

Abstract
A general method to study the phosphate group of phosphoenzymes with infrared difference spectroscopy by helper enzyme-induced isotope exchange was developed. This allows the selective monitoring of the phosphate P-O vibrations in large proteins, which provides detailed information on several band parameters. Here, isotopic exchange was achieved at the oxygen atoms of the catalytically important phosphate group that transiently binds to the sarcoplasmic reticulum Ca2+-ATPase (SERCA1a). [γ-18O3]ATP phosphorylated the ATPase, which produced phosphoenzyme that was initially isotopically labeled. The helper enzyme adenylate kinase regenerated the substrate ATP from ADP (added or generated upon ATP hydrolysis) with different isotopic composition than used initially. With time this produced the unlabeled phosphoenzyme. The method was tested on the ADP-insensitive phosphoenzyme state of the Ca2+-ATPase for which the vibrational frequencies of the phosphate group are known, and it was established that the helper enzyme is effective in mediating the isotope exchange process.
INTRODUCTION
The sarcoplasmic reticulum (SR) Ca2+-ATPase (1–8) is an integral membrane protein that can be found in the sarcoplasmic reticulum of muscle cells, where it pumps Ca2+ ions against their gradient to bring about muscle relaxation. The pumping cycle is generally discussed in terms of the E1/E2 model (9), as illustrated in Fig. 1. The Ca2E1 conformational state of the ATPase is phosphorylated by the γ-phosphate of ATP, which yields energy for the transport of Ca2+ ions across the SR membrane. The first phosphorylated intermediate Ca2E1P is reactive with ADP. Upon Ca2+ release into the SR, the phosphoenzyme undergoes a conformational change to the E2P state. This phosphoenzyme hydrolyzes, although it is not reactive with ADP. The entire cycle is reversible (10).
FIGURE 1.

A simplified scheme of the E1/E2 model for the pumping cycle of the SR Ca2+-ATPase. All reaction steps are reversible. The arrows indicate the direction of partial reactions when Ca2+ is pumped.
The transiently bound phosphate group is of interest because it controls the reaction cycle. In a previous study (11), three vibrational frequencies of this phosphate group of E2P were identified by monitoring isotope exchange with surrounding water. The isotope exchange results in spectral band shifts, which can be detected in the infrared difference spectrum. This has enabled the observation of the three P-O stretching vibrations out of 50,000 protein vibrations (11,12). They give rise to bands of the unlabeled phosphate group of E2P at 1194 cm−1, 1137 cm−1, and 1115 cm−1 in the infrared spectrum. From the identified frequencies, the bond lengths and bond energy of the P-O bonds of E2P have been calculated (11). These bond parameters have revealed a weakening of the bridging P-O bond, which contributes to the rapid hydrolysis rate of E2P.
To understand the different catalytic properties of the two phosphoenzymes Ca2E1P and E2P, their respective bond properties need to be compared. This requires knowledge of infrared band parameters of the Ca2E1P phosphate group at the same molecular level as obtained for E2P in the isotope exchange experiment. Some of the P-O bands of Ca2E1P have been identified (13) by comparison of spectra of labeled and unlabeled Ca2E1P. However, comparison of spectra from different samples is less sensitive than the observation of an isotope exchange reaction in one sample (12). In addition, interpretation of the spectra in Liu et al. (13) has been complicated by the fact that not only the isotopic composition of Ca2E1P altered the spectra, but also that of bound ATP. The method used to initiate an isotope exchange in E2P—an autocatalyzed isotope exchange with water—cannot be employed with Ca2E1P as this state is not reactive with water. This different reactivity of the phosphate group in E2P and Ca2E1P can be explained from the ATPase structure. In the partial reaction from Ca2E1P to E2P, the A-domain of the ATPase rotates so that its conserved TGES loop is positioned close to the phosphate group. The TGES loop of E2P binds a water molecule in a way that it can attack the phosphate group (14,15). It is also known from IR experiments that the bridging P-O bond is weaker in E2P and thus hydrolyzes faster (11,13,16).
Because Ca2E1P does not react with water, a new, more general approach for inducing isotope exchange at the phosphate oxygen atoms was developed and is the subject of this study. In this approach, isotope exchange is mediated by a helper enzyme, here adenylate kinase (ADK). ADK has previously been used by us as a helper enzyme for ADP removal and ATP regeneration to make measurements repeatable (17).
In this work, we tested the functionality of a helper enzyme as mediator of isotope exchange, using our knowledge of the E2P phosphate frequencies—particularly that of the band at 1194 cm−1, which is easily monitored. The method of helper enzyme-induced isotope exchange outlined here has the advantage of general functionality, because it can also be applied to other phosphoenzymes.
MATERIALS AND METHODS
Sample preparation
The SR Ca2+-ATPase was dialyzed for 90 min in a buffer containing 10 mM imidazole (pH 6.5 at ∼1°C), 20 μM CaCl2 and distilled H2O. Samples with 10 μl of SR suspension and additional compounds were first mixed to achieve a homogenous sample suspension. A total amount of 10 μl from the mixture was used for infrared sample preparation. First, an aliquot of 5 μl of the mixture was placed in the center of a BaF2 window with a trough of 5 μm and 8 mm diameter window and partially dried in a stream of nitrogen gas. A second aliquot of 5 μl of the mixture was placed on top of the first one and the sample was again dried with nitrogen gas. This protocol was established to concentrate the sample in the center of the window. After drying, the sample was rehydrated with ∼0.75 μl of 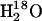 containing 15% Me2SO before immediately sealing it with a second flat window. The sample was placed in the spectrometer and left for equilibration for ∼45 min at 1°C before measuring.
containing 15% Me2SO before immediately sealing it with a second flat window. The sample was placed in the spectrometer and left for equilibration for ∼45 min at 1°C before measuring.
The approximate standard sample composition for the isotope exchange experiment is ∼0.8 mM Ca2+-ATPase, 70 mM imidazole (pH 6.5 at ∼1°C), 140 μM Ca2+, 3 mM caged ATP, 6 mM caged ADP, 10 mM Mg2+, 10 mM dithiothreitol, 0.5 mg/ml Ca2+ ionophore A23187, 0.5 mg/ml adenylate kinase, and 15% Me2SO.
Two types of control experiments were performed. Their sample composition was the same as that of the isotope exchange sample, but in one control experiment, ADK was omitted and in the other, labeled caged [γ-18O3]ATP was replaced with unlabeled caged ATP.
ADK (M5520) was purchased from Sigma (St. Louis, MO). Labeled water  at 95% was purchased from Larodan Fine Chemicals (Malmö, Sweden). Caged compounds were synthesized at the National Institute of Medical Research (London, UK). The caged [γ-18O3]ATP has 91% isotopic enrichment (12).
at 95% was purchased from Larodan Fine Chemicals (Malmö, Sweden). Caged compounds were synthesized at the National Institute of Medical Research (London, UK). The caged [γ-18O3]ATP has 91% isotopic enrichment (12).
Design of the isotope exchange experiment
ADK, also known as myokinase, transfers the γ-phosphate group from one of the ADP molecules to the other, thus producing an ATP molecule and an AMP molecule (18). In this work, we used this reaction for inducing an isotope exchange. The process of isotope exchange is illustrated in Fig. 2. It was started with the photolytic release of isotopically labeled [γ-18O3]ATP (see Fig. 3) from caged ATP. The labeled ATP phosphorylated the ATPase with labeled γ-phosphate. The phosphoenzyme transformed into the E2P intermediate, which consequently had its phosphate group labeled. Under our conditions, hydrolysis of E2P was the rate-limiting step and E2P accumulated under steady-state conditions. However, slow progression through the ATPase reaction cycle took place as long as ATP was present. This is indicated in Fig. 2 by putting Ca2E1 in brackets when it is only transiently adopted under steady-state conditions. Out of the unlabeled ADP generated by photolysis of caged ADP (see below) as well as by the ATPase upon hydrolysis of ATP, ADK produced new unlabeled ATP, which consequently yielded E2P with the phosphate group being unlabeled. This is the isotopic exchange, which is reflected in the infrared difference spectra by the appearance of a band at 1194 cm−1. This band is characteristic of the unlabeled phosphate group of E2P (11).
FIGURE 2.

Isotope exchange mediated by ADK. The asterisk indicates labeled phosphate groups. The bracket around Ca2E1 on the right-hand side indicates that this state is adopted transiently under steady-state conditions, but does not accumulate. See text for further explanation.
FIGURE 3.

Structure of [γ-18O3]ATP. The isotopically labeled oxygen atoms are shown bold.
Our samples contained caged ADP in addition to labeled caged [γ-18O3]ATP in the ADK experiments. Thus, there was ADP present that was derived from photolysis of caged ADP as well as from the hydrolysis of ATP by the ATPase. The concentration of ATP released from caged ATP was reduced to the approximate amount required for one cycle. ATP for subsequent cycles was then provided by activity of ADK. The addition of caged ADP and the low initial ATP concentration limited the isotope exchange process to the smallest possible time interval. In experiments without additional caged ADP, the amplitude of the 1194 cm−1 band was generally found to be smaller as compared to when caged ADP was added to the samples.
Precautions were made to ensure that the isotope exchange mediated by ADK was not confused with isotope exchange with water. Since the ATPase was phosphorylated initially with labeled ATP, labeled water 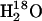 was used in the experiments. This prevents isotope exchange with water at the initially labeled phosphate group. After ADK-mediated isotope exchange generated the unlabeled phosphate group, isotope exchange with water could reduce the amount of unlabeled phosphoenzyme. Thus, running the experiments in
was used in the experiments. This prevents isotope exchange with water at the initially labeled phosphate group. After ADK-mediated isotope exchange generated the unlabeled phosphate group, isotope exchange with water could reduce the amount of unlabeled phosphoenzyme. Thus, running the experiments in 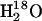 provides a clearcut attribution of the isotope exchange to that mediated by ADK but makes the task of observing it more difficult.
provides a clearcut attribution of the isotope exchange to that mediated by ADK but makes the task of observing it more difficult.
Fourier transform infrared measurements
Time-resolved Fourier-transform infrared (FTIR) spectra were obtained with a Bruker IFS 66 spectrometer equipped with a mercury cadmium tellurium detector (Bruker, Billerica, MA). Labeled and unlabeled ATP was released photolytically from the respective isotopomer of caged ATP. This transformed the ATPase from Ca2E1 to E2P, which accumulated under our steady-state conditions. Photolytic release was triggered by a Xenon flash tube, set at 700 V, which gave a photolysis yield of ∼15%. The Bruker OPUS software 4.2 was used for recording and processing the data; spectra were recorded in the rapid scan mode. Data were acquired with double-sided interferograms in a forward-backward mode at a spectral resolution of 4 cm−1 with the Blackman-Harris four-term apodization function. The time needed for one interferometer cycle was 65 ms.
The measurements started with the recording of a reference spectrum consisting of 2000 (∼130 s) scans, representing the state of the unperturbed sample. This was followed by the photolysis flash and the time was set to zero. Subsequent recording of one spectrum of 50 scans (∼3.25 s), 10 spectra of 150 scans (∼9.75 s) each, and 10 spectra of 1000 scans (∼65 s) each, took place. This recording took ∼12 min in total. From the spectra recorded after the photolysis flash and the reference spectrum recorded before the flash, difference spectra were calculated. Before the flash-induced experiment was started, baseline spectra were recorded to check the stability of the sample. The baseline spectra were recorded according to the same procedure as described above, but without execution of flash photolysis.
An absorbance spectrum was recorded for each sample to quantify the protein concentration. The difference spectra were normalized to a standard protein concentration before averaging to facilitate comparison between different samples. The normalization factor was calculated from the amide II band, here the difference in absorbance between 1546 cm−1 and 1492 cm−1 in the absorbance spectrum of the sample (after water subtraction with the criterion of a flat baseline at 2130 cm−1). The standard amide II absorbance was 260 mOD. The amide II band was chosen for normalization because it is less sensitive to errors in the water subtraction than the amide I band (19). This procedure gave very similar band amplitudes for the three sets of experiments with ADK. Those without ADK, however, produced smaller bands—possibly because less ATP than needed for saturating the protein bands was released.
Kinetic evaluation of spectra
To study the kinetics of the flash-induced reaction, some large bands characteristic of ATPase structure and with good signal/noise ratio were chosen for integration. The integration of band intensities was performed using integration method E in OPUS 4.2. The band area as a function of recording time was plotted in MatLab 7.0 (The MathWorks, Natick, MA). The selected bands were three protein bands in the amide I region and the band at 1194 cm−1, the latter of which served as a marker band of the isotope exchange process. The band areas of the protein bands were normalized to their respective value at 29 s, at which they had reached their plateau value. The 1194 cm−1 band area was in all experiments normalized after the maximum value of the 1194 cm−1 band in the control experiment with unlabeled caged ATP, which was also reached at 29 s. The latter normalization technique is chosen to visualize to which extent the unlabeled phosphate group of E2P accumulated in the isotope exchange experiments. This approach was sufficient for comparison of the three types of experiments with ADK. However, the control experiment without ADK yielded smaller band amplitudes in the amide I region than the other experiments. Using a multiplication factor of 1.4 provided amide I band amplitudes that were equal to those of the other experiments. Thus, the integrated band amplitudes from the experiment without ADK were multiplied by 1.4 before relating them to the experiment with unlabeled ATP. The spectra shown in Fig. 6 were not corrected by 1.4.
FIGURE 6.

Control experiment without ADK using caged [γ-18O3]ATP. (A) Difference spectra with time intervals for spectra recording as in Fig. 4. The labels refer to the peaks of the thin-line spectrum. (B) Double-difference spectrum calculated with the same time intervals as in Fig. 4.
RESULTS AND DISCUSSION
Investigation of adenylate kinase activity
The ability of ADK to produce ATP out of ADP in infrared samples has been investigated before in the presence as well as absence of Ca2+-ATPase (17). However, the functionality of ADK was also tested in ATPase samples, designed for E2P state accumulation. This was done in experiments (data not shown) with caged ADP as the only nucleotide in the sample.
The experiments demonstrated that ADK is active in the presence of ATPase. Due to ATP production by ADK, the ATPase became phosphorylated and E2P accumulated, as shown by the rise of typical bands for the Ca2E1 to E2P transition (at 1690, 1607, and 1194 cm−1) within the first 70 s.
Isotope exchange mediated by adenylate kinase
Bands characteristic of the transiently bound phosphate group of the SR Ca2+-ATPase in the E2P state, at 1194 cm−1, 1137 cm−1, and 1115 cm−1 in the infrared spectra, have previously been identified (11,12). In focus of this study was in particular the 1194 cm−1 band, which is easily detected. This band shifts to 1157 cm−1 upon isotopic labeling of all terminal oxygen atoms of the phosphate group (11).
Fig. 4 shows spectra that give evidence for ADK-mediated isotope exchange of the phosphate oxygen atoms of E2P. In these experiments, ATP released from caged ATP transformed the ATPase from the Ca2E1 state to the E2P state, which accumulated under our conditions. Because the ATPase was initially phosphorylated by [γ-18O3]ATP, E2P was initially labeled. [γ-18O3]ATP was gradually replaced by unlabeled ATP generated by ADK from ADP, which was either produced by the ATPase or photolytically released from caged ADP. The isotope exchange process should manifest in the spectra by the gradual appearance of a positive band at 1194 cm−1, representing the unlabeled phosphate group of E2P.
FIGURE 4.

Infrared difference spectra obtained in the isotope exchange experiment with ADK using caged [γ-18O3]ATP. The spectra reflect the reaction from the initial state Ca2E1 to E2P. After formation of E2P, the oxygen atoms of the phosphate group of the SR Ca2+-ATPase undergo 18O → 16O exchange. This can be monitored by the marker band at 1194 cm−1, which is characteristic of the unlabeled phosphate group. (A) The spectra are recorded 3.4–44 s (bold) and 65–106 s (thin line) after the flash. The labels refer to the peaks of the thin-line spectrum. (B) Double-difference spectrum of the absorbance recorded 65–106 s minus the absorbance recorded 3.4–44 s after the flash. (C) Isotope exchange spectrum obtained by exchange with water. The spectrum was taken from Fig. 2 E of Barth and Bezlyepkina (11) and multiplied by −1 to obtain a spectrum reflecting 18O → 16O exchange as in the present experiments.
Fig. 4 A shows difference spectra of the Ca2E1 to E2P reaction at different time intervals after the photolysis flash. The bold (3.4–44 s) and thin line (65–106 s) spectra show indeed an increase in band amplitude at 1194 cm−1 during a period when the E2P state is stable. This is demonstrated by the constant amplitude of the protein bands in the amide I region (1700–1610 cm−1), which is sensitive to the backbone structure. Accumulation of the E2P state is indicated by the large amplitude of the bands at 1690 cm−1 and 1607 cm−1 in the amide I region (20–22). The spectra illustrate that it is possible to follow the isotope exchange process by the gradual increase of the band at 1194 cm−1.
The isotope exchange process is more readily observed in double-difference spectra, which only show absorbance changes with respect to an earlier time of the process and thus facilitate monitoring the progress of a process over time. The double-difference spectrum in Fig. 4 B confirms the appearance of a band at 1194 cm−1. Also, the featureless amide I region confirms that the accumulation of E2P is constant to a few percent during that time. This rules out that the 1194 cm−1 band is caused by a conformational change of the ATPase and supports its assignment to isotope exchange by E2P. For comparison, Fig. 4 C shows the spectrum obtained in the previous isotope exchange experiment (11) where isotope exchange was with water. Similar bands are observed in both spectra. The band at 1244 cm−1 in Fig. 4 B indicates ATP consumption, that at 1054 cm−1 production of predominantly labeled Pi (downshifted from ∼1080 cm−1 (23) and that at 977 cm−1 production of AMP (17).
Bands selected for kinetic evaluation were the protein bands in the amide I region at 1690 cm−1, 1653 cm−1, and 1628 cm−1 that reflect changes in protein backbone conformation and the 1194 cm−1 band representing the unlabeled phosphate group of E2P. The 1690 cm−1 and the 1653 cm−1 bands were chosen because they are predominantly indicative of E2P. The band at 1628 cm−1 arises upon ATP binding. The 1194 cm−1 band is of primary interest since it monitors the progress of isotope exchange.
The time-dependency of these bands is presented in Fig. 5 A. As can be seen, the amide I bands rise quickly (within 3 s) to their maximum value and remain at a constant value for ∼100 s, i.e., as long as the E2P state is stable because there is sufficient ATP present in the sample. The subsequent gradual return to the initial Ca2E1 state is manifested by the decay of the protein bands after 100 s. During these first 100 s of the reaction, the band at 1194 cm−1 rises slowly and reaches its maximum at 140 s before it decays concomitantly with the other protein bands. This is in line with what is expected from the isotope exchange process. Once the band at 1194 cm−1 has reached its maximum value its behavior should not differ from the other bands of the E2P state.
FIGURE 5.

Time-course of selected infrared bands observed in isotope exchange experiments with ADK and control experiments. Labels refer to the wavenumber in cm−1 of the bands. The amplitudes were normalized as described in Materials and Methods. (A) Isotope exchange experiment with 0.5 mg/ml ADK using caged [γ-18O3]ATP. (B) Isotope exchange experiment with 1 mg/ml ADK using caged [γ-18O3]ATP. (C) Control experiment without ADK using caged [γ-18O3]ATP. (D) Control experiment with unlabeled ATP with 0.5 mg/ml ADK.
Isotope exchange experiments with twice the concentration of ADK were also performed, to elucidate whether the concentration of ADK affects the rate of isotope exchange. The time-dependency of the 1194 cm−1 band presented in Fig. 5 B demonstrates an increased rate of isotopic exchange, i.e., a faster progression to its maximum value, which is reached within ∼65 s. After ∼100 s, the 1194 cm−1 band and the protein bands decay simultaneously. This indicates that the rate of appearance of the 1194 cm−1 band depends on the concentration of ADK.
Control samples without adenylate kinase
To further confirm that the isotope exchange is dependent on the presence of adenylate kinase, control experiments without ADK were performed. In the absence of ADK, there should be no isotope exchange and thus, there should be no band at 1194 cm−1 of the unlabeled phosphate group in the spectra since labeled caged [γ-18O3]ATP is used to phosphorylate the ATPase. The difference spectra of the reaction are shown in Fig. 6 A. The shape of the spectra above 1200 cm−1 is very similar to that obtained in the presence of ADK, indicating that the structural change of the ATPase is not affected by the omission of ADK. However, in the absence of ADK, the protein bands appear to have smaller amplitudes, as compared to the amplitudes in the experiments with ADK, possibly because the amount of released ATP was not sufficient to generate saturating protein bands. The bold (3.4–44 s) and thin line (65–106 s) spectra were recorded at times when the concentration of E2P state was stable, as demonstrated by the constant amplitudes of the protein bands in the amide I region. In contrast to our expectation, both spectra show a band at 1194 cm−1. Its amplitude, however, is clearly smaller than in the experiments with ADK and there is only a very small increase in the 1194 cm−1 band amplitude between the two spectra. The appearance of a band at 1194 cm−1 in the first spectrum is likely due to the <100% labeling of the caged nucleotide. Its small increase can be explained by an ADK impurity in our SR preparation. Another possible explanation is that there is another band at 1194 cm−1, which is not related to the phosphate band but appears coincidently at the same position.
The double-difference spectrum in Fig. 6 B represents a time interval in which the E2P concentration was constant, as demonstrated by the absence of E2P bands in the amide I region. The spectrum shows only a very small band at 1194 cm−1, which can be compared to the prominent band at 1194 cm−1 in the double-difference spectra of the ADK experiments (see Fig. 4 B). The bands at 1035 cm−1 and 951 cm−1 are assigned to production of labeled Pi and ADP (17,21), respectively.
The small bands at 1124 (−), 1112 (+), and 1089 cm−1 (−) can be attributed to the hydrolysis of [γ-18O3]ATP. They were also observed in experiments with approximately four times less ATPase (to reduce the ATPase bands) without ADK. The bands arise from the differences in absorption of [γ-18O3]ATP and ADP. ADP has an absorbance maximum at 1107 cm−1 and a shoulder at 1126 cm−1. [γ-18O3]ATP has its maximum at 1083 cm−1 and a side band at 1114 cm−1. Subtracting the absorbance spectrum of [γ-18O3]ATP from that of ADP produced a difference spectrum with bands at 1130 (+), 1117 (−), 1008 (+), and 1071 cm−1 (−, broad). They are shifted by 7–18 cm−1 with respect to the bands observed in the hydrolysis spectrum of [γ-18O3]ATP because the latter was recorded in the presence of Mg2+, binding of which to the phosphate groups causes upshift of phosphate bands of ATP and ADP (24).
The bands at 1117 (−), 1008 (+), and 1071 cm−1 (−) appeared also in an AMP minus [γ-18O3]ATP difference spectrum and broad bands at 1130 (+) and 1070 cm−1 (−) in an ATP minus [γ-18O3]ATP spectrum. Thus they were also present in a spectrum that models the net reaction in our ADK samples: 2 [γ-18O3]ATP → ATP + AMP (ignoring Pi that absorbs below 1060 cm−1 when labeled) and were accordingly observed in hydrolysis spectra with ADK and approximately four times less ATPase at 1126 (−), 1111 (+), and 1094 cm−1 (−). Their presence in Fig. 4 B might therefore be due to the hydrolysis of [γ-18O3]ATP in ATPase/ADK samples. However, for the isotope exchange experiment shown in Fig. 4 C we have clear evidence that the bands at 1137 (+), 1124 (−), 1115 (+), and 1091 cm−1 (−) are associated with isotope exchange (11):
In experiments of 18O → 16O exchange of the phosphate oxygens with water they are observed in samples with higher and lower hydrolytic activity. A weighted subtraction of these spectra with different relative amplitudes of isotope exchange and hydrolysis bands does not cancel the bands at 1137, 1115, and 1091 cm−1 but reduces the amplitude of the 1091 cm−1 band.
The bands are observed with opposite signs upon 16O → 18O exchange with water. In these experiments the only isotopomer of ATP that is present is unlabeled ATP, the hydrolysis of which does not give rise to bands at 1137, 1124, 1115, and 1091 cm−1 (see Fig. 7 B). In addition, these experiments do not show hydrolytic activity in the time interval in which isotope exchange is observed.
Control experiments in which no isotope exchange is expected because the oxygen isotopes of water and of E2P are the same, do not show these bands when unlabeled ATP hydrolyzes or show them with much reduced amplitude when [γ-18O3]ATP hydrolyzes (with the exception of the 1091 cm−1 band).
Bands at 1139 and 1115 cm−1 are also observed upon isotope exchange of singly-labeled E2P phosphate groups to fully unlabeled groups in samples without hydrolytic activity in the time interval of isotope exchange.
FIGURE 7.

Control experiment with unlabeled caged ATP and 0.5 mg/ml ADK. (A) Difference spectra with time intervals for spectra recording as in Fig. 4. The labels refer to the peaks of the thin-line spectrum. (B) Double-difference spectrum calculated with the same time intervals as in Fig. 4.
In conclusion, we attribute the appearance of the bands at 1124, 1112, and 1089 cm−1 in both Figs. 4 C and 6 B to a coincidence of band positions in the spectra of E2P isotope exchange and of [γ-18O3]ATP hydrolysis. Returning to the main line of our present work, we wish to point out that our conclusions on ADK-mediated isotope exchange in this work rely on the observation of the 1194 cm−1 band, and are not affected by the above discussion.
The time-course in Fig. 5 C shows the time-dependence of the control experiment without ADK. The amplitude of the 1194 cm−1 band is much smaller than in the isotope exchange experiments in Fig. 5, A and B. To quantify this observation, we evaluated the increase in 1194 cm−1 band area between 19 s (third data point)—a time when E2P is fully formed—and 100 s, when the 1194 cm−1 band nearly reached its plateau in the experiment with ADK. This increase in the experiment without ADK is 21% of that in the experiments with standard concentration of ADK. In addition, the kinetics of the 1194 cm−1 band does not follow the decay kinetics of the protein bands. This shows that the behavior of the 1194 cm−1 band is independent of the protein bands, which implies that there might be an overlapping contribution from another reaction, most probably the hydrolysis reaction, which yields broad featureless bands in this region. Due to overlap with hydrolysis bands, it is impossible to make an integration of the 1194 cm−1 band that is completely without contribution from these bands. Summarizing the control experiment without ADK, the small extent of the increase of the band at 1194 cm−1 indicates that no significant isotope exchange takes place. This demonstrates that ADK is the mediator of isotope exchange in the experiments with ADK.
Control samples with unlabeled caged ATP
The use of unlabeled caged ATP instead of caged [γ-18O3]ATP in the samples led to a prominent band at 1194 cm−1 already in the first infrared spectrum, since the intermediate E2P with an unlabeled phosphate group was produced from the beginning (see Fig. 7). Band shape and band amplitudes above 1200 cm−1 are very similar to those in the spectra obtained with labeled ATP and ADK (Fig. 4). The bold (3.4–44 s) and thin line (65–106 s) spectra in Fig. 7 A show perfect overlap in the amide I region as well as of the 1194 cm−1 band, which implies that the E2P concentration is stable and that no isotope exchange takes place. The latter is not expected, since unlabeled caged ATP is used in this experiment. Labeled water, 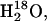 used in the experiment could, however, induce an ATPase-catalyzed isotope exchange between the phosphate group of E2P and the surrounding water. The constant amplitude of the 1194 cm−1 band, however, shows that under the conditions used in this work, no significant isotope exchange with 18O water takes place. The isotope exchange observed in the samples with labeled ATP and ADK is therefore mediated by ADK.
used in the experiment could, however, induce an ATPase-catalyzed isotope exchange between the phosphate group of E2P and the surrounding water. The constant amplitude of the 1194 cm−1 band, however, shows that under the conditions used in this work, no significant isotope exchange with 18O water takes place. The isotope exchange observed in the samples with labeled ATP and ADK is therefore mediated by ADK.
The conditions in our previous experiments of isotope exchange with water (11) were different from the present conditions, in particular the Mg2+ concentration was 20 mM instead of the 10 mM used here and the temperature was 10 C instead of 1°C. Under these conditions, E2P was stable for at least 15 min and isotope exchange with water was observed within 200 s. Under the present conditions, E2P was only stable for 100 s, and the lower Mg2+ concentration and lower temperature explain why these conditions are unfavorable for isotope exchange with water, which requires several dephosphorylation and rephosphorylation reactions. The Me2SO concentration was less in the previous experiments (10%) than in the present (15%), which cannot readily be understood since Me2SO is known to stabilize E2P (25,26). We note, however, that we needed a higher Me2SO concentration (20%) now to reproduce the previous experiments of isotope exchange with water. A reason for this might be differences in sample preparation and handling of minute volumes by different researchers making the Me2SO concentration in the infrared sample different from its nominal value.
The double-difference spectrum in Fig. 7 B verifies that there are no significant absorbance changes at 1194 cm−1 during the period when the E2P state is stable. The band at 1240 cm−1 can be attributed to consumption of ATP, that at 1076 cm−1 to production of unlabeled Pi and that at 976 cm−1 to production of AMP (27,28).
Fig. 5 D shows the time-dependence of the control experiment with unlabeled caged ATP. It is evident that the 1194 cm−1 band reaches its maximum value together with the protein bands, and stays stable for ∼100 s. This shows that the kinetic development of the 1194 cm−1 band does not differ from the behavior of the protein bands in the amide I region. When the cycle runs out of ATP and the ATPase gradually returns to the Ca2E1 state, there is also a synchronous decay of all bands. As can be seen by comparison of the time-dependencies in Fig. 5, A and D, it is apparent that the 1194 cm−1 band in the isotope exchange experiment only reaches ∼70% of the maximum amplitude of the band in the experiment with unlabeled caged ATP, implying that not all phosphate groups exchange. Each individual group that absorbs at 1194 cm−1 has all three terminal oxygen atoms exchanged because the band appears only when all oxygen atoms are unlabeled (11).
Finally, we wanted to demonstrate that our method has the potential of generating a high quality difference spectrum of an isotope exchange of the phosphate oxygen atoms. We used the experiments in which the ADK concentration was 1 mg/ml, twice that of the standard concentration. The isotope exchange bands of these experiments were relatively larger than the hydrolysis bands compared to the experiments under standard conditions. The bold spectrum in Fig. 8 A shows the absorbance change between 3.4–24 s and 24–75 s, the thin line spectrum the subsequent change between 75–96 s and 24–75 s. Most of the isotope exchange takes place in the first time interval (bold spectrum) whereas during the second time interval (thin line spectrum) the contribution from ATP hydrolysis (1248 cm−1) and production of predominantly labeled Pi (1057 cm−1) and of AMP (977 cm−1) dominates. In Fig. 8 B, these two spectra are subtracted to enhance the contribution of isotope exchange to the resulting spectrum. Similar spectra were obtained by using different time intervals for the subtracted spectra or by subtracting a spectrum obtained in the same time interval in experiments with the ADK standard concentration. The latter spectrum had relatively larger hydrolysis bands than that with twice the ADK concentration, thus enabling subtraction of the hydrolysis bands. All generated subtractions showed bands at 1194, 1154, and 1136 cm−1; the band at 1114 cm−1 is, for some, close to the noise limit. However, the spectrum in Fig. 8 B, which had the best signal/noise ratio, compares favorably with that obtained by isotope exchange with water shown in Fig. 4 C. This demonstrates that the two approaches for obtaining isotope exchange spectra are equivalent.
FIGURE 8.

Spectrum of ADK-mediated isotope exchange with 1 mg/ml ADK. (A) (Bold spectrum) Absorbance change between 3.4–24 s and 24–75 s; the flash artifact band at 1282 cm−1 was subtracted. (Thin-line spectrum) Absorbance change between 75–96 s and 24–75 s. Labels refer to the bold spectrum. (B) Subtraction of the two spectra shown in panel A.
The spectrum obtained with ADK-mediated exchange is clearly noisier than that obtained more directly by exchange with water. Also the signals in the amide I spectral region are larger because of partial overlap in time of the late phase of E2P formation with isotope exchange. By interactively subtracting a spectrum of E2P formation, like the one shown as a bold line in Fig. 4 A, the signals in the amide I region could be partially cancelled without affecting the appearance of the sharp bands in the phosphate region. However, the focus of this work was not the isotope exchange spectrum as such, but the validation of the approach of ADK-mediated isotope exchange. It is clear that with more averaging, spectra of similar quality can be obtained with ADK-mediated isotope exchange as with direct isotope exchange with water.
CONCLUSIONS
In this work, a novel method for observing isotope exchange at the phosphate oxygen atoms of a phosphoenzyme with infrared spectroscopy was developed. This yielded a difference spectrum focused on a small important group in a large protein because it was dominated by the isotopic shifts. The advantage of the method compared to separate experiments with labeled and unlabeled phosphoenzyme is that a spectrum of the isotope effect is obtained with one sample, thereby avoiding errors and sensitivity loss that are inevitable when different samples are compared. In this work ADK was used as a helper enzyme to mediate an isotope exchange at the phosphate oxygen atoms of the SR Ca2+-ATPase. The results demonstrate the functionality of ADK as a helper enzyme for isotope exchange. The approach used here could also be applied to other phosphoenzymes as well, using ADK or creatine phosphokinase and possibly other helper enzymes (kinases, phosphates) as mediators of the isotope exchange.
Acknowledgments
The authors thank J. E. T. Corrie and M. Webb (National Institute for Medical Research, London) for the preparation of labeled caged compounds and W. Hasselbach (Max-Planck Institut, Heidelberg) for the gift of Ca2+-ATPase.
This work was supported by Vetenskapsrådet and Knut och Alice Wallenberg Stiftelse.
Abbreviations used: ATP, P3-1-(2-nitrophenyl)ethyl ATP; Ca2E1, Ca2+ bound form of Ca2+-ATPase; Ca2E1P, ADP-sensitive phosphoenzyme; E2P, ADP-insensitive phosphoenzyme.
References
- 1.Hasselbach, W., and M. Makinose. 1961. The calcium pump of the relaxing granules of muscle and its dependence on ATP splitting. Biochem. Z. 333:518–528. [PubMed] [Google Scholar]
- 2.Andersen, J. P. 1989. Monomer-oligomer equilibrium of sarcoplasmic reticulum Ca-ATPase and the role of subunit interaction in the Ca2+ pump mechanism. Biochim. Biophys. Acta. 988:47–62. [DOI] [PubMed] [Google Scholar]
- 3.Hasselbach, W. 1981. Calcium activated ATPase of the sarcoplasmic reticulum membranes. In Membrane Transport. Elsevier, Amsterdam, The Netherlands. 183–208.
- 4.Inesi, G., and L. de Meis. 1985. Kinetic regulation of catalytic and transport activities in sarcoplasmic reticulum ATPase. In The Enzymes of Biological Membranes. Plenum Press, NY. 157–191.
- 5.Martonosi, A., G. Kracke, K. A. Taylor, L. Dux, and C. Peracchia. 1985. The regulation of the Ca2+ transport activity of sarcoplasmic reticulum. Soc. Gen. Phys. Ser. 39:57–85. [PubMed] [Google Scholar]
- 6.Mintz, E., and F. Guillain. 1997. Ca2+ transport by the sarcoplasmic reticulum ATPase. Biochim. Biophys. Acta. 1318:52–70. [DOI] [PubMed] [Google Scholar]
- 7.Lee, A., and J. East. 2001. What the structure of a calcium pump tells us about its mechanism. Biochem. J. 356:665–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toyoshima, C., and G. Inesi. 2004. Structural basis of ion pumping by Ca2+-ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 73:269–292. [DOI] [PubMed] [Google Scholar]
- 9.Meis, L. D., and A. L. Vianna. 1979. Energy interconversion by the Ca2+-dependent ATPase of the sarcoplasmic reticulum. Annu. Rev. Biochem. 48:275–292. [DOI] [PubMed] [Google Scholar]
- 10.Hasselbach, W. 1979. The reversibility of the sarcoplasmic calcium pump. Biochim. Biophys. Acta. 515:23–53. [DOI] [PubMed] [Google Scholar]
- 11.Barth, A., and N. Bezlyepkina. 2004. P-O bond destabilization accelerates phosphoenzyme hydrolysis of sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 279:51888–51896. [DOI] [PubMed] [Google Scholar]
- 12.Barth, A. 2002. Selective monitoring of 3 out of 50,000 protein vibrations. Biopolymers. 67:237–241. [DOI] [PubMed] [Google Scholar]
- 13.Liu, M., M. Krasteva, and A. Barth. 2005. Interactions of phosphate groups of ATP and Aspartyl phosphate with the sarcoplasmic reticulum Ca2+-ATPase: an FTIR study. Biophys. J. 89:4352–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toyoshima, C., H. Nomura, and T. Tsuda. 2004. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 432:361–368. [DOI] [PubMed] [Google Scholar]
- 15.Olesen, C., T. L.-M. Sorensen, R. C. Nielsen, J. V. Moller, and P. Nissen. 2004. Dephosphorylation of the calcium pump coupled to counterion occlusion. Science. 306:2251–2255. [DOI] [PubMed] [Google Scholar]
- 16.Andersson, J., and A. Barth. 2006. FTIR studies on the bond properties of the aspartyl phosphate moiety of the Ca2+-ATPase. Biopolymers. 82:353–357. [DOI] [PubMed] [Google Scholar]
- 17.Liu, M., E.-L. Karjalainen, and A. Barth. 2005. Use of helper enzymes for ADP removal in infrared spectroscopic experiments: application to Ca2+-ATPase. Biophys. J. 88:3615–3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atkinson, D. E. 1968. The energy charge of adenylate pool as a regulatory parameter. Biochemistry. 7:4030–4034. [DOI] [PubMed] [Google Scholar]
- 19.Barth, A., and W. Mäntele. 1998. ATP-Induced phosphorylation of the sarcoplasmic reticulum Ca2+-ATPase: molecular interpretation of infrared difference spectra. Biophys. J. 75:538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barth, A., W. Kreutz, and W. Mäntele. 1994. Changes of protein structure, nucleotide microenvironment, and Ca2+-binding states in the catalytic cycle of sarcoplasmic reticulum Ca2+-ATPase: investigation of nucleotide binding, phosphorylation and phosphoenzyme conversion by FTIR difference spectroscopy. Biochim. Biophys. Acta. 1194:75–91. [DOI] [PubMed] [Google Scholar]
- 21.Barth, A., F. von Germar, W. Kreutz, and W. Mäntele. 1996. Time-resolved infrared spectroscopy of the Ca2+-ATPase. J. Biol. Chem. 271:30637–30646. [DOI] [PubMed] [Google Scholar]
- 22.Barth, A. 1999. Phosphoenzyme conversion of the sarcoplasmic reticulum Ca2+-ATPase. J. Biol. Chem. 274:22170–22175. [DOI] [PubMed] [Google Scholar]
- 23.Barth, A., W. Mäntele, and W. Kreutz. 1991. Infrared spectroscopic signals arising from ligand binding and conformational changes in the catalytic cycle of sarcoplasmic reticulum Ca2+ ATPase. Biochim. Biophys. Acta. 1057:115–123. [DOI] [PubMed] [Google Scholar]
- 24.Takeuchi, H., H. Murata, and I. Harada. 1988. Interaction of adenosine 5′-triphosphate with Mg2+: vibrational study of coordination sites by use of 18O-labeled triphosphates. J. Am. Chem. Soc. 110:392–397. [Google Scholar]
- 25.Sande-Lemos, M. P., and L. de Meis. 1988. Regulation of ATP syntheses catalyzed by the calcium pump of SR. J. Biol. Chem. 263:3795–3798. [PubMed] [Google Scholar]
- 26.de Meis, L. 1989. Role of water in the energy of hydrolysis of phosphate compounds—energy transduction in biological membranes. Biochim. Biophys. Acta. 973:333–359. [DOI] [PubMed] [Google Scholar]
- 27.Barth, A., W. Kreutz, and W. Mäntele. 1990. Molecular changes in the sarcoplasmic reticulum calcium ATPase during catalytic activity. A Fourier-transform infrared (FTIR) study using photolysis of caged ATP to trigger the reaction cycle. FEBS Lett. 277:147–150. [DOI] [PubMed] [Google Scholar]
- 28.Thoenges, D., and A. Barth. 2002. Direct measurement of enzyme activity with infrared spectroscopy. J. Biomol. Screen. 7:353–357. [DOI] [PubMed] [Google Scholar]