

Abstract
Pressure-jump (p-jump)-induced relaxation kinetics was used to explore the energy landscape of protein folding/unfolding of Y115W, a fluorescent variant of ribonuclease A. Pressure-jumps of 40 MPa amplitude (5 ms dead-time) were conducted both to higher (unfolding) and to lower (folding) pressure, in the range from 100 to 500 MPa, between 30 and 50°C. Significant deviations from the expected symmetrical protein relaxation kinetics were observed. Whereas downward p-jumps resulted always in single exponential kinetics, the kinetics induced by upward p-jumps were biphasic in the low pressure range and monophasic at higher pressures. The relative amplitude of the slow phase decreased as a function of both pressure and temperature. At 50°C, only the fast phase remained. These results can be interpreted within the framework of a two-dimensional energy surface containing a pressure- and temperature-dependent barrier between two unfolded states differing in the isomeric state of the Asn-113–Pro-114 bond. Analysis of the activation volume of the fast kinetic phase revealed a temperature-dependent shift of the unfolding transition state to a larger volume. The observed compensation of this effect by glycerol offers an explanation for its protein stabilizing effect.
INTRODUCTION
The so-called conformational diseases, arising from the accumulation of misfolded proteins in or around cells, have challenged the main paradigms of structural biology. The most striking example is the prion phenomenon (1,2), where the observation of self-perpetuating protein conformational changes calls for a renewal of our understanding of molecular biology. Due to the recognition of the relationship between protein misfolding and human diseases, in particular of age-related neurodegenerative disorders such as Alzheimer's and Parkinson's disease (3,4), the field of protein folding has gained much interest in the past few years. Substantial progress toward an understanding of these complex processes has been made through a combination of novel experimental developments and theoretical advances (5). To characterize a protein folding reaction, several phenomenological models have been proposed (6), such as lattice simulations and statistical mechanical models. By comparison of such theoretical predictions with experiment, the so-called “new view” of protein folding has emerged in the last decade, which is based on energy “landscapes” (7,8). It postulates that a range of folding scenarios exists, containing many different paths that an unfolded polypeptide chain may explore before reaching its folded native state.
Generally, the kinetics of protein folding and unfolding are studied by a rapid change of denaturant concentration, or by temperature jump. Recently, pressure-induced protein unfolding/folding kinetics has received attention as an elegant alternative tool. This method consists of monitoring protein relaxations induced by sudden changes of pressure (9–16). Previously, this approach has been used extensively to study protein folding and unfolding under equilibrium conditions (17–19). Pressure-jumps provide unique information concerning packing and hydration properties of the transition state, inaccessible by other experimental techniques. Indeed, pressure induces conformational changes that reduce the overall volume of the system. In protein unfolding processes, formation of the ensemble of unfolded species, including also the protein hydration shell, is usually accompanied by a decrease in volume. This is believed to be caused by the combined effects of electrostriction of water molecules around newly exposed charged and polar groups, the decrease in partial volume of hydrophobic residues upon transfer from a nonpolar protein interior to water, and the elimination of packing defects (20). A jump in pressure, which can be performed in both directions (pressurization or depressurization), propagates rapidly without the difficulties inherent to the mixing of solutions, and can be used without significantly changing the solvent properties.
The model protein chosen in this work was bovine pancreatic ribonuclease A (RNase A; EC 3.1.27.5). This enzyme presents a well-known structure studied by x-ray crystallography (21) and NMR (22). In addition, the unfolding and refolding processes of the enzyme induced by chemical denaturant agents have been extensively studied by means of stopped-flow single- and double-jump and pulsed-labeled kinetic experiments by the groups of Baldwin (23,24) and Scheraga (25,26). Monitoring the unfolding of RNase A by a change of intrinsic fluorescence is possible although the protein does not possess any Trp residues in its sequence. However, to increase the signal, the previously constructed and characterized Y115W variant has been used (17). Its Trp residue is located on the solvent-exposed exterior of a β-turn, integrated in a hairpin subdomain, which is the most important chain folding initiation site (CFIS) of RNase A (27). Thus, Trp-115 may report as an intrinsic fluorescence probe on the CFIS as well as on the isomeric state of the neighboring Asn–Pro peptide bond. Characterization of the Y115W RNase A variant showed that the Y115W substitution does not significantly alter the activity and the stability of the enzyme at extreme conditions of temperature and pressure.
Here we use a p-jump technique to examine the dynamics of volume changes in the course of ribonuclease A folding and unfolding. We address the important question of whether folding and unfolding reaction paths are identical, a hypothesis that underlies many experimental and theoretical studies. Depending on experimental conditions, scenarios are found where the kinetics after upward and downward jumps is significantly different. The results can be interpreted by considering the dynamical aspect of two-dimensional energy surfaces. Furthermore, the p-jump approach proved to be useful to analyze the stabilizing effect of glycerol as a result of its interaction with the unfolding kinetic transition state. This approach appears to be promising to study the dynamics underlying protein structural changes.
EXPERIMENTAL PROCEDURES
Protein expression and purification
Y115W RNase A variant was previously constructed by site-directed mutagenesis as described by Torrent et al. (17). Protein expression and purification was performed accordingly.
NMR spectroscopy
One- and two-dimensional nuclear Overhauser enhancement (NOESY) (mixing time, 180 ms) and total correlation spectroscopy (TOCSY) 1H NMR spectra of Y115W RNase A were recorded at 35°C on a Bruker 600 MHz AMX spectrometer equipped with a cyroprobe for increased sensitivity. The protein concentration was 0.90 mM, and the sample contained 50 μM sodium 4,4-dimethyl-4-silapentane-1-sulphonate (DSS) as internal chemical shift standard, 0.20 M sodium phosphate buffer, and 100% D2O. The pH* of the solution was 6.7, where pH* is the pH meter reading without taking into account the deuterium isotope effect. The Trp proton resonances were assigned by comparing the relative intensities of their NOE signals (28).
Fluorescence measurements under high pressure
Fluorescence measurements were carried out using an Aminco Bowman Series 2 fluorescence-spectrophotometer (SLM Aminco, Foster City, CA), modified to accommodate a thermostated high pressure optical cell, allowing measurements up to 700 MPa. The lyophilized protein was dissolved to a concentration of 0.25 mg/mL in 50 mM MES buffer at pH 5.0, and filtered using a 0.22-μm filter. This buffer was selected for its relatively small pressure pH dependency (29). The protein solution was placed in a 5-mm diameter quartz cuvette, closed at the top with a flexible polyethylene film that was attached by a rubber O-ring.
For equilibrium studies tryptophan fluorescence was excited at 290 nm, using a bandwidth of 8 nm. Emission (accumulation of three scans) was collected between 310 and 440 nm with a bandwidth of 4 nm. Total fluorescence intensity between these two wavelengths was integrated to follow the unfolding/folding processes as a function of increasing/decreasing pressure, respectively. For kinetic studies, tryptophan fluorescence intensity was recorded at 350 nm (16 nm slit) and excited at 290 nm using a 4 nm slit. No photobleaching was observed under these conditions.
Pressure-induced equilibrium unfolding transitions
Following each pressure increment/decrement (steps of 20 MPa), the protein fluorescence was allowed to equilibrate before spectral recording. The equilibrium fluorescence intensity profiles versus pressure were fitted to Eq. 1
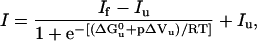 |
(1) |
where If and Iu are the fluorescence intensities of the folded and unfolded states, respectively, and I the observed fluorescence intensity at pressure p; 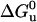 and ΔVu are the free-energy and volume change of unfolding at 0.1 MPa (1 atm), respectively.
and ΔVu are the free-energy and volume change of unfolding at 0.1 MPa (1 atm), respectively.
Pressure-jump-induced kinetics
Pressure-jumps consisted of sudden changes of pressure of ±40 MPa within a pressure range of 100–500 MPa. They were performed by using a homemade p-jump device connected to the high pressure optical cell placed in the abovementioned fluorescence spectrophotometer (30). Pressure-jumps (dead-time <5ms) were carried out by opening an electrically driven pneumatic valve localized between the high pressure optical cell and a ballast tank. The adiabatic temperature change associated to pressure-jumps did not exceed 0.4°C. After 20 s, the amplitude of the temperature change decreased to 0.2°C, and after 50 s, the initial temperature was recovered (30).
Determination of kinetic parameters from relaxation profiles
After each p-jump the relaxation profiles of the unfolding/folding reaction were fitted to single-exponential and when necessary to double (sequential) decays, according to Eqs. 2 and 3,
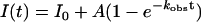 |
(2) |
 |
(3) |
where I(t) and I0 are the fluorescence intensities at time t and at time 0, A and B are the phase amplitudes, and kobs is the measured rate constant at the final pressure p.
The individual rate constants of the folding/unfolding reaction
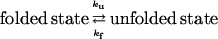 |
were determined from single exponential kinetics and from the fast phase in cases of two-exponential decays, according to Eqs. 4 and 5,
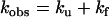 |
(4) |
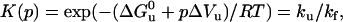 |
(5) |
where K(p) is the equilibrium constant at pressure p, 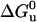 , and ΔVu are the free energy and volume change of unfolding obtained from equilibrium experiments and p is the final pressure of each jump. Linear plots of lnkf and lnku versus the final pressure of each jump allowed us to determine
, and ΔVu are the free energy and volume change of unfolding obtained from equilibrium experiments and p is the final pressure of each jump. Linear plots of lnkf and lnku versus the final pressure of each jump allowed us to determine 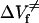 and
and 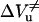 , the activation volumes for folding and unfolding, respectively, according to Eqs. 6 and 7.
, the activation volumes for folding and unfolding, respectively, according to Eqs. 6 and 7.
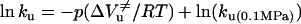 |
(6) |
 |
(7) |
The activation free energy  for folding and unfolding was obtained from Eqs. 8 and 9,
for folding and unfolding was obtained from Eqs. 8 and 9,
 |
(8) |
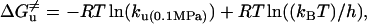 |
(9) |
where kB is the Boltzmann constant, R the gas constant, and h the Planck constant.
RESULTS
Structural characterization by NMR spectroscopy
The 1D 1H NMR spectrum of Y115W RNase A at 35°C is typical of folded RNase A (data not shown). The 2D TOCSY and NOESY spectra revealed one set of peaks arising from W115; these peaks were assigned and their chemical shift values are (in ppm): Hα 4.29, Hβ 3.15, Hβ′ 3.01, Hδ1 6.83, Hɛ3 8.20, Hζ2 7.70, Hζ3 7.27, and Hη2 7.32 (Fig. 1 a). The observation of only one set of resonances is taken as evidence that this residue adopts only one major conformation in the folded protein. The lack of NOE crosspeaks between Trp-115 and other residues' protons is consistent with the indole side chain being placed at a solvent exposed position.
FIGURE 1.

Two-dimensional NOESY NMR spectrum of RNase A Y115W in D2O, 0.20 M sodium phosphate, pH* 6.7, 35°C. (A) Downfield region. The dotted line traces the assignment of the Trp-115 peaks. (B) The dotted boxes mark the position expected for peaks arising from a trans Asn-113–Pro-114 peptide bond; these peaks are not seen strongly suggesting that this peptide bond is >90% in the cis conformation.
The Asn-113–Pro-114 peptide bond is cis in native, wild-type RNase A, but is trans in the three-dimensional dimer formed by swapping of the C-terminal β-strands (31). Under identical solution conditions, the RNase A dimer produces a strong pair of NOE signals arising from the N113 Hα and Pro-114 Hδ protons that are indicative of a trans conformation, and that are not observed in the monomeric wild-type RNase A (32). These signals are not observed in the NOESY spectrum of the Y115W variant, which strongly suggests that the Asn-113–Pro-114 peptide bond is in the cis conformation in the folded state.
Pressure-induced equilibrium unfolding
In a previous study (17) we showed that the Y115W amino acid replacement in the RNase A structure does not substantially perturb its heat and pressure stability. Upon increasing pressure, a more than threefold increase in fluorescence yield was obtained. This turned out to be a suitable intrinsic probe for fast kinetic measurements. The pressure-induced unfolding transition of Y115W RNase A was monitored between 30 and 50°C (Fig. 2). The equilibrium unfolding process was found to be fully reversible, and the thermodynamic parameters (Table 1) were determined within the framework of a two-state model. Under all conditions the fluorescent yield increased, as pressure was raised, without any significant spectral shift, indicating that the native polar environment of the Trp residue is maintained upon protein unfolding. This is consistent with the exposed position of the Tyr-115 residue in the wild-type enzyme, at the turn connecting β-strands 5 and 6 (21). From the thermodynamic data (shown partly in Table 1) it is apparent that both the stability of the Y115W variant (expressed by 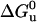 ) and the absolute value of the reaction volume (ΔVu) decrease linearly as a function of temperature. The expansibility (∂ΔV/∂T) was determined as Δα = 1.4 ml mol−1 K−1, comparable to that of the wild-type protein (33).
) and the absolute value of the reaction volume (ΔVu) decrease linearly as a function of temperature. The expansibility (∂ΔV/∂T) was determined as Δα = 1.4 ml mol−1 K−1, comparable to that of the wild-type protein (33).
FIGURE 2.

Normalized transition curves for the pressure-induced unfolding of the Y115W variant of RNase A at different temperatures. Experimental data for depressurization are coincident with those of pressurization. The solid lines are nonlinear regression fits of the experimental data, expressed as fraction of unfolded protein, Fu, based on a two-state model, for (○) 30°C, (▪) 35°C, (▴) 40°C, (•) 45°C, and (▵) 50°C.
TABLE 1.
Thermodynamic parameters of Y115W RNase A variant calculated from pressure-induced unfolding curves at pH 5.0
| Experimental conditions |  |
ΔVu ml mol−1 | p1/2 MPa | kobs at p1/2 s−1 |  |
 |
|---|---|---|---|---|---|---|
| 30°C | 34.55 (1.43) | −73.81 (3.28) | 468 (1.39) | 4.20 × 10−3 (1.0 × 10−4) | −58.17 (2.98) | 15.17 (2.98) |
| 50°C | 10.25 (0.29) | −45.51 (1.15) | 225 (0.85) | 9.49 × 10−2 (2.10 × 10−3) | −17.50 (0.53) | 27.84 (0.53) |
| 50°C, 10% dextran | 13.56 (2.59) | −55.38 (10.34) | 245 (1.10) | 9.29 × 10−2 (1.7 × 10−3) | −20.42 (1.06) | 25.46 (1.06) |
| 50°C, 30% glycerol | 25.82 (0.96) | −62.01 (2.54) | 416 (1.55) | 3.19 × 10−2 (2.0 × 10−4) | −44.28 (2.12) | 17.50 (2.12) |
Numbers in parentheses are the mean ± SE of the data. The activation volumes were determined from the rapid phase of upward p-jumps.
p-Jump-induced folding and unfolding kinetics
As expected from equilibrium measurements, p-jumps of 40 MPa in magnitude in either direction (upward and downward), produced an increase or decrease in fluorescence intensity, respectively. The timescale of the relaxation kinetics was of the order of minutes. The monitored spectral changes were observed to be fully reversible, as in equilibrium experiments. The amplitude of the fluorescence change observed after each p-jump was in excellent agreement with the spectral change observed in equilibrium measurements. The kinetics induced by upward and downward p-jumps were compared under identical final conditions of temperature and pressure. Dissimilar kinetics were observed. Whereas downward p-jumps resulted always in monophasic kinetics, upward p-jumps led to biphasic kinetics at pressures lower than p1/2, the pressure at half-transition. A typical example of these kinetics is shown in Fig. 3. As shown in Fig. 4, the relative amplitude of the slow phase decreased as a function of both pressure and temperature. Above p1/2, the slow phase was no longer detectable. Similarly, at 50°C, only the fast phase subsisted. Fig. 5 illustrates the pressure profile of kobs at two selected temperatures. Obviously, at 50°C the monophasic upward and downward p-jump profiles were distinguished by only very small differences. These were constant throughout the pressure range and may be ascribed to small temperature differences due to adiabatic compression and decompression. In contrast, at 35°C, a second kinetic phase was observed in upward p-jumps in the lower pressure range (below p1/2). We have carried out upward and downward p-jumps in the whole pressure range, every 5°C between 30 and 50°C. Without exception, we always found the same pattern distinguishing upward and downward p-jumps at temperatures below 50°C.
FIGURE 3.

p-Jump induced Y115W RNase A folding and unfolding relaxation kinetics at 30°C. Comparison of upward and downward p-jumps leading to identical final pressures. The kinetics after a downward p-jump is fitted by a single exponential (A), that after an upward p-jump by a double (B) and a single exponential (C, dashed line). The corresponding residuals are shown on the right side.
FIGURE 4.

Normalized amplitude of the slow kinetic phase after upward p-jumps, as a function of pressure, at 35°C (○), 40°C (▴), and 45°C (•).
FIGURE 5.

Pressure dependence of kobs, determined from upward (solid symbols) and downward (open symbols) p-jumps at 35°C (▴▵), 50°C (•○), and 50°C in presence of 30% glycerol (▪□). The slow phase observed after positive pressure-jumps at 35 ° is shown by inverted triangles. At each condition, the data from two independent experimental series were superimposed in a way to obtain data points every 20 MPa.
We investigated whether the absence of the slow phase in downward pressure-jump experiments might be explained by its reduced amplitude, as compared to that of upward pressure-jumps. This could be the case in downward pressure-jumps starting from an initial pressure higher than p1/2. However, downward pressure-jumps within a range well below p1/2 (under conditions where the amplitude of the slow phase after upward pressure-jumps is higher than 50%) lead still to single exponential fast kinetics. We must therefore conclude that the kinetic folding/unfolding mechanism is different after upward and downward pressure-jumps.
At each temperature, the pressure profile of kobs of the fast phase is characterized by a u-shape, i.e., it exhibits a Chevron plot-like behavior, which becomes more and more pronounced when the temperature is decreased. The origin of the u-shape is explained by the pressure dependence of the individual rate constants kf and ku, as described below.
Pressure dependence of the individual folding and unfolding rate constants
As explained in Experimental Procedures, the individual rate constants of the fast phase, kf and ku were determined at each final pressure from kobs and K(p). In all cases, kf decreases and ku increases linearly as a function of pressure (Fig. 6). Obviously, kf predominates at pressures below the equilibrium midpoint unfolding transition, while ku predominates at higher pressure. This picture was not affected by the direction of the pressure-jumps.
FIGURE 6.

Effect of pressure at 30°C (A), 50°C (B), and 50°C in presence of 30% glycerol (C) on the individual folding, kf, (solid symbols) and unfolding, ku, (open symbols) rate constants. The dependence of the individual rate constants on initial conditions is shown for positive (♦⋄ (A), •○ (B), ▪□ (C)), and negative pressure-jumps (
 (A), ▴▵ (B)), ▾∇ (C). Each plot results from the superimposition of two independent experimental series.
(A), ▴▵ (B)), ▾∇ (C). Each plot results from the superimposition of two independent experimental series.
Solvent effects
In the presence of glycerol (30%), both 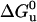 and the absolute value of ΔVu were strongly decreased (Table 1). As a consequence, the thermodynamic parameters determined at 50°C resembled those determined at 30°C in the absence of glycerol. In contrast, the presence of 10% dextran (molecular weight, 40,000) had no significant effect on the equilibrium parameters. Further information, not accessible under equilibrium conditions, was obtained by a kinetic analysis. As shown in Fig. 5, in the presence of glycerol, upward and downward p-jump experiments resulted in identical relaxation times. The kobs pressure profile was, however, strongly shifted to higher pressures (see also Table 1). In contrast, dextran did not significantly affect the kobs values.
and the absolute value of ΔVu were strongly decreased (Table 1). As a consequence, the thermodynamic parameters determined at 50°C resembled those determined at 30°C in the absence of glycerol. In contrast, the presence of 10% dextran (molecular weight, 40,000) had no significant effect on the equilibrium parameters. Further information, not accessible under equilibrium conditions, was obtained by a kinetic analysis. As shown in Fig. 5, in the presence of glycerol, upward and downward p-jump experiments resulted in identical relaxation times. The kobs pressure profile was, however, strongly shifted to higher pressures (see also Table 1). In contrast, dextran did not significantly affect the kobs values.
DISCUSSION
Dependence of relaxation rate constants on final and initial conditions
An intriguing result of this paper is the observation that the p-jump-induced unfolding/refolding kinetics depended on the direction of the pressure-jump; i.e., it depended on the initial physical chemical condition. This is against expectation, because in the majority of cases, protein kinetics depends only on final conditions. Furthermore, the observed differences in folding and unfolding kinetics cannot be explained by a hysteresis behavior as reported for some proteins. Indeed, the pressure-induced folding and unfolding transition curves under equilibrium conditions can be superimposed, and they are identical to those constructed after pressure-jumps. In contrast, a mechanistic understanding of the present results may be provided by a closer kinetic analysis of possible scenarios in relaxation reactions.
Bimolecular reactions
Typical examples of bimolecular reactions are those reflecting the binding/dissociation of a ligand (L) to or from a protein (P). k1 and k−1 are the forward and backward reactions rates, respectively. If the equilibrium reaction
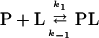 |
is displaced in the relaxation process, the observed rate constant kobs does depend indeed on the initial conditions, because the respective initial concentrations of (P) and (L) depend on the initial equilibrium conditions. For example, under pseudo-first-order conditions
 |
(10) |
Monomolecular reactions
The situation is completely different in monomolecular reactions, such as transitions from a protein conformation (P) to (Q).
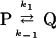 |
Here the observed rate constant is composed only by the two individual rate constants
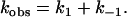 |
(11) |
Because these are concentration independent, kobs depends exclusively on the final conditions of the equilibrium at the end of the relaxation process (34), given by
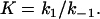 |
(12) |
Sequential and branched reactions
Would a more complex reaction, for example, a sequential reaction (A→B→…→X→Y) or a branched reaction (A→B; A→C), explain the dissimilar kinetics after upward and downward p-jumps? The answer is no. In all these cases, kobs will depend in a complex way on the different rate constants. In turn, these can be expressed as a function of a more or less complex equilibrium constant, which again, is determined by the final physical chemical conditions, only.
Hypothetical reaction model
Clearly, the structural transition studied in this paper is a monomolecular reaction. Therefore, Eqs. 11 and 12 should apply. However, in the low pressure domain we observe two kinetic phases for upward p-jumps, and only one phase for downward p-jumps. A possible solution for this apparent contradiction may be offered by the reaction scheme depicted in Fig. 7. As we will discuss in the next section, this model is in line with the actual kinetic and structural knowledge of ribonuclease A. In this section, we will limit ourselves to a formal kinetic analysis, however. We hypothesize the existence of one folded state, F, and two unfolded states, denoted U1 and U2. Stricto sensu, the latter two states should not be considered as completely unfolded because amide proton exchange experiments showed that pressure-unfolded ribonuclease A retains some secondary structure, at least at 10°C (35). However, the residual structure in the pressure-unfolded state appears to be limited and of low stability. Indeed, the protection factors observed by Zhang et al. were very small. In fact only 14 amide protons had protection factors >10 and none had a protection factor >80. Such a modest residual structure should not be confused with the extensive and robust structure present in the quasinative intermediate state, In, studied by Cook et al. (36), which has protection factors only fourfold lower than the native protein itself as reported by Brems and Baldwin (37), which are on the order of 100,000 (38). Therefore, and for simplicity, these states are denoted “unfolded” in the following.
FIGURE 7.

Schematic free energy landscape of the Y115W variant of RNase as a function of pressure, including one folded state (F) and two unfolded states (U1 and U2). p-Jumps are indicated by vertical arrows. A slow relaxation of the energy surface to a new physical chemical condition is indicated by horizontal arrows. (A) Upward p-jumps resulting in biphasic kinetics below p1/2. (B) Downward p-jumps resulting in monophasic kinetics.
In upward p-jumps (Fig. 7 A), initially, at low pressure (A), all the protein is in state F, and the two unfolded states are separated by an energy barrier. The upward p-jump provokes a relative energetic change of the three states, and the protein relaxes in a fast phase (Φ1) from F to U1 (B). In the beginning, the reaction cannot proceed further, because U1 is separated from U2 by a high energy barrier. However, at the higher pressure, this energy barrier relaxes in turn to the new physical chemical condition: it decreases slowly, allowing the protein to occupy partly U2 (C). This second reaction is slow (Φ2) because the decrease of the energy barrier is slow. Continuing now our p-jumps to pressures higher than p1/2 (D), only one kinetic phase is observed (Φ1 and Φ2 are indistinguishable because the energetic barrier between the unfolded states has vanished).
In downward p-jumps (Fig. 7 B), most of the protein is initially, at high pressure, in state U2 (D). The protein relaxes within a single phase (C) to occupy partly U1 and F (Φ1 and Φ2 are indistinguishable and the two unfolded states are not separated by a high energy barrier). This situation does not change when we continue with our p-jumps to still lower pressures (E). The protein relaxes again within a single observable phase to state F. Then, the energy barrier between the unfolded states will slowly build up again (A). However, this is of no kinetic importance because then the protein already occupies the folded state F.
This reaction model, though hypothetical, is in accord with our kinetic observations. A pressure-dependent energy barrier, which adapts only slowly to the new pressure after a pressure-jump, explains why we observe two kinetic phases in upward p-jumps below p1/2, but only one phase at higher pressures, and also in downward p-jumps. The analysis of the respective amplitudes of the two kinetic phases confirms this model. Regardless of the temperature, the relative amplitude of the slow phase (Φ2 in our model) decreases as a function of pressure, until it vanishes when p1/2 is reached. This results from the steady decrease at higher pressures of the energy barrier existing between the unfolded states (in the limit of a negligible energy barrier, the slow phase is no more detectable). Furthermore, at a given pressure, its relative amplitude decreases as a function of temperature (to finally become unobservable at 50°C). This can readily be explained by an overcoming of the energy barrier between U1 and U2 at high temperature according to the Arrhenius relation.
Similar kinetic discrepancies at identical final pressures may be expected by performing p-jumps of different pressure amplitudes. However, we preferred not to perform these because different pressure amplitudes result in different adiabatic heating/cooling effects, which might have biased the significance of the results. Such temperature artifacts are inevitable in p-jumps (30), and therefore we conducted our experiments by applying always constant pressure amplitudes (40 MPa). However, relaxation kinetics using different perturbation amplitudes have been used by Leeson et al. (39) in temperature-jump experiments with CspA protein connected to an infrared spectroscopy detection method. Although the experimental setup of the temperature and p-jump experiments was quite different, it is interesting to note, that the temperature-jump approach resulted in similar conclusions, implying perturbation-induced changes of the protein energy landscape. Thus, temperature- and p-jump relaxation methods appear as highly complementary experimental approaches. Indeed, the temperature-jump study concluded by calling for performing jumps in opposite directions. This is not feasible with the temperature-jump, but easy to perform with the p-jump method.
Relevance of the reaction model with actual kinetic and structural knowledge of ribonuclease A
Clearly, the above model fits the complex folding/unfolding kinetics presented here. In this section we will check its validity with respect to structural data of ribonuclease A. In a previous report, the two kinetic phases were explained by two unfolding pathways originating from two distinct folded states (30). However, this hypothesis had to be dismissed in view of our NMR data, indicating that the protein adopts only one major conformation in its folded state. This leaves us with a minimal reaction model containing only one folded (F) and two unfolded (U1 and U2) states.
The identity of U1 and U2 may become clear by a comparison with the work of Scheraga and co-workers (25), who used chemical denaturants to induce unfolding of RNase A. Their method, single- and double-jump stopped-flow, as well as pulsed-labeling kinetic experiments, monitored the progress of the reaction by detecting changes of fluorescence of the tyrosyl residues of the enzyme (25,26). Similarly to our results, they identified two kinetic phases. They showed that the fast-phase monitors conformational unfolding whereas the slow-phase monitors the cis-trans isomerization arising mainly from two proline residues that are cis in the native structure. Specifically, Tyr-115 was shown to report locally on the isomerization of the Asn-113–Pro-114 peptide bond. Further evidence for this very local reporting came from the use of double mutants by Juminaga et al. (25), showing that the fluorescence of Y115 does not sense the isomerization of the nearby P117. This suggests strongly that the fluorescence of the tryptophan residue replacing Tyr-115 in this study, does also report on the isomeric state of the Asn-113–Pro-114 peptide bond. According to the NOESY spectrum, the Asn-113–Pro-114 peptide bond of the Y115W variant is—like wild-type RNase A—in the cis conformation in the folded state. Hence, the unfolded states U1 and U2 appear to reflect the cis and trans isomers of the Asn-113–Pro-114 peptide bond.
Hence, the two kinetic phases observed in upward p-jumps can be understood as a rapid protein unfolding, from F to U1, followed by a slower cis-trans isomerization from U1 to U2. In the first step, the native isomeric state of Pro-114 (cis) is conserved. In the second step, the cis-trans isomerization is possible due to a pressure-induced decrease of the energy barrier between the cis (U1) and trans (U2) isomers of Pro-114. The isomerization of other prolines will probably also occur. However, our probe (Trp-115) is sensitive only to the isomerization of Asn-113–Pro-114 peptide bond and to the folded state of the protein. The nonobservation of two kinetic phases in downward pressure-jumps is explained by an initially low energy barrier between U1 and U2. This appears justified in light of a recent report of Bhat et al. (40), showing that the isomeric state of Pro-114 has little effect on the folding kinetics of RNase A. Although substitution of Pro-114 by Ala or Gly was found to destabilize RNase A by 3 kcal/mol (41), previous NMR (42), x-ray (43), folding (44–46), and computational modeling (47) data show that RNase A refolds equally from both isomers. Thus, cis Pro-114 can be classified as a Type II (stabilizing but not essential) proline according to the nomenclature introduced in the pioneering theoretical study by Levitt (48). In their study of the cis-trans equilibrium of glycyl-l-proline, Cheng and Bovey (49) observed that cis-trans ratio increases as the temperature is increased. This observation is perfectly in line with our observation of a diminishing amplitude for the slow phase (cis to trans) as the temperature is increased because at higher temperatures, there is less trans isomer present at equilibrium. Moreover, Cheng and Bovey (49) observed that the rate of cis-trans interconversion also increases markedly with temperature. Applied to this model, this means that under unfolding conditions (high pressure/temperature) the remaining energy barrier between the cis and trans isomers of the Asn-113–Pro-114 peptide bond is low enough to allow their rapid interchange.
Would the weak, fragile structure present in the pressure denatured state (see preceding section) have a significant effect on Pro-114 isomerization occurring during refolding? This is most unlikely. According to Wedemeyer et al. (26), “the cis-trans isomerization of the Asn-113–Pro-114 peptide bond makes relatively minor, localized changes that do not affect the structure of the adjacent hydrophobic core of residues 58–110.” Therefore, conversely, any structure in this hydrophobic core region is unlikely to affect the Pro isomerization. Moreover, the cis-trans isomeric state of Pro-114 only weakly affects the folding and native structure of RNase A (40).
These results are interesting with respect to the role of the chain-folding initiation site (CFIS). When unfolding is promoted, the CFIS would be one of the last subdomains to unfold and the cis-trans isomerization of the Asn-113–Pro-114 peptide bond takes place only after a first rapid conformational unfolding. In this view, after an increase of pressure, the two-dimensional energy surface must first equilibrate to the new physical chemical condition, before the isomerization of the proline peptide bond can take place. To the contrary, when folding is induced (downward pressure-jump), one of the first subdomains to fold would be the CFIS, independently of the isomeric state of the Asn-113–Pro-114 peptide bond because the energy barrier between both isomers is very low.
An intriguing feature of this work is the pressure dependence of the slow phase (U1 to U2), which, according to our model, reflects the pressure-dependent slow decrease of the energy barrier between cis and trans Pro-114. At high pressure the rate of this phase is increased, and its amplitude decreased. No literature data are available to explain this pressure effect. Further experiments, namely high-pressure modeling studies, are necessary to understand the effect of pressure on proline isomerization.
The stabilizing effect of glycerol via its action on the transition state
As shown in Table 1, glycerol slows down significantly the folding/unfolding reaction rates. As observed for many other proteins (50), it thus exerts a stabilizing effect against pressure-induced protein structural changes. This study offers a possibility to better understand this effect via a closer analysis of the reaction transition state. Here we are applying this analysis to the fast kinetic phase observed in upward p-jumps only. Very similar conclusions (not shown) can be drawn from the downward p-jumps. The slow phase, however, contained too few and too scattered data points to permit a thermodynamic analysis.
As shown in Fig. 8, the unfolding transition state always occupies a volume in between those of the folded and the unfolded states. However, as shown in the inset, the contribution of the unfolding activation volume 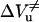 to the reaction volume ΔVu decreases strongly as a function of temperature. Interestingly, the presence of 30% glycerol compensates this effect: at 50°C the ratio of
to the reaction volume ΔVu decreases strongly as a function of temperature. Interestingly, the presence of 30% glycerol compensates this effect: at 50°C the ratio of 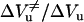 is comparable to that at 30°C in the absence of glycerol.
is comparable to that at 30°C in the absence of glycerol.
FIGURE 8.

Free energy (G)/volume (V) diagram of the p-jump induced unfolding reaction (fast kinetic phase) at 30°C (•), 50°C (▵), 50°C in the presence of 30% glycerol (○), and 50°C in the presence of 10% dextran (▴). The reaction goes from the folded state (F) to the unfolded state (U) via the transition state (#). The free energy and the volume of the unfolded state were set to zero. The inset shows the ratio between the activation volume, ΔV‡, and the reaction volume, ΔV, as a function of temperature in water (•), in the presence of 10% dextran (○), and in the presence of 30% glycerol (▾). The error bars are contained within the size of the symbols.
From these data, together with the individual unfolding rate constants ku, it was possible to construct the free energy (G)/volume (V) diagram of Fig. 8. Because our experiments do not permit determination of absolute values of G and V, these were set arbitrarily to zero for the unfolded states. Under this constraint, the free energy of the transition state is ∼80 kJ higher than that of the unfolded state, regardless of the temperature and solvent composition. However, as expected, the free energy of the folded state is significantly lower at 30°C than at 50°C. In the presence of 30% glycerol at 50°C, the stability lies in between those of 30°C and 50°C in aqueous solvent. In water, the volume of the transition state is close to that of the unfolded state at 30°C, and close to that of the folded state at 50°C. Most remarkably, in the presence of 30% glycerol at 50°C, the volume of the transition state is close to that at 30°C in water.
This combined free energy/volume analysis of the unfolding transition state offers a new way to understand, at least partly, the stabilizing effect of glycerol. The transition state in the pressure-induced unfolding reaction is certainly a highly labile species presenting a strongly decreased volume with respect to the folded state. This volume decrease is probably due to a collapse of voids (existing in the folded structure) and to a higher packing density of water molecules around exposed charged and/or polar residues. This transient structural reorganization has an energetic cost, which increases as a function of volume change. At high temperature, here at 50°C, the volume decrease from the folded to the transition state is less important, and the activation free energy—and thus the structural stability—is decreased accordingly. Glycerol, acting as an osmolyte, may be seen as compensating this temperature effect by decreasing the volume of the transition state via its reducing action on the hydration shell of the transition state. The result is a transition state, resembling in its volume and energy properties to that observed in its absence at lower temperature. However, the stabilizing effect of glycerol does not seem to be related to an increased solvent viscosity. In fact, as shown in Fig. 1, no stabilizing effect was observed when glycerol was replaced by dextran, another viscogenous compound. Moreover, as shown in the inset of Fig. 8, the presence of dextran did not affect the volume of the transition state. This possible explanation of the structure stabilizing effect of glycerol via its action on the volume of the unfolding transition state is certainly attractive. Nevertheless, to assess its general validity, more experimental p-jump data from other proteins are now needed.
CONCLUSION
After several decades of abundant literature concerning the effects of chemical agents, temperature, and pressure on protein structure and stability under equilibrium conditions, the dynamic aspect of protein folding and unfolding is increasingly gaining interest (51). This is certainly due to the recognition that protein folding/unfolding is a complex reaction that can be described properly only in terms of multidimensional energy surfaces (7,52,53). Apart from computational approaches, the p-jump method appears as an interesting tool, avoiding some of the constraints of the T-jump method (39). Taking RNase A as a model, the p-jump approach results are consistent with pressure and temperature-dependent dynamic features of a two-dimensional energy surface. Furthermore, the method permitted to investigate the importance of the kinetic transition state in its interaction with hydration water. It would be very interesting now, to apply the power of this method to other proteins that tend to unfold under certain pathological conditions. Especially the mechanism of the structural conversion of amyloidogenic proteins is far from being understood (54–58). As these proteins appear to be particularly sensitive toward pressure, it is foreseeable that the investigation of their pressure-induced dynamics will be fertile.
Acknowledgments
The authors are grateful for the expert technical assistance of C. Valentin.
This work was undertaken in the framework of the European COST-D30 action. It was supported by the grants BMC2003-08458-CO2-02 and CTQ2004-08275-C02-02 from the Ministerio de Educación y Ciencia (Spain), and SGR01-00196 from the Direcció General de Recerca, Generalitat de Catalunya. We are also indebted to “Fundació M. F. de Roviralta” of Barcelona for equipment purchasing grants. J.F. acknowledges a fellowship from the University of Girona.
References
- 1.Prusiner, S. B. 1982. Novel proteinaceous infectious particles cause scrapie. Science. 216:136–144. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner, S. B., M. R. Scott, S. J. DeArmond, and F. E. Cohen. 1998. Prion protein biology. Cell. 93:337–348. [DOI] [PubMed] [Google Scholar]
- 3.Carrell, R. W., and D. A. Lomas. 1997. Conformational disease. Lancet. 350:134–138. [DOI] [PubMed] [Google Scholar]
- 4.Taylor, J. P., J. Hardy, and K. H. Fischbeck. 2002. Toxic proteins in neurodegenerative disease. Science. 296:1991–1995. [DOI] [PubMed] [Google Scholar]
- 5.Dobson, C. M., A. Sali, and M. Karplus. 1998. Protein folding: a perspective from theory and experiment. Angew. Chem. Int. Ed. Engl. 37:868–893. [DOI] [PubMed] [Google Scholar]
- 6.Karplus, M., and D. L. Weaver. 1994. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci. 3:650–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dill, K. A., and H. S. Chan. 1997. From Levinthal to pathways to funnels. Nat. Struct. Biol. 4:10–19. [DOI] [PubMed] [Google Scholar]
- 8.Bryngelson, J. D., J. N. Onuchic, N. D. Socci, and P. G. Wolynes. 1995. Funnels, pathways, and the energy landscape of protein folding: a synthesis. Proteins. 21:167–195. [DOI] [PubMed] [Google Scholar]
- 9.Vidugiris, G. J., J. L. Markley, and C. A. Royer. 1995. Evidence for a molten globule-like transition state in protein folding from determination of activation volumes. Biochemistry. 34:4909–4912. [DOI] [PubMed] [Google Scholar]
- 10.Jacob, M., G. Holtermann, D. Perl, J. Reinstein, T. Schindler, M. A. Geeves, and F. X. Schmid. 1999. Microsecond folding of the cold shock protein measured by a pressure-jump technique. Biochemistry. 38:2882–2891. [DOI] [PubMed] [Google Scholar]
- 11.Herberhold, H., and R. Winter. 2002. Temperature- and pressure-induced unfolding and refolding of ubiquitin: a static and kinetic Fourier transform infrared spectroscopy study. Biochemistry. 41:2396–2401. [DOI] [PubMed] [Google Scholar]
- 12.Woenckhaus, J., R. Kohling, P. Thiyagarajan, K. C. Littrell, S. Seifert, C. A. Royer, and R. Winter. 2001. Pressure-jump small-angle x-ray scattering detected kinetics of staphylococcal nuclease folding. Biophys. J. 80:1518–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Panick, G., and R. Winter. 2000. Pressure-induced unfolding/refolding of ribonuclease A: static and kinetic Fourier transform infrared spectroscopy study. Biochemistry. 39:1862–1869. [DOI] [PubMed] [Google Scholar]
- 14.Desai, G., G. Panick, M. Zein, R. Winter, and C. A. Royer. 1999. Pressure-jump studies of the folding/unfolding of Trp repressor. J. Mol. Biol. 288:461–475. [DOI] [PubMed] [Google Scholar]
- 15.Herberhold, H., S. Marchal, R. Lange, C. H. Scheyhing, R. F. Vogel, and R. Winter. 2003. Characterization of the pressure-induced intermediate and unfolded state of red-shifted green fluorescent protein—a static and kinetic FTIR, UV/VIS and fluorescence spectroscopy study. J. Mol. Biol. 330:1153–1164. [DOI] [PubMed] [Google Scholar]
- 16.Tan, C. Y., C. H. Xu, J. Wong, J. R. Shen, S. Sakuma, Y. Yamamoto, R. Lange, C. Balny, and K. C. Ruan. 2005. Pressure equilibrium and jump study on unfolding of 23-kDa protein from spinach photosystem II. Biophys. J. 88:1264–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torrent, J., J. P. Connelly, M. G. Coll, M. Ribó, R. Lange, and M. Vilanova. 1999. Pressure versus heat-induced unfolding of ribonuclease A: the case of hydrophobic interactions within a chain-folding initiation site. Biochemistry. 38:15952–15961. [DOI] [PubMed] [Google Scholar]
- 18.Font, J., A. Benito, J. Torrent, R. Lange, M. Ribó, and M. Vilanova. 2006. Pressure- and temperature-induced unfolding studies: thermodynamics of core hydrophobicity and packing of ribonuclease A. Biol. Chem. 387:285–296. [DOI] [PubMed] [Google Scholar]
- 19.Torrent, J., P. Rubens, M. Ribó, K. Heremans, and M. Vilanova. 2001. Pressure versus temperature unfolding of ribonuclease A: an FTIR spectroscopic characterization of 10 variants at the carboxy-terminal site. Protein Sci. 10:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva, J. L., D. Foguel, and C. A. Royer. 2001. Pressure provides new insights into protein folding, dynamics and structure. Trends Biochem. Sci. 26:612–618. [DOI] [PubMed] [Google Scholar]
- 21.Wlodawer, A., L. A. Svensson, L. Sjolin, and G. L. Gilliland. 1988. Structure of phosphate-free ribonuclease A refined at 1.26 A. Biochemistry. 27:2705–2717. [DOI] [PubMed] [Google Scholar]
- 22.Santoro, J., C. Gonzalez, M. Bruix, J. L. Neira, J. L. Nieto, J. Herranz, and M. Rico. 1993. High-resolution three-dimensional structure of ribonuclease A in solution by nuclear magnetic resonance spectroscopy. J. Mol. Biol. 229:722–734. [DOI] [PubMed] [Google Scholar]
- 23.Udgaonkar, J. B., and R. L. Baldwin. 1988. NMR evidence for an early framework intermediate on the folding pathway of ribonuclease A. Nature. 335:694–699. [DOI] [PubMed] [Google Scholar]
- 24.Schultz, D. A., F. X. Schmid, and R. L. Baldwin. 1992. Cis proline mutants of ribonuclease A. II. Elimination of the slow-folding forms by mutation. Protein Sci. 1:917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juminaga, D., W. J. Wedemeyer, and H. A. Scheraga. 1998. Proline isomerization in bovine pancreatic ribonuclease A. 1. Unfolding conditions. Biochemistry. 37:11614–11620. [DOI] [PubMed] [Google Scholar]
- 26.Wedemeyer, W. J., E. Welker, and H. A. Scheraga. 2002. Proline cis-trans isomerization and protein folding. Biochemistry. 41:14637–14644. [DOI] [PubMed] [Google Scholar]
- 27.Matheson, R. R., and H. A. Scheraga. 1978. A method for predicting nucleation sites for protein folding based on hydrophobic contacts. Macromolecules. 11:819–829. [Google Scholar]
- 28.Wüthrich, K. 1986. NMR of Proteins and Nucleic Acids. Wiley, New York.
- 29.Kitamura, Y., and T. Itoh. 1987. Reaction volume of protonic ionization for buffering agents: prediction of pressure-dependence of pH and pOH. J. Solution Chem. 16:715–725. [Google Scholar]
- 30.Torrent, J., J. Font, H. Herberhold, S. Marchal, M. Ribó, K. Ruan, R. Winter, M. Vilanova, and R. Lange. 2006. The use of pressure-jump relaxation kinetics to study protein folding landscapes. Biochim. Biophys. Acta. 1764:489–496. [DOI] [PubMed] [Google Scholar]
- 31.Liu, Y., G. Gotte, M. Libonati, and D. Eisenberg. 2001. A domain-swapped RNase A dimer with implications for amyloid formation. Nat. Struct. Biol. 8:211–214. [DOI] [PubMed] [Google Scholar]
- 32.López-Alonso, J. P., M. Bruix, J. Font, M. Ribó, M. Vilanova, M. Rico, G. Gotte, M. Libonati, C. Gonzalez, and D. V. Laurents. 2006. Formation, structure and dissociation of the ribonuclease S domain-swapped dimmer. J. Biol. Chem. 281:9400–9406. [DOI] [PubMed] [Google Scholar]
- 33.Chatani, E., K. Nonomura, R. Hayashi, C. Balny, and R. Lange. 2002. Comparison of heat- and pressure-induced unfolding of ribonuclease A: the critical role of Phe46 which appears to belong to a new hydrophobic chain-folding initiation site. Biochemistry. 41:4567–4574. [DOI] [PubMed] [Google Scholar]
- 34.Gutfreund, H. 1971. Transients and relaxation kinetics of enzyme reactions. Annu. Rev. Biochem. 40:315–344. [DOI] [PubMed] [Google Scholar]
- 35.Zhang, J., X. Peng, A. Jonas, and J. Jonas. 1995. NMR study of the cold, heat, and pressure unfolding of ribonuclease A. Biochemistry. 34:8631–8641. [DOI] [PubMed] [Google Scholar]
- 36.Cook, K. H., F. X. Schmid, and R. L. Baldwin. 1979. Role of proline isomerization in folding of ribonuclease A at low temperatures. Proc. Natl. Acad. Sci. USA. 76:6157–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brems, D. N., and R. L. Baldwin. 1985. Protection of amide protons in folding intermediates of ribonuclease A measured by pH-pulse exchange curves. Biochemistry. 24:1689–1693. [DOI] [PubMed] [Google Scholar]
- 38.Laurents, D. V., M. Bruix, M. Jamin, and R. L. Baldwin. 1998. A pulse-chase-competition experiment to determine if a folding intermediate is on or off-pathway: application to ribonuclease A. J. Mol. Biol. 283:669–678. [DOI] [PubMed] [Google Scholar]
- 39.Leeson, D. T., F. Gai, H. M. Rodriguez, L. M. Gregoret, and R. B. Dyer. 2000. Protein folding and unfolding on a complex energy landscape. Proc. Natl. Acad. Sci. USA. 97:2527–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bhat, R., W. J. Wedemeyer, and H. A. Scheraga. 2003. Proline isomerization in bovine pancreatic ribonuclease A. 2. Folding conditions. Biochemistry. 42:5722–5728. [DOI] [PubMed] [Google Scholar]
- 41.Schultz, D. A., and R. L. Baldwin. 1992. Cis proline mutants of ribonuclease A. I. Thermal stability. Protein Sci. 1:910–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiong, Y., D. Juminaga, G. V. Swapna, W. J. Wedemeyer, H. A. Scheraga, and G. T. Montelione. 2000. Solution NMR evidence for a cis Tyr-Ala peptide group in the structure of [Pro93Ala] bovine pancreatic ribonuclease A. Protein Sci. 9:421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pearson, M. A., P. A. Karplus, R. W. Dodge, J. H. Laity, and H. A. Scheraga. 1998. Crystal structures of two mutants that have implications for the folding of bovine pancreatic ribonuclease A. Protein Sci. 7:1255–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Juminaga, D., W. J. Wedemeyer, R. Garduno-Juarez, M. A. McDonald, and H. A. Scheraga. 1997. Tyrosyl interactions in the folding and unfolding of bovine pancreatic ribonuclease A: a study of tyrosine-to-phenylalanine mutants. Biochemistry. 36:10131–10145. [DOI] [PubMed] [Google Scholar]
- 45.Dodge, R. W., and H. A. Scheraga. 1996. Folding and unfolding kinetics of the proline-to-alanine mutants of bovine pancreatic ribonuclease A. Biochemistry. 35:1548–1559. [DOI] [PubMed] [Google Scholar]
- 46.Houry, W. A., and H. A. Scheraga. 1996. Nature of the unfolded state of ribonuclease A: effect of cis-trans X-Pro peptide bond isomerization. Biochemistry. 35:11719–11733. [DOI] [PubMed] [Google Scholar]
- 47.Pincus, M. R., F. Geewitz, H. Wako, and H. A. Scheraga. 1983. Cis trans isomerization of proline in the peptide (His 105 Val 124) of ribonuclease A containing the primary nucleation site. J. Protein Chem. 2:131–146. [Google Scholar]
- 48.Levitt, M. 1981. Effect of proline residues on protein folding. J. Mol. Biol. 145:251–263. [DOI] [PubMed] [Google Scholar]
- 49.Cheng, H. N., and F. A. Bovey. 1977. Cis-trans equilibrium and kinetic studies of acetyl-L-proline and glycyl-L-proline. Biopolymers. 16:1465–1472. [DOI] [PubMed] [Google Scholar]
- 50.Jacob, M., and F. X. Schmid. 1999. Protein folding as a diffusional process. Biochemistry. 38:13773–13779. [DOI] [PubMed] [Google Scholar]
- 51.Kitahara, R., S. Yokoyama, and K. Akasaka. 2005. NMR snapshots of a fluctuating protein structure: ubiquitin at 30 bar-3 kbar. J. Mol. Biol. 347:277–285. [DOI] [PubMed] [Google Scholar]
- 52.Smeller, L., F. Meersman, and K. Heremans. 2006. Refolding studies using pressure. The energy landscape of lysozyme in the pressure-temperature plane. Biochim. Biophys. Acta. 1764:497–505. [DOI] [PubMed] [Google Scholar]
- 53.Winter, R., and W. Dzwolak. 2004. Temperature-pressure configurational landscape of lipid bilayers and proteins. Cell Mol. Biol. (Noisy-le-grand). 50:397–417. [PubMed] [Google Scholar]
- 54.Torrent, J., M. T. Alvarez-Martinez, J. P. Liautard, and R. Lange. 2006. Modulation of prion protein structure by pressure and temperature. Biochim. Biophys. Acta. 1764:546–551. [DOI] [PubMed] [Google Scholar]
- 55.Torrent, J., M. T. Alvarez-Martinez, J. P. Liautard, C. Balny, and R. Lange. 2005. The role of the 132–160 region in prion protein conformational transitions. Protein Sci. 14:956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Torrent, J., M. T. Alvarez-Martinez, M. C. Harricane, F. Heitz, J. P. Liautard, C. Balny, and R. Lange. 2004. High pressure induces scrapie-like prion protein misfolding and amyloid fibril formation. Biochemistry. 43:7162–7170. [DOI] [PubMed] [Google Scholar]
- 57.Torrent, J., M. T. Alvarez-Martinez, F. Heitz, J. P. Liautard, C. Balny, and R. Lange. 2003. Alternative prion structural changes revealed by high pressure. Biochemistry. 42:1318–1325. [DOI] [PubMed] [Google Scholar]
- 58.Marchal, S., E. Shehi, M. C. Harricane, P. Fusi, F. Heitz, P. Tortora, and R. Lange. 2003. Structural instability and fibrillar aggregation of non-expanded human ataxin-3 revealed under high pressure and temperature. J. Biol. Chem. 278:31554–31563. [DOI] [PubMed] [Google Scholar]