

Abstract
Nerve growth factor (NGF) mimics play an important role for therapies that target the receptor tyrosine kinase A (trkA). The N-terminal fragment of the NGF (N-term@NGF) was previously demonstrated to be an important determinant for affinity and specificity in the binding to trkA. Here we use a variety of computational tools (contact surface analysis and free energy predictions) to identify residues playing a key role for the binding to the receptor. Molecular dynamics simulations are then used to investigate the stability of complexes between trkA and peptides mimicking N-term@NGF. Steered molecular dynamics calculations are finally performed to investigate the process of detaching the peptide from the receptor. Three disruptive events are observed, the first involving the breaking of all intermolecular interactions except two salt bridges, which break subsequently.
INTRODUCTION
Neurotrophins constitute a family of structurally related proteins, which promote and regulate the survival of neurons in the vertebrate nervous system (1–3). This family includes the nerve growth factor (NGF) as well as the brain-derived neurotrophic factor, Neurotrophin-3 and Neurotrophin-4/5 (4). Mature neurotrophins are homodimers that are derived by proteolytic cleavage from precursor proteins (5). These proteins bind with similar affinity to the receptor p75NTR, a member of the tumor-necrosis-factor receptor superfamily, and selectively to the members of the receptor tyrosine kinase (trk) family, namely trkA, trkB, and trkC, which are involved in the activation of different intracellular signal transduction cascades (6–9). Specifically, NGF binds with the highest affinity to trkA (10–12), which mediates the survival-promoting and neurite-growth-promoting effects of NGF during development (13–16).
Because of their crucial role for nervous system development, neurotrophins may reduce neuronal degeneration in neurodegenerative diseases, such as Alzheimer's (17–24). However, a major obstacle to the neurotrophin-based therapies is their delivery: significant doses of proteins must be achieved in the target region to really counteract disease processes and, simultaneously, this delivery must be hampered in other regions to prevent side-effects (25–27). A promising strategy involves the use of peptide mimics, i.e., molecules with either agonist or competitive antagonist effect and retaining the most essential elements of neurotrophic action, which may be useful in the treatment of several neurodegenerative diseases and nerve injuries (28–31). For NGF, such peptides have been shown to exhibit neurotrophic effects by binding to trkA (32–38).
The ligand/receptor interface involves two patches: one comprises residues of NGF core region and loops of the C-term of the fifth extracellular domain of trkA (trkA-d5), the other includes the N-terminal fragment of NGF (N-term@NGF) and the ABED β-sheet of trkA-d5 (39,40) (Fig. 1 a). The latter is known to determine the affinity and specificity of the NGF binding to trkA, as indicated by experimental data (15,41–46) and further suggested by molecular dynamics (MD) calculations (47).
FIGURE 1.

(a) Cα trace of the NGF/trkA-d5 complex as obtained by the MD simulation of Settanni et al. (47). Each of the two NGF identical subunits (black) binds to a trkA-d5 subunit (gray). The trace is thicker for the residues involved in NGF/trkA-d5 molecular recognition. The trkA-d5/N-term@NGF regions are further marked with two ellipses. (b) Cα trace of the complex formed by trkA-d5 and H4PIFHRGEFS13 (c1). A second peptide in c2 differs from that in c1 for the replacement of His-8 with Gly and the removal of Ser-13. Both c1 and c2 are investigated here by MD simulations. (c) TrkA-d5/peptide interactions in c1 and c2. The van der Waals and H-bond interactions are depicted as continuous and dashed lines, respectively. His-4 is accommodated in a receptor's pocket lined by His-291, Pro-302, and His-343; at the bottom, His-4 hydrogen bonds with Phe-303 and Gly-344. The Pro-5 side chain interacts with His-343 and Leu-333. Ile-6 is positioned in another receptor's pocket lined by Val-294, Met-296, and Leu-333; at the bottom, Ile-6 side chain forms van der Waals interactions with the receptor surface-exposed disulfide region between Cys-345 and Cys-300. Phe-7 interacts with Val-294, Arg-9 with Glu-334, Gly-10 with Met-296, Glu-11 with Arg-347, Phe-12 with Met-296, and Ser-13 with Met-296. His-8 does not interact with trkA-d5. Glu-339, which is boxed, interacts with Arg-9 only in c2. (d) Cα trace of the trkA-d5/NGF complex without S2SHPIFHRGEFS13 residues (c3). Also, this complex undergoes MD simulation. The initial structure (thick trace) is compared with the last c3 MD snapshot (thin trace) (87).
Here we attempt at identifying a short peptide as a trkA-d5 high-affinity mimic of the N-term@NGF. Our work is based on the x-ray (39) and the MD (47) structures of the NGF/trkA-d5 complex (Fig. 1 a). First, we identify NGF residues exhibiting significant contact surface, and thus nonpolar interaction, with the receptor; among those, we locate the residues forming significant electrostatic interaction and/or binding-energy hot-spots, as defined in Kortemme and Baker (48) and Kortemme et al. (49). Based on this analysis, we conclude that residues from 4 to 13 (H4PIFHRGEFS13, according to the pdb 1WWW numeration (39,50)), at the N-term, may have high affinity for trkA-d5.
Subsequently, under the assumption that the mode of binding of the peptide is similar to that of the original protein, we investigate the conformational flexibility of the adduct formed by the H4PIFHRGEFS13 peptide with trkA-d5 using MD (Fig. 1 b). The stability of the “complementary” complex, i.e., the complex in which the same peptide is removed from NGF, is also explored by MD (Fig. 1 d). Our calculations confirm that most residues belonging to the 4–13 peptide are important for trkA-d5 binding, His-8 and Ser-13 being the only groups not stabilizing the complex. Thus, we modify the peptide mimic by deleting Ser-13 and mutating His-8 to Gly, which ensures a larger flexibility of the peptide. MD simulations carried out on the resulting peptide (H4PIFGRGEF12) suggest that the latter, despite its shorter length, forms an additional salt bridge with the receptor, besides those originally formed by Arg-9 and Glu-11. Thus, we identify this peptide as a high-affinity mimic of the N-term@NGF. As a final step, we investigate the binding process by steered MD (51–59), and the aforementioned salt bridges turn out to be the most persistent interactions between the ligand and the receptor.
METHODS
All calculations discussed here are based on the last snapshot of our previous MD simulation of the NGF/trkA-d5 complex (see Supplementary Material) (47). Calculations based on the x-ray structure (39) yield very similar results, which are not commented on here (Table 2).
TABLE 2.
TrkA-d5/N-term@NGF interactions
| NGF Residues | CS (nm2)
|
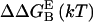 f.c. f.c. |
ΔΔ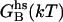
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| 1WWW | f.c. | c1 | c2 | 1WWW | f.c. | c1 | c2 | ||
| His-4 | 1.03 | 1.06 | 1.00 | 1.19 | 2.99 | 5.40 | 4.60 | 5.18 | 3.44 |
| Pro-5 | 0.47 | 0.51 | 0.57 | 0.36 | 1.47 | – | – | – | – |
| Ile-6 | 0.79 | 0.83 | 0.95 | 0.72 | −0.55 | 2.10 | 2.06 | 2.95 | 2.47 |
| Phe-7 | 0.39 | 0.49 | 0.54 | 0.25 | −0.84 | 1.27 | – | 1.44 | 1.39 |
| His-8(Gly) | 0 | 0 | 0 | 0 | 0 | – | 0.49 | – | – |
| Arg-9 | 0.44 | 0.57 | 0.53 | 1.06 | 2.52 | 0.29 | 2.11 | 2.06 | 2.99 |
| Gly-10 | 0.02 | 0.08 | 0.04 | 0.07 | – | – | – | – | – |
| Glu-11 | 0.65 | 0.68 | 0.90 | 0.83 | 6.33 | 0.69 | 2.50 | 1.83 | 2.82 |
| Phe-12 | 0.30 | 0.29 | 0.45 | 0.38 | −0.22 | 0.70 | 4.18 | 0.70 | 1.93 |
| Ser-13 | 0.34 | 0.39 | 0.10 | * | −2.13 | 0.35 | 0.35 | −0.22 | * |
Contact surface (CS) (61), electrostatic alanine scanning calculations (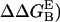 (65), and binding energy hot-spot predictions
(65), and binding energy hot-spot predictions 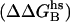 (48,49) are reported for NGF residues from His-4 to Ser-13. The NGF/trkA-d5 full-complex (f.c.) is the last MD snapshot of the previously performed simulation (47) and the c1 and c2 conformations are those after 5.7 ns of MD.
(48,49) are reported for NGF residues from His-4 to Ser-13. The NGF/trkA-d5 full-complex (f.c.) is the last MD snapshot of the previously performed simulation (47) and the c1 and c2 conformations are those after 5.7 ns of MD.
In c2, Ser-13 is absent.
We focus on structural (item 1, below) and energetic (items 2 and 3, below) features associated with the binding of chain Y of trkA-d5 to NGF homodimer in complex with the other receptor chain X:
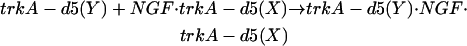 |
We calculate the difference of the solvent-accessible surfaces of NGF in the presence and in the absence of trkA-d5(Y), here called contact-surface (CS). CS provides a rough estimate of the nonpolar contribution to the binding free energy (60). We use the center-probe area definition (61) and set the solvent probe radius to 0.14 nm (62,63). The residues providing the largest CS in the NGF N-term region turn out to be located between His-4 and Ser-13 (sequence H4PIFHRGEFS13).
- The Poisson-Boltzmann equation (64) is applied to calculate the electrostatic contribution to the free energy of binding
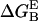 (65):
(65):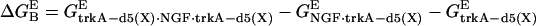
- Each molecule is assumed to adopt the same conformation in the complex and in the isolated form. Then, the change of
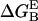 upon mutation of each residue R (except Gly) to Ala is calculated from the formula
upon mutation of each residue R (except Gly) to Ala is calculated from the formula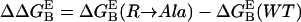
- The structures of the mutated protein are obtained by deleting all the side-chain atoms of residue R except the Cβ, which is saturated. Then, the structures undergo an energy minimization, using the standard pairwise generalized Born/surface area model of solvation (66,67), as implemented in the AMBER7 program (68). We use the AMBER99 force field (69,70), assuming 100 mM monovalent ionic concentration, protein dielectric constant εin = 2, and solvent dielectric constant εwat = 78.5.
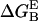 is calculated using the APBS program (71) under the following conditions: the nonlinear Poisson-Boltzmann equation is solved; the temperature is set to 300 K; a 100 mM monovalent ion concentration is assumed; the Connolly molecular surface definition (72) is used; a two-runs focusing procedure is applied.
is calculated using the APBS program (71) under the following conditions: the nonlinear Poisson-Boltzmann equation is solved; the temperature is set to 300 K; a 100 mM monovalent ion concentration is assumed; the Connolly molecular surface definition (72) is used; a two-runs focusing procedure is applied.- Initially we run the APBS program with boundary conditions defined using a Debye-Hückel model for multiple charged spheres, along with a grid spacing of 0.067 nm. Then, we run the program with boundary conditions derived from the first run, with grid spacing as small as 0.047 nm. Test-calculation with a finer grid spacing, i.e., 0.045 nm, shows that the calculated free energies differ by ∼0.2 kJ/mol (≃0.1 kT).
We calculate which NGF residues at the NGF/trkA-d5(Y) interface contribute significantly to the complex stability using Baker's computational alanine scanning procedure (48,49). The predictor locates the residues at the interface, which may significantly destabilize the complex when mutated to alanine, estimating both the van der Waals and the electrostatic contributions to the free energy of binding. Binding energy hot-spots are defined as residues whose
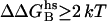 . The prediction is performed on the entire NGF/trkA-d5 and on the N-term@NGF mimic/trkA-d5 complexes.
. The prediction is performed on the entire NGF/trkA-d5 and on the N-term@NGF mimic/trkA-d5 complexes.
Molecular dynamics calculations
We perform MD simulations on:
The complex formed by trkA-d5 and the N-term@NGF derived peptide H4PIFHRGEFS13 (c1, Fig. 1 b). His-4 and Ser-13 are terminated by acetyl and N-methyl groups, respectively.
The complex formed by trkA-d5 and the peptide of sequence H4PIFGRGEF12 (c2).
The trkA-d5/NGF complex without such peptide, along with Ser-2 and Ser-3 (c3, Fig. 1 d).
c1, c2, and c3 are solvated in rectangular water boxes (Table 1). The same computational protocol is used for c1, c2, and c3. We use the AMBER99 force field (69,70) and TIP3P (73) for the biomolecules and water, respectively. Calculations are performed using the GROMACS program (74). Periodic boundary conditions are applied. Particle mesh Ewald is used to evaluate long-range electrostatics (75–77). A cutoff of 1 nm is used for the real part of the electrostatics and for the van der Waals interactions. Constraints are applied to the bonds using the LINCS algorithm (78). The integration time step is set to 1.5 fs.
TABLE 1.
Selected features of the three complexes (c1, c2, and c3; see also Fig. 1, b and d) and of the unbound peptide systems investigated in this work
| Box edges (nm)
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Complex name | Amino acids | N-methyl groups | Acetyl groups | Water molecules | Sodium ions | x | y | z |
| c1 | 111 | 2 | 2 | 9689 | 4 | 7.6 | 7.2 | 5.8 |
| c2 | 110 | 2 | 2 | 9538 | 4 | 7.3 | 7.2 | 6.3 |
| c3 | 407 | 4 | 4 | 24165 | 2 | 12.8 | 9.5 | 6.7 |
| Unbound peptide | 9 | 1 | 1 | 2987 | 0 | 5.1 | 4.3 | 4.2 |
Equilibration runs are carried out using the Berendsen thermostat and barostat (79). The solvent undergoes ∼50 ps of MD simulations keeping the protein atoms fixed to achieve the correct solvent density. Subsequently, the systems are energy-minimized and gradually heated by eight MD runs with increasing temperature (22.5 ps each, from 0 to 10, 30, 70, 100, 150, 200, 250, and 300 K). The 5.7 ns of MD simulations in the NPT ensemble (T = 300 K and P = 1 atm) are then performed using the Nosé-Hoover thermostat (80,81) and the Parrinello-Rahman procedure (82,83); the pressure coupling is isotropic in all directions.
Root mean square deviations (RMSD) are calculated for Cα atoms using the x-ray structure (39) as the reference conformation. RMSD plots for c1 and c2 fluctuate at ∼0.15 nm within the first 0.6 ns, which are discarded; on the contrary, the same plot calculated for c3 displays increasing RMSD (Fig. 2). The maximal distance allowed for the existence of an H-bond between hydrogen and acceptor atom is 0.25 nm, while the maximal supplement of the donor-hydrogen-acceptor angle is 60°, 0° corresponding to the extended conformation. Two oppositely charged residues are assumed to form a salt-bridge interaction when at least one H-bond can be detected between their side chains.
FIGURE 2.

RMSDs (black line) and CS between ligand and the receptor (gray line) calculated for the three complexes investigated here by MD. The inset shows the RMSDs of NGF and trkA-d5 chains X and Y in c3 (continuous black line, dashed black line, and continuous gray line, respectively).
Steered molecular dynamics
Three nanoseconds of steered molecular dynamics (SMD) (51–59) are carried out in the NPT ensemble with the same setup, based on the c2 MD last snapshot, except that the peptide is harmonically restrained (force constant is 1000 kJ/(mol × nm2) (51,54,56,59)). The restraint is initially applied at the peptide mass center and moves away from the receptor mass center with a velocity of 0.5 nm/ns. An additional 4 ns long SMD simulation is performed with the same setup but at 0.25 nm/ns restraint-point velocity. Then, 5.4 ns of MD in the NPT ensemble, with the same setup as above, is performed. Finally, the unbound peptide is immersed in a smaller water box (“unbound peptide” in Table 1) and 25 ns of MD simulation in the NPT ensemble is performed.
RESULTS AND DISCUSSION
A calculation of the contact surface (CS) (61) between trkA-d5 and NGF suggests that the NGF residues from His-4 to Ser-13 form significant contacts with the receptor (CS > 0.20 nm2), except His-8 and Gly-10. In particular, His-4 and Ile-6 form the most extensive contacts with the receptor, being accommodated in specific pockets (Fig. 1 c and Table 2). Being based on a solvent-accessible surfaces calculation, CS-based methods provide a rough estimate of the nonpolar interaction (60) and thus of the residues which are responsible for the most relevant hydrophobic contributions to the overall free energy of binding. Only a few other NGF residues have such significant CS: Ser-19, Trp-21, Tyr-52, Phe-54, Arg-59, Ile-145, Lys-146, Gly-147, Thr-197, His-198, Arg-217, and Val-225. This indicates again the relevance of the N-term@NGF for ligand/receptor interaction (15,41–47).
To dissect the most important electrostatic interactions between N-term@NGF and trkA-d5, we perform electrostatic alanine scanning calculations (65). We calculate the electrostatic free energy of receptor binding to the ligand in which the residue R is mutated to Ala and then we subtract the corresponding free energy calculated for the native peptide. The larger the resulting 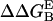 , the greater the electrostatic contribution of residue R side chain to the binding free energy. Here we distinguish H-bonds from salt bridges, which are characterized by a larger desolvation penalty upon formation and by a much stronger electrostatic attraction, involving two oppositely charged groups (84). His-4, Arg-9, and Glu-11 turn out to provide the most negative electrostatic free energies, presumably because of H-bond and salt-bridge interactions with the receptor (namely, with residues Phe-303, Gly-344, Glu-334, and Arg-347; see Fig. 1 c and Table 2). A similar picture is obtained by applying the Baker's binding-energy hot-spot predictor (48,49): within the N-term region, His-4, Ile-6, Arg-9, Glu-11, and Phe-12 significantly contribute to the complex stability
, the greater the electrostatic contribution of residue R side chain to the binding free energy. Here we distinguish H-bonds from salt bridges, which are characterized by a larger desolvation penalty upon formation and by a much stronger electrostatic attraction, involving two oppositely charged groups (84). His-4, Arg-9, and Glu-11 turn out to provide the most negative electrostatic free energies, presumably because of H-bond and salt-bridge interactions with the receptor (namely, with residues Phe-303, Gly-344, Glu-334, and Arg-347; see Fig. 1 c and Table 2). A similar picture is obtained by applying the Baker's binding-energy hot-spot predictor (48,49): within the N-term region, His-4, Ile-6, Arg-9, Glu-11, and Phe-12 significantly contribute to the complex stability 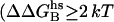 , Table 2).
, Table 2).
We conclude that NGF residues from His-4 to Ser-13 have good affinity for trkA-d5, with the exception of His-8. Thus, we may expect that the peptide which has the same sequence (H4PIFHRGEFS13) might have good affinity for the receptor. To investigate the stability of this peptide with trkA-d5 (c1, Fig. 1 b), we perform an MD simulation along with a complementary system, which comprises both receptors trkA-d5 and the whole NGF without the N-terms (c3, Fig. 1 d). As for the peptide, we assume that its mode of binding is similar to that of full-length NGF.
The results of the simulation of c1 are summarized in Fig. 2 and in Table 3. The RMSD fluctuates around a relatively small value (0.15 nm, Fig. 2) and the peptide/receptor contacts are maintained as in the full complex, with the notable exception of Ser-13: this residue is much more solvated here than in the full complex (Tables 2 and 3) and it is not interacting with trkA-d5 as in the full complex. Our calculations further confirm that His-8 does not form interactions with the receptor. This suggests that His-8 can be substituted by a simpler residue like Gly or Ala. Since Gly allows for a higher flexibility of the peptide, which in turn might assist trkA-d5 binding, we substitute here His-8 with Gly.
TABLE 3.
Ligand-receptor interactions in c1 and c2 complexes
| NGF | trkA-d5 | c1 | c2 |
|---|---|---|---|
| H-bonds (%) | |||
| His-4 Nε | Phe-303 O | 87 | 99 |
| His-4 Nε | Gly-344 O | 22 | 3 |
| Arg-9 sc | Glu-334 O | 4 | 72 |
| Salt bridges (%) | |||
| Arg-9 sc | Glu-334 sc | 84 | 1 |
| Arg-9 sc | Glu-339 sc | 0 | 40 |
| Glu-11 sc | Arg-347 sc | 100 | 100 |
| Hydrophobic contacts (nm) | |||
| His-4 | Phe-303 | 0.29 (0.02) | 0.28 (0.01) |
| His-4 | Gly-344 | 0.33 (0.04) | 0.35 (0.03) |
| His-4 | His-291 | 0.48 (0.13) | 0.38 (0.04) |
| His-4 | Pro-302 | 0.36 (0.02) | 0.36 (0.02) |
| His-4 | His-343 | 0.37 (0.03) | 0.38 (0.03) |
| Pro-5 | His-343 | 0.36 (0.02) | 0.36 (0.02) |
| Pro-5 | Leu-333 | 0.40 (0.03) | 0.40 (0.04) |
| Ile-6 | Leu-333 | 0.40 (0.05) | 0.38 (0.03) |
| Ile-6 | Cys-345 | 0.38 (0.03) | 0.38 (0.03) |
| Ile-6 | Cys-300 | 0.41 (0.04) | 0.44 (0.04) |
| Ile-6 | Met-296 | 0.35 (0.03) | 0.41 (0.05) |
| Ile-6 | Val-294 | 0.40 (0.04) | 0.47 (0.05) |
| Phe-7 | Val-294 | 0.43 (0.08) | 0.41 (0.05) |
| Arg-9 | Glu-334 | 0.29 (0.05) | 0.33 (0.10) |
| Arg-9 | Glu-339 | 1.14 (0.21) | 0.45 (0.21) |
| Gly-10 | Met-296 | 0.44 (0.06) | 0.63 (0.10) |
| Glu-11 | Arg-347 | 0.27 (0.01) | 0.27 (0.01) |
| Phe-12 | Met-296 | 0.34 (0.02) | 0.37 (0.04) |
| Ser-13 | Met-296 | 0.42 (0.09) | * |
H-bonds, salt bridges, and minimum distances between nonhydrogen atoms of couples of interacting residues, as described in Fig. 1 c. The term sc means side chain.
In c2, Ser-13 is absent.
We probe then the stability of the peptide in which His-8 is mutated to Gly and Ser-13 is absent (H4PIFGRGEF12, c2). This peptide is expected to interact with the receptor as strongly as H4PIFHRGEFS13, being one amino acid shorter. The peptide interacts with trkA-d5 as in c1, except Arg-9@NGF: the salt bridge formed by this residue with Glu-334@trkA is almost always broken and the H-bond formed with the backbone of Glu-334@trkA is strengthened. Moreover, a new salt bridge, not present in c1, is observed between Arg-9@NGF and Glu-339@trkA but only in 40% of the trajectory (Fig. 1 c and Table 3): this interaction is therefore weaker than the salt bridge between Arg-9@NGF and Glu-334@trkA in c1 (84%). However, it is compensated by the formation of an H-bond between Arg-9@NGF and Glu-334@trkA backbone (Table 3). Both terminal residues are forming significant interaction with the receptor, unlike in c1. The complex is stable, with the RMSD fluctuating at ∼0.15 nm (Fig. 2). The Baker's binding-energy hot-spots predictor (48,49), applied to the last c2 snapshot, suggests that all of the peptide residues, except Pro-5, Gly-8, and Gly10, are significantly contributing to the complex stability (Table 2). We conclude that the larger conformational flexibility of the peptide caused by the His-8 to Gly mutation might assist the formation of a new salt bridge interaction.
Very different results are obtained for the complex between trkA-d5 and the NGF lacking the N-term segments (c3 in Fig. 1 d). In this case, although we start from the bound conformation of the full complex, the receptor slowly detaches from the N-terms deleted neurotrophin, as evinced from the RMSD that grows during the simulation and the CS between NGF homodimer and the receptors that decreases, indicating an increase of the solvation of the different subunits during the dynamics (Fig. 1 d and Fig. 2). The calculation is stopped after 5.7 ns of MD, as the previously described simulations. Although the results strongly suggest a dissociation of the complex, observing such an event requires a timescale that is currently not covered by MD simulations.
We conclude that the interactions of the peptide in c2 with the receptor appear to be far stronger than those formed by the rest of the neurotrophin.
Steered molecular dynamics
Steered molecular dynamics (SMD) is based on the final MD snapshot of c2 (Fig. 3 b). Although the investigation of the entire molecular recognition process is beyond the domain of applicability of molecular simulation methods, useful qualitative insights on the ligand/receptor binding process can be obtained by steering the ligand away from the receptor and observing the applied force profile (51–59).
FIGURE 3.

Steered molecular dynamics of c2 moving the restraint point with a velocity of 0.5 nm/ns. (a) Distance between the mass centers of trkA-d5 and of the ACEHPIFGRGEFNME peptide (solid line) and force on the peptide (dashed line) plotted as a function of the simulation time. The peptide/receptor interactions are broken after 1.1 ns of SMD, except the salt bridges formed by the peptide residues Arg-9 and Glu-11, which break down at ∼1.45 ns and ∼2.15 ns of SMD, respectively. The inset shows the number of water oxygens within 0.45 nm of nonhydrogen atoms of the peptide residues, as a function of the simulation time. (b,c) Structure of c2 at the beginning (b) and at the end (c) of the simulation: the Cα trace of the receptor is represented (green at 5.7 ns of MD, red after 3 ns of SMD and further 5.4 ns of MD), along with the sticks representation of the peptide, colored by atom type. (d) Separated fit of the peptide and of the receptor structures in the initial (green) and the final (red) conformations (87).
Here we apply such an approach to the case of c2, by monitoring the external force and the distance between the mass centers of ligand and receptor as a function of the simulated time (Fig. 3 a). Until 0.8 ns the distance is almost constant, no ligand/receptor interactions are broken, and the force grows linearly as fast as 1.12 pN/ps. Then, at ∼1 ns, the force is large enough to break almost simultaneously all intermolecular interactions except the salt bridges formed by Arg-9@NGF and Glu-11@NGF. The breaking of all these interactions determines a sharp decrease of the force, an increase of the receptor/peptide mass centers distance and of the solvation of the NGF residues (Fig. 3 a). The force is now much smaller and does not change on the disruption, at ∼1.45 ns, of the salt bridge formed by Arg-9, which is observed in c1 and in c2 to be the least stable among the two peptide/receptor salt bridges (Table 3). In contrast, it decreases suddenly on the rupture of the last interaction, the salt bridge formed by Glu-11, at ∼2.15 ns (Fig. 3 a). The hydration of the ligand increases at each of these disruptive events (inset in Fig. 3 a). To check the reliability of our results, a second SMD simulation is performed by applying an external force that increases slower: a qualitatively similar unbinding process is observed (Fig. 4).
FIGURE 4.

Steered molecular dynamics of c2. The restraint point is moved at a velocity of 0.25 nm/ns, which is half the velocity of the steered MD in Fig. 3 a. Distance between the mass centers of trkA-d5 and of the peptide (solid line) and force on the peptide (dashed line) plotted as a function of the simulation time. The force increases with a rate of ∼0.61 pN/ps within the first 1.2 ns. All peptide/receptor interactions are broken after 1.5 ns of SMD except those formed by the peptide residues Arg-9 and Glu-11, which break down at ∼1.75 ns and ∼3.3 ns of SMD, respectively. The inset shows the number of water oxygens within 0.45 nm of nonhydrogen atoms of the peptide residues, as a function of the simulation time.
After a total of 3 ns of SMD, the peptide is unbound from the receptor and further 5.4 ns of MD are performed, without applying external force to the peptide (Fig. 3 c). The mean RMSD calculated for the receptor is 0.14 (0.02) nm and the conformation of the unbound trkA-d5 is very similar to that of the receptor bound to NGF (85); the mean RMSD for the peptide is 0.30 (0.05) nm (Fig. 3 d).
By performing 25 ns of MD simulation of the peptide, we observe that the peptide is folding into itself progressively: the RMSD is 0.26 nm at 0 ns and 0.20 nm at 25 ns. Arg-9 and Glu-11 do not form an intrapeptide salt bridge either here or in the previous complexes; two intrapeptide H-bonds, between Ile-6 N and His-4 Nδ and between Phe-7 N and His-4 O are maintained during the dynamics. The side chains are oriented as in the bound peptide, with the exception of Glu-11 side chain, that interacts with the backbone of the peptide C-term part.
CONCLUDING REMARKS
Our simulations suggest that the ACEHPIFGRGEFNME peptide may provide high affinity to the trkA-d5 receptor, as all the residues form interactions with the receptor except the Gly-8 and Gly-10 residues; however, the latter provides conformational flexibility, allowing the peptide to form an additional interaction with the receptor.
SMD calculations suggest that the strengths of the contacts between the peptide and its target are similar, with the exception of the salt bridges formed by Arg-9 and Glu-11 with trkA-d5 (Fig. 1 c). These interactions are the most persistent in the SMD, possibly because of the large flexibility and length of the side chains involved, which can rearrange so as to maintain the electrostatic interactions despite the increase of the peptide/receptor distance, with respect to all the other residues at the interface.
The challenge is now to perform in vitro and in vivo experiments which may quantify the actual efficacy of this peptide as an agonist or competitive antagonist (25,86).
SUPPLEMENTARY MATERIAL
An online supplement to this article is available at BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Dr. Aneta Bozena Jezierska for critically reading the manuscript.
This work was financially supported by the Italian Ministero dell'Istruzione, dell'Università e della Ricerca, grant No. FIRB RBNE03FH5Y-007.
References
- 1.Lewin, G. R., and Y. A. Barde. 1996. Physiology of the neurotrophins. Annu. Rev. Neurosci. 19:289–317. [DOI] [PubMed] [Google Scholar]
- 2.Bibel, M., and Y. A. Barde. 2000. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 14:2919–2937. [DOI] [PubMed] [Google Scholar]
- 3.Kalb, R. 2005. The protean actions of neurotrophins and their receptors on the life and death of neurons. Trends Neurosci. 28:5–11. [DOI] [PubMed] [Google Scholar]
- 4.Dechant, G., and H. Neumann. 2002. Neurotrophins. Adv. Exp. Med. Biol. 513:303–334. [DOI] [PubMed] [Google Scholar]
- 5.Lee, R., P. Kermani, K. K. Teng, and B. L. Hempstead. 2001. Regulation of cell survival by secreted proneurotrophins. Science. 294:1945–1948. [DOI] [PubMed] [Google Scholar]
- 6.Frade, J. M., and Y. A. Barde. 1998. Nerve growth factor: two receptors, multiple functions. Bioessays. 20:137–145. [DOI] [PubMed] [Google Scholar]
- 7.Chao, M., P. Casaccia-Bonnefil, B. Carter, A. Chittka, H. Kong, and S. O. Yoon. 1998. Neurotrophin receptors: mediators of life and death. Brain Res. Brain Res. Rev. 26:295–301. [DOI] [PubMed] [Google Scholar]
- 8.Patapoutian, A., and L. F. Reichardt. 2001. Trk receptors: mediators of neurotrophin action. Curr. Opin. Neurobiol. 11:272–280. [DOI] [PubMed] [Google Scholar]
- 9.Huang, E. J., and L. F. Reichardt. 2003. Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72:609–642. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan, D. R., B. L. Hempstead, D. Martin-Zanca, M. V. Chao, and L. F. Parada. 1991. The trk proto-oncogene product: a signal transducing receptor for nerve growth factor. Science. 252:554–558. [DOI] [PubMed] [Google Scholar]
- 11.Klein, R., S. Q. Jing, V. Nanduri, E. O'Rourke, and M. Barbacid. 1991. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell. 65:189–197. [DOI] [PubMed] [Google Scholar]
- 12.Barbacid, M. 1994. The Trk family of neurotrophin receptors. J. Neurobiol. 25:1386–1403. [DOI] [PubMed] [Google Scholar]
- 13.Levi-Montalcini, R. 1966. The nerve growth factor: its mode of action on sensory and sympathetic nerve cells. Harvey Lect. 60:217–259. [PubMed] [Google Scholar]
- 14.Bradshaw, R. A., T. L. Blundell, R. Lapatto, N. Q. McDonald, and J. Murray-Rust. 1993. Nerve growth factor revisited. Trends Biochem. Sci. 18:48–52. [DOI] [PubMed] [Google Scholar]
- 15.Wiesmann, C., and A. M. De Vos. 2001. Nerve growth factor: structure and function. Cell. Mol. Life Sci. 58:748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brancucci, A., N. Kuczewski, S. Covaceuszach, A. Cattaneo, and L. Domenici. 2004. Nerve growth factor favours long-term depression over long-term potentiation in layer II–III neurones of rat visual cortex. J. Physiol. 559:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hefti, F. 1997. Pharmacology of neurotrophic factors. Annu. Rev. Pharmacol. Toxicol. 37:239–267. [DOI] [PubMed] [Google Scholar]
- 18.Hagg, T., M. Manthorpe, H. L. Vahlsing, and S. Varon. 1988. Delayed treatment with nerve growth factor reverses the apparent loss of cholinergic neurons after acute brain damage. Exp. Neurol. 101:303–312. [DOI] [PubMed] [Google Scholar]
- 19.Apfel, S. C., J. A. Kessler, B. T. Adornato, W. J. Litchy, C. Sanders, and C. A. Rask. 1998. Recombinant human nerve growth factor in the treatment of diabetic polyneuropathy. NGF study group. Neurology. 51:695–702. [DOI] [PubMed] [Google Scholar]
- 20.Jonhagen, M. E. 2000. Nerve growth factor treatment in dementia. Alzheimer Dis. Assoc. Disord. 14(Suppl 1):31–38. [DOI] [PubMed] [Google Scholar]
- 21.Tuszynski, M. H., L. Thal, M. Pay, D. P. Salmon, H. S. U. R. Bakay, P. Patel, A. Blesch, H. L. Vahlsing, G. Ho, G. Tong, S. G. Potkin, J. Fallon, L. Hansen, E. J. Mufson, J. H. Kordower, C. Gall, and J. Conner. 2005. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat. Med. 11:551–555. [DOI] [PubMed] [Google Scholar]
- 22.Capsoni, S., G. Ugolini, A. Comparini, F. Ruberti, N. Berardi, and A. Cattaneo. 2000. Alzheimer-like neurodegeneration in aged antinerve growth factor transgenic mice. Proc. Natl. Acad. Sci. USA. 97:6826–6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Capsoni, S., S. Giannotta, and A. Cattaneo. 2002. Nerve growth factor and galantamine ameliorate early signs of neurodegeneration in antinerve growth factor mice. Proc. Natl. Acad. Sci. USA. 99:12432–12437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.De Rosa, R., A. A. Garcia, C. Braschi, S. Capsoni, L. Maffei, N. Berardi, and A. Cattaneo. 2005. Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. USA. 102:3811–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halvorson, K. G., K. Kubota, M. A. Sevcik, T. H. Lindsay, J. E. Sotillo, J. R. Ghilardi, T. J. Rosol, L. Boustany, D. L. Shelton, and P. W. Mantyh. 2005. A blocking antibody to nerve growth factor attenuates skeletal pain induced by prostate tumor cells growing in bone. Cancer Res. 65:9426–9435. [DOI] [PubMed] [Google Scholar]
- 26.Poduslo, J. F., and G. L. Curran. 1996. Permeability at the blood-brain and blood-nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res. Mol. Brain Res. 36:280–286. [DOI] [PubMed] [Google Scholar]
- 27.Hefti, F. F., A. Rosenthal, P. A. Walicke, S. Wyatt, G. Vergara, D. L. Shelton, and A. M. Davies. 2005. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol. Sci. 27:85–91. [DOI] [PubMed] [Google Scholar]
- 28.Saragovi, H. U., M. I. Greene, R. A. Chrusciel, and M. Kahn. 1992. Loops and secondary structure mimetics: development and applications in basic science and rational drug design. Biotechnol. NY. 10:773–778. [DOI] [PubMed] [Google Scholar]
- 29.Saragovi, H. U., and K. Gehring. 2000. Development of pharmacological agents for targeting neurotrophins and their receptors. Trends Pharmacol. Sci. 21:93–98. [DOI] [PubMed] [Google Scholar]
- 30.Pollack, S. J., and S. J. Harper. 2002. Small molecule Trk receptor agonists and other neurotrophic factor mimetics. Curr. Drug Targets CNS Neurol. Disord. 1:59–80. [DOI] [PubMed] [Google Scholar]
- 31.Massa, S. M., Y. Xie, and F. M. Longo. 2003. Alzheimer's therapeutics: neurotrophin domain small molecule mimetics. J. Mol. Neurosci. 20:323–326. [DOI] [PubMed] [Google Scholar]
- 32.LeSauteur, L., L. Wei, B. F. Gibbs, and H. U. Saragovi. 1995. Small peptide mimics of nerve growth factor bind trkA receptors and affect biological responses. J. Biol. Chem. 270:6564–6569. [DOI] [PubMed] [Google Scholar]
- 33.LeSauteur, L., N. K. Cheung, R. Lisbona, and H. U. Saragovi. 1996. Small molecule nerve growth factor analogs image receptors in vivo. Nat. Biotechnol. 14:1120–1122. [DOI] [PubMed] [Google Scholar]
- 34.Xie, Y., M. A. Tisi, T. T. Yeo, and F. M. Longo. 2000. Nerve growth factor (NGF) loop 4 dimeric mimetics activate ERK and AKT and promote NGF-like neurotrophic effects. J. Biol. Chem. 275:29868–29874. [DOI] [PubMed] [Google Scholar]
- 35.Beglova, N., S. Maliartchouk, I. Ekiel, M. C. Zaccaro, H. U. Saragovi, and K. Gehring. 2000. Design and solution structure of functional peptide mimetics of nerve growth factor. J. Med. Chem. 43:3530–3540. [DOI] [PubMed] [Google Scholar]
- 36.Maliartchouk, S., Y. Feng, L. Ivanisevic, T. Debeir, A. C. Cuello, K. Burgess, and H. U. Saragovi. 2000. A designed peptidomimetic agonistic ligand of TrkA nerve growth factor receptors. Mol. Pharmacol. 57:385–391. [PubMed] [Google Scholar]
- 37.Maliartchouk, S., T. Debeir, N. Beglova, A. C. Cuello, K. Gehring, and H. U. Saragovi. 2000. Genuine monovalent ligands of TrkA nerve growth factor receptors reveal a novel pharmacological mechanism of action. J. Biol. Chem. 275:9946–9956. [DOI] [PubMed] [Google Scholar]
- 38.Zaccaro, M. C., H. B. Lee, M. Pattarawarapan, Z. Xia, A. Caron, P. J. L'Heureux, Y. Bengio, K. Burgess, and H. U. Saragovi. 2005. Selective small molecule peptidomimetic ligands of TrkC and TrkA receptors afford discrete or complete neurotrophic activities. Chem. Biol. 12:1015–1028. [DOI] [PubMed] [Google Scholar]
- 39.Wiesmann, C., M. H. Ultsch, S. H. Bass, and A. M. de Vos. 1999. Crystal structure of nerve growth factor in complex with the ligand-binding domain of the TrkA receptor. Nature. 401:184–188. [DOI] [PubMed] [Google Scholar]
- 40.Urfer, R., P. Tsoulfas, L. O'Connell, J. Hongo, W. Zhao, and L. G. Presta. 1998. High resolution mapping of the binding site of TrkA for nerve growth factor and TrkC for neurotrophin-3 on the second immunoglobulin-like domain of the Trk receptors. J. Biol. Chem. 273:5829–5840. [DOI] [PubMed] [Google Scholar]
- 41.Kahle, P., L. E. Burton, C. H. Schmelzer, and C. Hertel. 1992. The amino terminus of nerve growth factor is involved in the interaction with the receptor tyrosine kinase p140trkA. J. Biol. Chem. 267:22707–22710. [PubMed] [Google Scholar]
- 42.Ibanez, C. F., L. L. Ilag, J. Murray-Rust, and H. Persson. 1993. An extended surface of binding to Trk tyrosine kinase receptors in NGF and BDNF allows the engineering of a multifunctional pan-neurotrophin. EMBO J. 12:2281–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shih, A., G. R. Laramee, C. H. Schmelzer, L. E. Burton, and J. W. Winslow. 1994. Mutagenesis identifies amino-terminal residues of nerve growth factor necessary for Trk receptor binding and biological activity. J. Biol. Chem. 269:27679–27686. [PubMed] [Google Scholar]
- 44.Treanor, J. J., C. Schmelzer, B. Knusel, J. W. Winslow, D. L. Shelton, F. Hefti, K. Nikolics, and L. E. Burton. 1995. Heterodimeric neurotrophins induce phosphorylation of Trk receptors and promote neuronal differentiation in PC12 cells. J. Biol. Chem. 270:23104–23110. [DOI] [PubMed] [Google Scholar]
- 45.McDonald, N. Q., and M. V. Chao. 1995. Structural determinants of neurotrophin action. J. Biol. Chem. 270:19669–19672. [DOI] [PubMed] [Google Scholar]
- 46.Banfield, M. J., R. L. Naylor, A. G. Robertson, S. J. Allen, D. Dawbarn, and R. L. Brady. 2001. Specificity in Trk receptor:neurotrophin interactions: the crystal structure of TrkB-d5 in complex with neurotrophin-4/5. Structure. 9:1191–1199. [DOI] [PubMed] [Google Scholar]
- 47.Settanni, G., A. Cattaneo, and P. Carloni. 2003. Molecular dynamics simulations of the NGF-TrkA domain 5 complex and comparison with biological data. Biophys. J. 84:2282–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kortemme, T., and D. Baker. 2002. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA. 99:14116–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kortemme, T., D. E. Kim, and D. Baker. 2004. Computational alanine scanning of protein-protein interfaces. Sci. STKE. 2004:pl2. [DOI] [PubMed] [Google Scholar]
- 50.Berman, H. M., J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov, and P. E. Bourne. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grubmuller, H., B. Heymann, and P. Tavan. 1996. Ligand binding: molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 52.Isralewitz, B., S. Izrailev, and K. Schulten. 1997. Binding pathway of retinal to bacterio-opsin: a prediction by molecular dynamics simulations. Biophys. J. 73:2972–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Izrailev, S., S. Stepaniants, M. Balsera, Y. Oono, and K. Schulten. 1997. Molecular dynamics study of unbinding of the avidin-biotin complex. Biophys. J. 72:1568–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marrink, S. J., O. Berger, P. Tieleman, and F. Jahnig. 1998. Adhesion forces of lipids in a phospholipid membrane studied by molecular dynamics simulations. Biophys. J. 74:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Isralewitz, B., M. Gao, and K. Schulten. 2001. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 11:224–230. [DOI] [PubMed] [Google Scholar]
- 56.Kosztin, D., S. Izrailev, and K. Schulten. 1999. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys. J. 76:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bayas, M. V., K. Schulten, and D. Leckband. 2003. Forced detachment of the CD2–CD58 complex. Biophys. J. 84:2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grubmuller, H. 2005. Force probe molecular dynamics simulations. Methods Mol. Biol. 305:493–515. [DOI] [PubMed] [Google Scholar]
- 59.Lorenzo, A. C., and P. M. Bisch. 2005. Analyzing different parameters of steered molecular dynamics for small membrane interacting molecules. J. Mol. Graph. Model. 24:59–71. [DOI] [PubMed] [Google Scholar]
- 60.Sitkoff, D., K. A. Sharp, and B. Honig. 1994. Accurate calculation of hydration free energies using macroscopic solvent models. J. Phys. Chem. 98:1978–1988. [Google Scholar]
- 61.Lee, B., and F. M. Richards. 1971. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55:379–400. [DOI] [PubMed] [Google Scholar]
- 62.Eisenhaber, F., P. Lijnzaad, P. Argos, C. Sander, and M. Scharf. 1995. The double cube lattice method: efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16:273–284. [Google Scholar]
- 63.Vriend, G. 1990. WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8:52–56. [DOI] [PubMed] [Google Scholar]
- 64.Fogolari, F., A. Brigo, and H. Molinari. 2002. The Poisson-Boltzmann equation for biomolecular electrostatics: a tool for structural biology. J. Mol. Recognit. 15:377–392. [DOI] [PubMed] [Google Scholar]
- 65.Massova, I., and P. A. Kollman. 1999. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J. Am. Chem. Soc. 121:8133–8143. [Google Scholar]
- 66.Bashford, D., and D. A. Case. 2000. Generalized Born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 51:129–152. [DOI] [PubMed] [Google Scholar]
- 67.Tsui, V., and D. A. Case. 2001. Theory and applications of the generalized Born solvation model in macromolecular simulations department of molecular biology. Biopolym. Nucl. Acid Sci. 56:275–291. [DOI] [PubMed] [Google Scholar]
- 68.Tsui, V., and D. A. Case. 2000. Molecular dynamics simulations of nucleic acids with a generalized Born solvation model. J. Am. Chem. Soc. 122:2489–2498. [Google Scholar]
- 69.Wang, J., P. Cieplak, and P. A. Kollman. 2000. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21:1049–1074. [Google Scholar]
- 70.Ponder, J. W., and D. A. Case. 2003. Force fields for protein simulations. Adv. Protein Chem. 66:27–85. [DOI] [PubMed] [Google Scholar]
- 71.Baker, N. A., D. Sept, S. Joseph, M. J. Holst, and J. A. McCammon. 2001. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA. 98:10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Connolly, M. L. 1983. Solvent-accessible surfaces of proteins and nucleic acids. Science. 221:709–713. [DOI] [PubMed] [Google Scholar]
- 73.Jorgensen, W. L., J. Chandrasekhar, J. Madura, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 74.Lindahl, E., B. Hess, and D. van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 75.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 76.Essmann, U., L. Perera, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8593. [Google Scholar]
- 77.Sagui, C., and T. A. Darden. 1999. Molecular dynamics simulations of biomolecules: long-range electrostatic effects. Annu. Rev. Biophys. Biomol. Struct. 28:155–179. [DOI] [PubMed] [Google Scholar]
- 78.Hess, B., H. Bekker, H. J. C. Berendsen, and J. G. E. M. Fraaije. 1997. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18:1463–1472. [Google Scholar]
- 79.Berendsen, H. J. C., J. P. M. Postma, A. DiNola, and J. R. Haak. 1984. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81:3684–3690. [Google Scholar]
- 80.Nosé, S. 1984. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 52:255–268. [Google Scholar]
- 81.Hoover, W. G. 1985. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 82.Parrinello, M., and A. Rahman. 1981. Polymorphic transitions in single crystals: a new molecular dynamics approach. J. Appl. Phys. 52:7182–7190. [Google Scholar]
- 83.Nosé, S., and M. L. Klein. 1983. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50:1055–1076. [Google Scholar]
- 84.Fersht, A. 1999. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, New York.
- 85.Ultsch, M. H., C. Wiesmann, L. C. Simmons, J. Henrich, M. Yang, D. Reilly, S. H. Bass, and A. M. de Vos. 1999. Crystal structures of the neurotrophin-binding domain of TrkA, TrkB and TrkC. J. Mol. Biol. 290:149–159. [DOI] [PubMed] [Google Scholar]
- 86.Covaceuszach, S., A. Cattaneo, and D. Lamba. 2005. Neutralization of NGF-TrkA receptor interaction by the novel antagonistic anti-TrkA monoclonal antibody MNAC13: a structural insight. Proteins. 58:717–727. [DOI] [PubMed] [Google Scholar]
- 87.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—Visual Molecular Dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]