 2.

Core histone (H2B-EGFP) dynamics suggests their multimeric state. (a) Autocorrelation curves of EGFP in the cell nucleus and H2B-EGFP in the cytoplasm and in the nucleus. (Inset) Mean diffusion constants for the above cases. (b) Fluorescence images of the Drosophila salivary gland cell nucleus (polytene chromosomes) and HeLa nucleus and the associated autocorrelation curves of H2B-EGFP proteins in HeLa nucleus (★), salivary gland cell nucleus (⋄), and salivary gland cell nucleus with 600 mM NaCl (•). (Inset) Probability histograms of the diffusion correlation timescales for H2B-EGFP in HeLa nucleus and salivary gland cell nucleus and comparison of the probability histograms with H2B-EGFP diffusion in salivary gland cells in 600 mM NaCl concentration.
Core histone mobility is conserved between mammalian and polytene cells
To test whether the core histone mobility is a generic feature, we measured H2B mobility in the polytene chromosomes of salivary glands derived from transgenic H2B-EGFP Drosophila lines (24). In this system as well, H2B undergoes hindered diffusion with β ∼ 0.67. Here, two distinct regimes in the distribution of correlation timescales τ1 = 368 ± 68 μs and τ2 = 863 ± 217 μs with the associated diffusion constants D1 = 15.8 ± 3.2 μm2/s and D2 = 6.8 ± 1.3 μm2/s were observed (Fig. 2 b). The magnitude of these timescales is consistent with those observed in HeLa cells. Thus τ1 represents the monomeric fraction of H2B-EGFP whereas τ2 represents the multimeric H2B-EGFP. The larger presence of monomeric fraction of core histones in the polytene system, in comparison to HeLa cell nucleus, is perhaps due to the nature of chromatin organization in Drosophila salivary gland cells.
Spatial heterogeneity of chromatin architecture determines core histone mobility
We use salt-induced disassembly of nucleosomes to alter the heterogeneity of chromatin architecture. Upon addition of 600 mM NaCl (25), the chromatin architecture is affected, which is reflected in the change of β-value. Here the correlation curve fits perfectly with single species normal 3D unhindered diffusion with a single mean correlation time scale of 301 ± 104 μs with the corresponding D = 20.9 ± 6.9 μm2/s (Fig. 2 b). The lower correlation time of H2B-EGFP upon addition of 600 mM NaCl is possibly due to the dissociation of both free and bound multimeric H2B-EGFP into monomers. With the addition of salt, the change in β-value from 0.67 to 1 and the decrease in the standard deviation of correlation timescale suggest that diffusion of core histone proteins is dependent on the spatial heterogeneity of chromatin architecture within the cell nucleus.
Linker histone dynamics
Linker histone diffusion is distinct from core histones and reveals two timescales
Linker histones bind at the entry and exit sites of DNA on the nucleosome to form a stable higher order chromatin structure and this interaction has been suggested to be highly dynamic (14,15). We find that the autocorrelation behavior of the linker histone protein H1.1-EGFP, measured using FCS, is significantly different from that of the core histone proteins Fig. 3 a. Here the FCS curves do not fit with single species 3D unhindered diffusion or with the anomalous diffusion. Clearly there is a second distinct timescale in the autocorrelation function, which may be attributed to the dynamic interactions of the linker histones with the chromatin fiber, consistent with earlier FRAP studies (14,15). To obtain the underlying diffusion timescales, we have fitted the data with a two species diffusion model described in the Methods section. The fits to the data show two distinct timescales (τD1 = 298.3 ± 58.8 μs) with diffusion constant D1 = 19.5 ± 3.5 μm2/s commensurate with 3D diffusion, and (τD2 = 26.5 ± 12.8 ms) D2 = 0.3 ± 0.1 μm2/s possibly arising due to H1.1-EGFP interaction with DNA.
FIGURE 3.

Linker histone shows distinct interaction timescales inside the cell nucleus. (a) Fluorescence images of H1.1-EGFP and tailless H1.1-EGFP transfected HeLa nucleus (scale bar, 5 μm) and the associated autocorrelation function curves for H1.1-EGFP (★), tailless H1.1-EGFP (○). (Inset) Fluorescence recovery curves for tailless H1.1-EGFP, H1.1-EGFP, EGFP, and core histones, H2B-EGFP and H4-EGFP. (b) Different subtypes of H1 proteins show distinct interaction timescales. Autocorrelation curves for different H1 subtypes, H1.1, H1.2, H1.4, H1.5 inside the HeLa cell nucleus. (Inset) Normalized FRAP data for different subtypes of H1 proteins, H1.1, H1.2, H1.4, and H1.5. (c) Probability histograms of different subtypes of H1 proteins H1.1-EGFP, H1.2-EGFP, H1.4-EGFP, H1.5-EGFP. (Inset to panel c (i)) Mean and standard deviations of the fractions of the noninteracting species in different H1 subtypes. (Inset to panel c (ii)) Autocorrelation function curves for H1.5-EGFP, in the cytoplasm and inside the cell nucleus.
Tail residues determine the interaction timescales of linker histone diffusion
To gain more insight into the origin of the second timescale (τ2), the amino acid residues (1–40 and 121–216) corresponding to the tail sequence of H1.1 were deleted. Deleting the tail residues of H1.1-EGFP histones abolished the second diffusion timescale and the resultant FCS curves (Fig. 3 a) fit well with single species unhindered 3D diffusion with D = 20.2 ± 5.2 μm2/s. H1.1 tails could both interact with DNA as well as with adjacent histones on the chromatin assembly suggesting that these interactions are the source of the second timescale (τ2) (19). FRAP experiments presented in inset to Fig. 3 a for H1.1-EGFP, tailless H1.1-EGFP in comparison with free EGFP, H4-EGFP, and H2B-EGFP strongly support our conclusions. To test the origin of the second timescale (τ2) of the linker histones, we measured the correlation timescale of strongly interacting H1.5-EGFP in the cytoplasm in transiently transfected cells overexpressing H1.5-EGFP. FCS curves (Fig. 3 c, inset) showed a single species subdiffusive autocorrelation behavior with mean correlation timescale 376.6 ± 132.4 μs (with 〈β〉 = 0.72 reflecting the cytoplasmic anomalous diffusion factor). This indicates that the second timescale (τ2) of the linker histones within the nucleus arises primarily due to its interaction with the chromatin assembly.
Linker histone subtypes show distinct changes in their mobility and localization
To see if the various linker histone subtypes show similar diffusion characteristics, we repeated our investigations on linker histone subtypes (H1.2, H1.4, and H1.5). These histones also showed two distinct timescales of diffusion where the mean second timescale (τ2) corresponded to 28 ms, 34 ms, 34 ms for H1.2, H1.4, H1.5, respectively (Fig. 3, b and c). A fit of the interaction timescale (τ2) with the normal distribution shows that the peak values of the distribution function is statistically distinct for H1.1, and H1.2 compared to H1.4 and H1.5 (Fig. 3 c). The C-terminal tails of H1.1 and H1.2 are smaller, with less number of interacting residues, than that of H1.4 and H1.5. This is possibly reflected in the shorter diffusion timescale commensurate with their tail lengths that define the extent of interactions. The data on the fraction of interacting linker histone subtypes is shown in the inset to Fig. 3 c.
To outline the differences in mobility between different linker histone subtypes, both FRAP and FCS experiments were performed within the cell nucleus. In FRAP experiments, the bleaching cross section was ∼3 μm and the mean recovery curve is an average of the FRAP from 10 different cells. The FRAP data shows that the fluorescence recovery of the linker histones are markedly faster than the core particles (H2B and H4). This is in line with previous observations in the literature (13–15) and is characteristic of the dynamic nature of interactions associated with linker histones. A comparison of the FRAP data between different subtypes of H1 proteins shows that the H1.1, H1.2, H1.4, H1.5 recover to about 96%, 87%, 83%, 83% in a time duration of 160 s. In contrast, core histones H2B and H4 recover to <20% over a 30-min timescale. The colocalization experiments between the various linker histone subtypes, show differences in their spatial localization within the cell nucleus, possibly a reflection of their functional roles (Supplementary Material).
Functional perturbations such as ATP depletion and cell death differentially affect core and linker histone dynamics
Alterations in core and linker histone diffusion during ATP depletion
To probe whether the multimeric state of the H2B is ATP dependent, we carried out FCS measurements on H2B-EGFP in ATP-depleted cells. Fig. 4 a shows the distribution of correlation timescale in H2B-EGFP cells under normal and ATP-depleted conditions. Upon ATP depletion, there is a significant decrease in both the mean correlation timescale τD = 533.5 ± 248.3 μs (D = 12.7 ± 5.3 μm2/s) and its distribution reflecting the emergence of a fast timescale. This could be possibly due to an enriched monomeric fraction upon ATP depletion, suggesting that the maintenance of the multimeric state is an energy-dependent process. Similar results are obtained for another core histone particle, H4-EGFP (Supplementary Material). To check if the linker histone diffusion is ATP dependent, FCS measurements were carried out on ATP-depleted HeLa cells. These showed that unlike core histones, the mobility of linker histones remains invariant upon ATP depletion (Fig. 4 a).
FIGURE 4.

(a) Fluorescence images of H2B-EGFP transfected HeLa nucleus before and after ATP depletion. (Scale bar, 5 μm). (i) Probability histograms of correlation timescales of H2B-EGFP diffusion inside HeLa nucleus before and after ATP depletion. The bar graphs of the mean correlation timescales are also shown in the inset. (ii) Probability histograms of H1.5-EGFP correlation timescale in milliseconds, before and after ATP depletion. (b) Time course of histone protein dynamics within cells upon induction of apoptosis. (i) Autocorrelation function curves for H2B-EGFP before and after 4 h of addition of staurosporine in HeLa cells. (Inset) Fluorescence images of H2B-EGFP transfected HeLa cells before and after addition of staurosporine. (Scale bar, 5 μm). (ii) FRAP curves for H1.1-EGFP in HeLa cells, in normal conditions and after staurosporine treatment.
Alterations in core and linker histone diffusion during cell death
To elucidate the effect of large-scale changes in the chromatin structure on diffusive behavior of histones, we performed FCS experiments on apoptotic HeLa cells where apoptosis was induced by addition of 10 μM staurosporine. Integral chromatin proteins, like H2B-EGFP and H4-EGFP, showed complete loss of correlation signal under these conditions (Fig. 4 b). However, less tightly associated proteins like the linker histones (H1.1) and noninteracting proteins such as EGFP, showed only a partial loss of correlation in FCS experiments (data not shown). This indicates that the free fraction of the core histones (H2B-EGFP or H4-EGFP) becomes highly immobile, possibly as a result of chromatin condensation characteristic of the apoptotic state. In this case, the diffusion constant could not be extracted for the core histones, as there was a total loss of correlation signal on induction of apoptosis. Fluorescence imaging shows that the inherent fluorescence intensity levels do not change significantly upon treatment with staurosporine, indicating minimal leakage of the free core histones from the nucleus under these conditions. The fluorescence confocal image (Fig. 4 b, inset) shows punctated bright structures upon induction of apoptosis, indicating the heterogeneity in chromatin compaction. Further to elucidate changes in linker histone mobility in apoptotic cells, FRAP experiments were carried out on HeLa cells expressing strongly interacting H1.5-EGFP under normal and apoptotic conditions. FRAP recovery is significantly slower under conditions of apoptosis relative to normal cells (Fig. 4 b).
These findings using a combination of FCS and FRAP techniques on both linker and core histones, suggest that core histones, are more adversely affected by induction of apoptosis in comparison with those of linker histones or a free molecule that interact less with the chromatin. The measured diffusion constants from all the experiments are presented in Table 1.
TABLE 1.
Diffusion constants for different FCS experiments: EGFP, TMR-dextrans, ACN-EGFP, and EGFP-tagged core and linker histones
| Nucleus (ATP+) | Nucleus (ATP−) | Cytoplasm | Sorted chromosome | |
|---|---|---|---|---|
| EGFP | 26.4 ± 2.7 | 26.0 ± 7.9 | X | X |
| TMR-D | 19.8 ± 4.9 | X | X | 14.8 ± 8.2 |
| ACN-EGFP | 7.1 ± 3.2 | 8.7 ± 4.7 | X | X |
| H2B-EGFP | 7.3 ± 1.9 | 12.7 ± 5.3 | 22.2 ± 5.4 | X |
| H4-EGFP | 7.3 ± 3 | 10.4 ± 4.8 | X | X |
| H1 subtypes | D1 | D2 | ATP(−) D1 | ATP(−) D2 | Cytoplasm |
|---|---|---|---|---|---|
| H1.1 (H1a) | 19.5 ± 3.5 | 0.3 ± 1 | X | X | X |
| H1.5 (H1b) | 25.1 ± 4.7 | 0.2 ± 1 | 24.8 ± 5.7 | 0.2 ± 0.1 | 16.6 ± 5.5 |
| H1.2 (H1c) | 24.9 ± 3.9 | 0.2 ± 1 | X | X | X |
| H1.4 (H1e) | 23.6 ± 4.3 | 0.2 ± 1 | X | X | X |
| H1.1 (tailless) | 20.2 ± 5.2 | X | X | X | X |
Unit (μm2/s).
Numerical simulation reveals possible mechanism of core and linker histone diffusion
Core histone mobility
The FCS experiments show that the core histone mobility deviates from single species normal 3D diffusion but undergoes subdiffusive transport. To understand the origins of this subdiffusion, numerical simulations (26) were carried out that incorporated all the relevant experimental details. In the simulation, Brownian particles are made to undergo normal 3D diffusion through a homogenous mesh that mimicked the chromatin assembly. Each of these particles is made to undergo 10,000 random steps in 3D such that at any given instant an average of at least one molecule crosses the confocal detection. Such a detection, in the simulation, is realized by imposing a Gaussian excitation profile with the width (σ) of the profile defined by 50 units. Equivalent to the FCS experiment, the Brownian particle crossing through this Gaussian profile contributes to the intensity variation in the time series and therefore the autocorrelation curve may be calculated. Particles starting from any random site diffuse in 3D with step size of 10 units through the mesh. In each random step, if the particle encounters the mesh coordinates, a time delay is introduced by reversing the particle to its previous coordinates. The parameters in the simulation are the step size (δX), transmission probability, the confocal volume, and the mesh size (Supplementary Material). Here the transmission probability is defined as the probability of crossing the barrier as the particle reaches the mesh structure. By keeping the step size (10 units), confocal volume (σ = 50 units), and transmission probability (5%) fixed, we vary the mesh size to visualize the mechanism of core histone diffusion. Mesh sizes were chosen to be 0 (no mesh), 10 and 50 units. In addition, to mimic the dynamic nature of the chromatin assembly, we introduce 20% variability in the mean value of the mesh size. As expected, the autocorrelation function (ACF) of a Brownian particle with mesh size (0 units) undergoes normal unhindered 3D diffusion. But as the mesh size is varied, the effect of confinement starts being reflected in the particle undergoing subdiffusive transport (Fig. 5 a). In the inset, we plot the mean square displacement of the particle as a function of time for an average of 100 particles with step size 10 units, diffusing through a mesh of size 0, 11, 12, and 15units. The effect of confinement is maximum when the mesh size is comparable to the step size (〈ΔX2〉 ∝ tβ, where β < 1). As the mesh size is increased relative to the step size, the particle again undergoes normal 3D unhindered diffusion (where β approaches 1). Taken together with our experimental findings, these simulations suggest that as the size of a particle becomes comparable to the mesh size its diffusion tends to be confined within the local chromatin mesh, leading to subdiffusive transport.
FIGURE 5.

Numerical simulation study of core and linker histone dynamics. (a) Autocorrelation function generated by numerical simulation for single species of 3D diffusion with confined mesh structure with varying sizes (m = 0, 10, 50). (Inset (i)) Typical single particle trajectory of 2D random walk generated within a confined mesh structure. (Inset (ii)) Mean square displacement versus time for different mesh sizes (m = 0, 11, 12, 15). (b) Autocorrelation function generated by numerical simulation for 3D diffusion with randomly distributed interacting sites (N = 0, 1500, 2000, and 3000). (Inset) Typical single particle trajectory of 2D random walk.
Linker histone mobility
To understand the microscopic origins of the two distinct timescales obtained in the case of linker histones, we carried out a similar numerical simulation on the linker histones (26). Each of these particles is made to undergo 40,000 random steps, with step size of 4 units in 3D such that at any given instant, on average at least one molecule crosses the confocal detection with width (σ = 30 units). In this simulation, the linker histone proteins are treated as Brownian particles that undergo both 3D random diffusion and interact with dynamic pinning sites depicting chromatin binding sites. To mimic the environment experienced by the linker histones, we incorporated the presence of binding sites in the simulation. Here we assume a single particle diffusing in an environment with a variable number of interacting sites (N = 0, 1500, 2000, 3000) in the confocal volume, where the particle binds with a mean residence time of 4000 units with a Gaussian distribution of width 40%. The results of this simulation are plotted in Fig. 5 b, showing the presence of distinct second timescale in the autocorrelation curve. In addition, when the number of interacting sites was varied and all the other parameters fixed, a clear increase in the second timescale was observed. By comparing the simulation with the experimental results, the maximum number of binding sites within the confocal volume is found to be ∼2000.
DISCUSSIONS
Implications of core histone dynamics
Histone protein dynamics within the cell nucleus has become increasingly important in the context of chromatin remodeling, epigenetic states, and various genetic processes (27). Our results suggest that core histones are in a multimeric form whereas the linker histones are in monomeric form within the cell nucleus. The diffusion of multimeric core histones is sensitive to the architecture of the chromatin assembly. Previous measurements suggested that the reconstituted nucleosomes, with a hydrodynamic diameter of 10 nm that diffused through a polymer mesh, underwent anamolous diffusion (28). The chromatin architecture within the cell nucleus is reminiscent of a dynamic polymer matrix characterized by a mesh size. Molecules smaller than ∼3 nm hydrodynamic radius (EGFP, RH ∼ 1.7 nm) undergo unhindered diffusion whereas larger molecules are confined by the mesh. The physical origins of anomalous diffusion arise due to such confinement, where additional time is spent during each random Brownian step. In such cases, the mean square displacement is not proportional to time but proportional to tβ, where β (β < 1) is a measure of the confinement (29). This multimerization process is found to be ATP dependent because upon ATP depletion the multimeric core histones (H2B-EGFP) become monomeric, outlined in the schematic in Fig. 6. Interestingly, the depletion of ATP leads to the enrichment of monomeric, unbound core histones within the nucleus. This illustrates the existence of a dynamic equilibrium between the chromatin-bound and free fraction of core histones that is tuned by energy-dependent processes. A similar-sized molecule, such as a transcription cofactor activated notch, also undergoes confined diffusion within the nucleus (Supplementary Material). The core histone mobility was invariant across organisms, whether in mammalian cell lines or polytene chromosomes, suggesting similar evolutionarily conserved local chromatin architecture in these two systems. Because the histone proteins are released from the chromatin assembly either in transcription or replication, their ordered repacking is crucial to maintain epigenetic states. The possible existence of histones in their multimeric form and their local confined diffusion within the cell nucleus suggests higher fidelity of dynamic exchange of histones to maintain epigenetic memory (6).
FIGURE 6.

Schematic model of the mobility of core (i) and linker (ii) histones within the cell nucleus. Core histones (H2B-EGFP) remain in the multimeric form (red) in normal physiological condition. Upon ATP depletion the multimeric core histones (H2B-EGFP) become monomeric (green). Linker histones diffuse through the chromosomal mesh structure by diffusion with interaction (red, interacting; green, noninteracting). For tailless linker histones the motion is purely diffusive and there is no interaction.
Implications of linker histone dynamics
In contrast, the mobility of the linker histones is strongly defined by both normal diffusion as well as their interactions with chromatin. Indeed the mobility of the linker histones in the cytoplasm confirmed that the second timescale in the diffusion behavior arises due to the interactions with the chromatin assembly. Furthermore deletion of the histone tail residues on H1.1 establishes that the tail residues are involved in these interactions (19) (Fig. 6). In the case of linker histones, the unhindered diffusion timescale and the interaction timescale are insensitive to ATP depletion suggesting that interactions are primarily governed by energy-independent molecular processes. The various subtypes of linker histones are found to undergo similar diffusive processes where the minor variations in the interaction timescales could possibly correspond to differential tail lengths of the histones. This interaction timescale ∼30 ms, measured as the mean correlation timescale, may suggest a mechanism to introduce dynamic local conformational fluctuations in chromatin assembly. Here, the DNA at entry and exit points on the nucleosome can be continuously modulated by the binding and unbinding of the linker histones thus providing a mechanism by which regulatory proteins may spontaneously access specific DNA sequences (30,31).
The mobility of histone proteins that are differentially associated with chromatin is also differentially affected upon induction of apoptosis. The highly interacting core histones become completely immobile in apoptotic nuclei, while the mobility of less interacting linker histones or noninteracting protein is only partially affected. The simulation results adequately describe our experimental findings that the nanoscale chromatin architecture defines the dynamics of core and linker histones within the cell nucleus. The differential binding timescales of core and linker histones, in addition to other chromatin binding proteins (such as high mobility group proteins (HMG) (32) and heterochromatin binding proteins HP1 (33)), perhaps provides an appropriate substrate for dynamic self-organization of regulatory information on DNA within living cells (34). Work in progress in our lab, using single molecule microscopy, is currently addressing the importance of histone dynamics in the context of their epigenetic states and their role in transcription control.
Acknowledgments
We thank Satyajit Mayor, Madan Rao, Apurva Sarin, and K. Vijay Raghavan for useful discussions and NCBS for the imaging facility funded by the Wellcome Trust. We also thank Yamuna Krishnan for critical reading of the manuscript.
References
- 1.Spector, D. L. 2003. The dynamics of chromosome organization and gene regulation. Annu. Rev. Biochem. 72:573–608. [DOI] [PubMed] [Google Scholar]
- 2.Schalch, T., S. Duda, D. F. Sargent, and T. J. Richmond. 2005. X-ray structure of a tetranucleosome and its implications for the chromatin fibre. Nature. 436:138–142. [DOI] [PubMed] [Google Scholar]
- 3.Zlatanova, J., P. Caiafa, and K. van Holde. 2000. Linker histone binding and displacement: versatile mechanism for transcriptional regulation. FASEB J. 14:1697–1704. [DOI] [PubMed] [Google Scholar]
- 4.Cosgrove, M. S., J. D. Boeke, and C. Wolberger. 2004. Regulated nucleosome mobility and the histone code. Nat. Struct. Mol. Biol. 11:1037–1043. [DOI] [PubMed] [Google Scholar]
- 5.Jenuwein, T., and C. D. Allis. 2001. Translating the histone code. Science. 293:1074–1080. [DOI] [PubMed] [Google Scholar]
- 6.Ahmad, K., and S. Henikoff. 2002. Epigenetic consequences of nucleosome dynamics. Cell. 111:281–284. [DOI] [PubMed] [Google Scholar]
- 7.Khorasanizadeh, S. 2004. The nucleosome: from genomic organization to genomic regulation. Cell. 116:259–272. [DOI] [PubMed] [Google Scholar]
- 8.Hansen, J. C. 2002. Conformational dynamics of the chromatin fiber in solution: determinants, mechanisms, and functions. Annu. Rev. Biophys. Biomol. Struct. 31:361–392. [DOI] [PubMed] [Google Scholar]
- 9.Becker, M., A. Becker, F. Miyara, Z. Han, M. Kihara, D. T. Brown, G. L. Hager, K. Latham, E. Y. Adashi, and T. Misteli. 2005. Differential in-vivo binding dynamics of somatic and oocyte-specific linker histones in oocytes and during ES cell nuclear transfer. Mol. Biol. Cell. 16:3887–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bustin, M., F. Catez, and J. H. Lim. 2005. The dynamics of histone H1 function in chromatin. Mol. Cell. 17:617–620. [DOI] [PubMed] [Google Scholar]
- 11.Gasser, S. M. 2002. Visualizing chromatin dynamics in interphase nuclei. Science. 296:1412–1416. [DOI] [PubMed] [Google Scholar]
- 12.Kanda, T., K. F. Sullivan, and G. M. Wahi. 1998. Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8:377–385. [DOI] [PubMed] [Google Scholar]
- 13.Kimura, H., and P. Cook. 2001. Kinetics of core histones in living human cells: little exchange of H3 & H4 and some rapid exchange of H2B. J. Cell Biol. 153:1341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Misteli, T., A. Gunjan, R. Hock, M. Bustin, and D. T. Brown. 2000. Dynamic binding of histone H1 to chromatin in living cells. Nature. 408:877–881. [DOI] [PubMed] [Google Scholar]
- 15.Lever, M. A., J. P. Th'ng, X. Sun, and M. J. Hendzel. 2000. Rapid exchange of histone H1.1 on chromatin in the living human cells. Nature. 408:873–876. [DOI] [PubMed] [Google Scholar]
- 16.Turner, B. M. 2002. Cellular memory and the histone code. Cell. 111:285–291. [DOI] [PubMed] [Google Scholar]
- 17.Thiriet, C., and J. J. Hayes. 2005. Replication-independent core histone dynamics at transcriptionally active loci in vivo. Genes Dev. 19:677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruno, M., A. Flaus, C. Stockdale, C. Rencurel, H. Ferreira, and T. O. Hughes. 2003. Histone H2A/H2B dimer exchange by ATP-dependent chromatin remodeling activities. Mol. Cell. 12:1599–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Th'ng, J. P., R. Sung, M. Ye, and M. J. Hendzel. 2005. H1 family histones in the nucleus: control of binding and localization by the c-terminal domain. J. Biol. Chem. 280:27809–27814. [DOI] [PubMed] [Google Scholar]
- 20.Contreras, A., and R. E. Herrera. 2003. The dynamic mobility of histone H1 is regulated by cyclin/CDK phosphorylation. Mol. Cell. Biol. 23:8626–8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marshall, W. F. 2002. Order and disorder in the nucleus. Curr. Biol. 12:185–192. [DOI] [PubMed] [Google Scholar]
- 22.Chen, Y., J. D. Muller, Q. Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koberna, K., D. Stanek, J. Malinsky, M. Eltsov, A. Pliss, V. Ctrnacta, S. Cermanova, and I. Raska. 1999. Nuclear organization studied with the help of a hypotonic shift: its use permits hydrophilic molecules to enter into living cells. Chromosoma. 108:325–335. [DOI] [PubMed] [Google Scholar]
- 24.Pardue, M. L. 1994. Looking at Polytene Chromosomes. Methods Cell Biol. 44:333–351. [DOI] [PubMed] [Google Scholar]
- 25.Mangenot, S., A. Leforestier, P. Vachette, D. Durand, and F. Livolant. 2002. Salt induced conformation and interaction changes of nucleosome core particles. Biophys. J. 82:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss, M., M. Elsner, F. Kartberg, and T. Nilsson. 2004. Anomalous subdiffusion is a measure for cytoplasmic crowding in living cells. Biophys. J. 87:3518–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura, H. 2005. Histone dynamics in living cells revealed by photobleaching. DNA Repair. 4:939–950. [DOI] [PubMed] [Google Scholar]
- 28.Mangenot, S., S. Keller, and J. Radler. 2003. Transport of nucleosome core particles in semidilute DNA solutions. Biophys. J. 85:1817–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weidemann, T., M. Wachsmuth, T. A. Knoch, G. Muller, W. Waldeck, and J. Langowski. 2003. Counting nucleosome in living cells with a combination of fluorescence correlation spectroscopy and confocal imaging. J. Mol. Biol. 334:229–240. [DOI] [PubMed] [Google Scholar]
- 30.Tomschik, M., H. Zheng, K. van Holde, J. Zlatanova, and S. H. Leuba. 2005. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA. 102:3278–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li, G., M. Levitus, C. Bustamante, and J. Widom. 2005. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 12:46–53. [DOI] [PubMed] [Google Scholar]
- 32.Phair, R. D., P. Scaffidi, C. Elbi, J. Vecerova, A. Dey, K. Ozato, D. T. Brown, G. Hager, M. Bustin, and T. Misteli. 2004. Global nature of dynamic protein-chromatin interactions in vivo: three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 24:6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmiedeberg, L., K. Weisshart, S. Diekmann, G. Meyer zu Hoerste, and P. Hemmerich. 2004. High and low-mobility populations of HP1 in heterochromatin of mammalian cells. Mol. Biol. Cell. 15:2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorski, S., and T. Misteli. 2005. Systems biology in the cell nucleus. J. Cell Sci. 118:4083–4092. [DOI] [PubMed] [Google Scholar]
- 35.Periasamy, N., and A. S. Verkman. 1998. Analysis of fluorophore diffusion by continuous distribution of diffusion coefficients: application to photo-bleaching measurements of multicomponent and anomalous diffusion. Biophys. J. 75:557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]