

Abstract
Pathogenic Yersinia have a pronounced tropism for lymphatic tissues and harbor a virulence plasmid that encodes a type III secretion system, pTTSS, that transports Yops into host cells. Yops are critical virulence factors that prevent phagocytosis by macrophages and neutrophils and Yersinia mutants lacking one or more Yops are defective for survival in lymphatic tissues, liver, and gastrointestinal tract. However, here we demonstrate that Y. pseudotuberculosis (Yptb) mutants lacking the pTTSS survived as well as or better than wild-type (WT) Yptb in the mesenteric lymph nodes (MLN). Infection with pTTSS mutants caused lymphadenitis with little necrosis, whereas infection with WT Yptb provoked lymphadenitis with multiple necrotic suppurative foci. Gentamicin protection assays and microscopic examination of the MLN revealed that pTTSS mutants resided extracellularly adjacent to B and T lymphocytes in the cortex and paracortex. WT Yptb was found extracellularly adjacent to neutrophils and macrophages in necrotic areas and adjacent to B and T lymphocytes in less-inflamed areas. To determine whether lymphocytes protected pTTSS mutants from phagocytic cells, Rag1−/− mice were infected with pTTSS mutants or WT Yptb. pTTSS mutants but not WT, were impaired for survival in MLN of Rag1−/− mice, suggesting that lymphocyte-rich regions constitute a protective niche for pTTSS mutants. Finally, we show that invasin and the chromosomally encoded TTSS were not required for Yptb survival in MLN. In summary, chromosomally encoded factors are sufficient for Yptb replication in the cortex and paracortex of MLN; the pTTSS enables Yersinia to survive within phagocyte-rich areas of lymph nodes, and spread to other tissues.
Synopsis
The pathogenic bacteria, Yersinia, synthesize an apparatus called a type III secretion system, which transports bacterial proteins, Yops, from the bacteria into important immune cells, such as macrophages and neutrophils. Normally, macrophages and neutrophils control bacterial infections by ingesting the bacteria; however, the Yops inactivate these immune cells, which in turn, enable Yersinia to replicate extracellularly and cause disease in many types of tissues. Pathogenic Yersinia are frequently found in lymph nodes of infected hosts, and the Yops are important for the bacteria to replicate and cause disease in lymph nodes since Yersinia mutants that lack Yops do not colonize lymph nodes efficiently. Surprisingly, the authors found that Yersinia pseudotuberculosis lacking the type III secretion system colonizes the mesenteric lymph nodes and survives extracellularly next to lymphocytes. However, in mice lacking lymphocytes, the type III secretion mutants did not survive although wild-type Yersinia did. The authors' findings reveal that other bacterial factors are sufficient for mesenteric lymph node (MLN) colonization of Yersinia and that lymphocytes provide a protective niche for Yersinia strains lacking the type III secretion system. Potentially, these avirulent mutant strains, which persist for at least 5 d in the mesenteric lymph nodes, could be used as live attenuated vaccines to protect against Yersinia infections, or as carriers of other antigens.
Introduction
One hallmark of Yersinia infections is colonization of the lymph nodes. After subcutaneous delivery of Y. pestis by the bite of an infected flea, Y. pestis usually migrates to draining lymph nodes and causes a fulminant infection leading to severe swelling of these organs, termed buboes, and then spreads systemically [1]. Y. pestis is thought to have evolved from the enteric pathogen, Y. pseudotuberculosis (Yptb), about 1,500–20,000 y ago [2]. However, Y. pestis acquired two additional plasmids important for its life cycle [3]. Despite the high degree of genetic similarity between the two species [2], infections with Yptb in humans occur after ingestion of contaminated food or water and are generally mild and self-limiting [1]. However, a frequent clinical manifestation of Yptb infections is mesenteric lymphadenitis [1,4]. Infection in humans with the more distantly related Y. enterocolitica (Ye) [2,5] generally produces gastrointestinal pathology as well as mesenteric lymphadenitis [1,4]. Thus, although enteric Yersinia species and Y. pestis use different means of transmission and cause different syndromes, all three species have a pronounced tropism for lymphoid tissues [1,4].
All three pathogenic Yersinia species carry a highly homologous 70-Kb virulence plasmid, termed pIB1 in Yptb, pYV in Ye, and pCD1 in Y. pestis [6–8]. These plasmids are key virulence factors that encode the structural (Ysc), regulatory (Lcr), and effector (Yops) proteins, of a type III secretion system (TTSS) [9,10]. The structural genes of the pIB1-encoded TTSS (pTTSS) are primarily encoded in two operons, yscAL and yscNU. These two operons are separated by a bicistronic operon containing virG, which encodes a lipoprotein [11], and the lcrF, which encodes the transcriptional activator of the ysc and yop promoters [12,13]. The pTTSS allows Yersinia to secrete and translocate Yops into host cells [9]. It is thought that Yops are critical in allowing Yersinia spp. to reside extracellularly in infected tissues and to resist clearance by normal innate immune functions by a number of mechanisms, including apoptosis, down-regulation of pro-inflammatory responses to Yersinia, and prevention of phagocytosis [9,14–23]. For instance, YopJ triggers apoptosis in macrophages in culture and Mac1+ cells in the mesenteric lymph nodes (MLN) of mice [15–17,24]. In cultured cells, YopH, YopO, YopE, and YopT have all been implicated in preventing phagocytosis by macrophages and neutrophils [18–21]. Moreover, YopE, YopH, and YopJ decrease the levels of pro-inflammatory cytokines and chemokines [17,22,23,25]. Thus, Yops disrupt normal processes of cultured cells and inhibit innate host defense mechanisms in infected tissues.
The importance of Yops for the virulence of pathogenic Yersinia spp. has been studied in many different animal models and by different routes of infection [24,26–31]. In Yptb, yop mutants are attenuated for virulence after intragastric, intraperitoneal, or intravenous inoculation [24,28,29,32], and several yop mutants are defective for colonizing intestinal and lymphoid tissues of mice [24,27]. In addition to the pTTSS, Y. pestis, Yptb, and highly pathogenic Ye subspecies each have a chromosomally encoded TTSS, cTTSS [33,34]. The cTTSS in Ye is important for virulence [35–37], but differs considerably from the cTTSS encoded by Y. pestis and Yptb, and the role of the Y. pestis and Yptb cTTSS has not been extensively studied [33,38].
Our previous work demonstrated that Yptb strains lacking one or more Yops were deficient in colonization of the gastrointestinal (GI) tract, Peyer's patches (PP), and MLN [27]. In control studies, we tested the ability of strains lacking the entire pTTSS to colonize different tissues after intragastric inoculation of mice, expecting that these strains would be severely attenuated. Surprisingly, pTTSS mutants colonized the MLN as efficiently as or better than wild-type (WT), although they were defective in colonizing the GI tract, spleen, and liver. Further investigation into the behavior of WT and pTTSS mutants revealed that both were found predominantly in the cortex and paracortex of the MLN, and both remained extracellular regardless of the presence of the pTTSS. Our results indicate that chromosomally encoded factors are necessary and sufficient for Yptb to replicate within B and T lymphocyte zones of MLN.
Results
pTTSS Mutants Colonize the MLN and PP, but Not the GI Tract or Spleen
To determine whether the pTTSS was required for colonization of various tissues after intragastric inoculation, several Yptb mutants with deletions in the operons encoding the pTTSS were created. Deletions of the yscBL operon, the yscNU operon, or both operons and the virGlcrF operon, designated yscBU, were generated. A strain lacking the pIB1 virulence plasmid was also tested to determine whether other plasmid-encoded genes were important for colonization. All four pTTSS mutants, yscBL, yscNU, yscBU, and pIB1 − were used to intragastrically inoculate BALB/c mice, and their colonization levels were compared to the isogenic WT strain, YPIIIpIB1. At 6 h, 2 d, or 5 d post-inoculation, the lumen contents of the ileum, cecum, and ascending colon, the PP, cecal lymph node (CLN), MLN, and spleen were dissected, and the colony forming units per gram (cfu/g) tissue determined. At 6 h post-inoculation, no difference in colonization of any tissue was observed between the yscBU mutant and WT YPIIIpIB1 (Figure 1 A–1E, and unpublished data). At 2 d post-inoculation, the pTTSS mutants were severely attenuated for colonization throughout the GI tract, PP, CLN (Figure 1F–1J, and unpublished data). In contrast, the level of colonization of the MLN was similar between WT, and three of the four TTSS mutants, yscBL, yscNU, and pIB1− indicating that the TTSS was not required for colonization of MLN. At 5 d post-inoculation, the defect in colonization of the pTTSS mutants was evident throughout the GI tract and spleen, whereas colonization levels of all pTTSS mutants in the MLN, PP, and CLN were similar to WT or in some cases higher than WT (Figure 1K–1O and unpublished data).
Figure 1. pTTSS Is Not Required for MLN Colonization.

BALB/c or C57BL6/J mice were intragastrically inoculated with 2 × 108 WT YPIII, YPIII yscBL, YPIII yscNU, YPIII yscBU, or YPIII pIB1−, and colonization levels of the ileum (A), (F), and (K), cecum (B), (G), and (L), PP (C), (H), and (M), MLN (D), (I), and (N), and spleen (E), (J), and (O) were determined at 6 h (A–E), 2 d (F–J), and 5 d (K–O) post-inoculation. BALB/c mice were intragastrically inoculated with 2 × 108 mouse commensal E. coli, and colonization levels of tissues were studied at 6 h (A–E) and 2 d post-inoculation (F–J). BALB/c mice were intragastrically inoculated with 2 × 109 of WT IP2666, IP2666 yscNU, or IP2666 pIB1−, and colonization levels were determined at 4 d post-inoculation ( [K–O], middle section).
Data are from 4–12 mice from at least two different experiments. All data was combined for each strain, tissue, and time point. Each square represents the log cfu/g tissue from one mouse; open squares indicate that less than 10 cfu were recovered per tissue, and bars represent the geometric mean. Asterisks (*) and black circles (•) indicate statistically significant differences between the WT and the mutants, calculated by the t test (*p < 0.01) or by Mann-Whitney (•U < 0.05), respectively.
To determine whether pTTSS mutants colonized other strains of mice efficiently, C57Bl6/J mice were infected with WT YPIIIpIB1, yscBU, or pIB1− (Figure 1K–1O). As observed in BALB/c mice, the pTTSS mutants colonized the PP and MLN at levels comparable to WT YPIII and were deficient in colonizing the ileum, cecum, and spleen. Therefore, the pTTSS or other elements encoded on the pIB1 plasmid are not required for Yptb survival and replication in the PP or MLN at 5 d post-inoculation in BALB/c or C57Bl6/J mice.
To confirm that the ability of Yptb to colonize the MLN in the absence of pTTSS was not due to a unique property of the YPIII strain, similar studies were performed using the Yptb strain, IP2666. IP2666, unlike YPIII, has a functional copy of PhoP which enables IP2666 Yptb to replicate intracellularly in bone marrow-derived macrophages [38]. Because IP2666 is slightly more virulent than YPIII during intragastric infections of mice (L. Logsdon and J. Mecsas, unpublished data), mice were sacrificed at day 4 post-inoculation (Figure 1K–1O). Surprisingly, the IP2666 pTTSS mutants, yscNU, and pIB1−, colonized the MLN at levels 10-fold higher than WT IP2666 (Figure 1K–1O). As observed for YPIII, the IP2666 pTTSS mutants were recovered in significantly fewer numbers in the ileum and spleen; however, about 10-fold more IP2666 pTTSS than YPIII pTTSS were recovered in the spleen, consistent with the increased virulence of the IP2666 strain (Figure 1O).
Since other Gram-negative bacteria reside in the lymphoid tissues for long periods of time [39], and commensal bacteria remain alive in dendritic cells in the MLN for several days [40], a mouse-commensal Escherichia coli strain isolated from mouse feces (see Materials and Methods) was used to intragastrically infect BALB/c mice. At 6 h post-inoculation, E. coli colonized the GI tract and lymphatic tissues at levels comparable to or higher than Yptb (Figure 1A–1E). However, by 2 d post-inoculation, E. coli were undetectable in the MLN and were almost cleared from the PP and the CLN (Figure 1F–1J and unpublished data). These results indicate that the ability of Yptb to survive in the MLN for up to 5 d is not a general property of all Gram-negative enteric bacteria. In conclusion, Yptb colonization of the MLN occurs in the absence of the pTTSS or other pIB1-encoded genes, demonstrating that chromosomally encoded factors are sufficient for MLN colonization.
pTTSS Mutants Cause a Milder Inflammatory Response in the MLN Than WT Yersinia
To investigate the host response to infection by WT and pTTSS mutants in the MLN, infected MLN were analyzed by histology. The IP2666 strain was selected for this experiment, since the IP2666 strain was more virulent than the YPIII strain. BALB/c mice were inoculated intragastrically with 2 × 109 WT IP2666 or the pIB1− mutant, and the MLN were harvested 4 d post-inoculation and prepared for histology. Gross anatomical analysis of the MLN showed that both strains caused enlargement of the MLN, i.e., hyperplasia, compared to uninfected MLN (Table 1 and Figure 2). Histological analysis of uninfected MLN showed normal follicular lymphocytic areas (Figure 2A) adjacent to a compact paracortex (Figure 2C and 2D) with no evidence of necrotic lesions or lymphoid hyperplasia (Figure 2A–2D). In most mice infected with WT IP2666, one or more MLN had multiple necrotic foci located at the edge of the MLN and close to the germinal centers (Table 1 and Figure 2E) or in the paracortex (Table 1). Areas of necrosis contained bacteria (Figure 2G, arrow), neutrophils, eosinophilic amorphous material, and many apoptotic nuclei (Figure 2H). These findings contrasted notably with MLN from mice infected with the pIB1− mutant, which had lymphoid hyperplasia and occasionally a necrotic cell but no evidence of acute necrosis (Table 1 and Figure 2I–2L). Although WT-infected MLN presented with neutrophil infiltration primarily around the necrotic foci, the pIB1 −-infected MLN generally presented with diffused infiltration of neutrophils in the paracortex. Despite the fact that IP2666 pTTSS mutants were recovered at 10 times the level of WT IP2666 (Figure 1N), no extensive necrosis was observed, suggesting that one or more components of the pTTSS trigger an acute inflammatory response leading to necrosis, whereas pTTSS mutants persist, causing milder inflammatory response.
Table 1.
MLN Histopathological Analysis
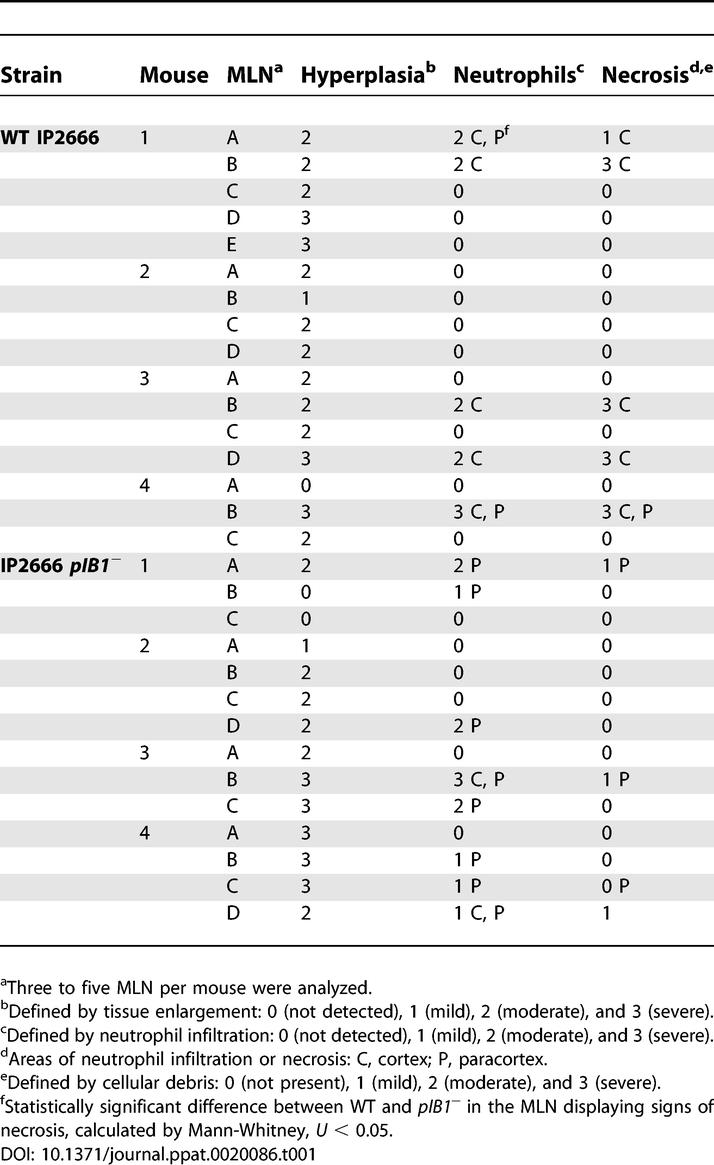
Figure 2. MLN Histopathology during WT or pIB1− Infection.

BALB/c mice were intragastrically inoculated with 2 × 109 WT or IP2666 pIB1−, and MLN were processed for H&E staining 4 d later. MLN sections from uninfected (Uninf.) (A–D), WT infected (E–H), and pIB1− infected (I–L) are shown at 15×, 40×, 90×, and 450× magnification. White boxes indicate magnified areas in the next slide. White arrow points to the bacterial foci, black arrow to neutrophils. Scale bars correspond to 133 μm for 15× magnification, 50 μm for 40× magnification, 22 μm for 90× magnification, and 4.4 μm for 450× magnification. Pictures shown are representative of multiple fields and samples from 16 MLN infected with WT or 14 MLN infected with pIB1−.
C, cortex; gc, germinal center; M, medulla; P, paracortex.
The pIB1 − Strain Localizes in Lymphocyte-Rich Areas
Immunohistochemistry studies were undertaken to determine the microenvironment of WT and pTTSS mutants in the MLN. WT IP2666 microcolonies were found in necrotic areas (Figure 3), in lymphocyte-rich areas of the cortex and paracortex that had no evidence of acute necrosis although apoptotic nuclei were often observed close to the bacteria (Figure 3G–3I), and in the medulla (unpublished data). pIB1 − microcolonies were found in lymphocyte areas of the cortex and paracortex, but not in the medulla (Figure 3J–3L and unpublished data). In the cases in which infiltration of phagocytes were observed, pIB1− mutants were not detected in these areas (unpublished data), unlike in necrotic areas containing WT bacteria.
Figure 3. Yptb Localizes in the Cortex and Paracortex of the MLN.

BALB/c mice were intragastrically inoculated with 2 × 109 WT or IP2666 pIB1−. At 4 d post-inoculation, MLN were harvested and stained for Yptb followed by hematoxylin staining. Picture sections of uninfected (A–C), WT infected (D–I), and pIB1− infected (J–L) MLN were taken at 150×, 900×, and 4,500× magnification. White boxes indicate magnified areas in the next slide. Arrows point to Yptb microcolonies. Scale bars correspond to 133 μm for 15× magnification, 22 μm for 90× magnification, and 4.4 μm for 450× magnification. Pictures shown are representative of multiple fields and samples from MLN infected with WT or pIB1−.
C, cortex; gc, germinal center; M, medulla; P, paracortex.
The cortex and paracortex are primarily composed of lymphocytes [41,42]. In order to confirm the cell types adjacent to Yptb, immunofluorescence studies were performed. Serial MLN sections from infected BALB/c mice were stained with anti-Yptb and either a mixture of antibodies that recognize lymphocytes (CD4, CD8, and B220) (Figure 4A–4F) or an antibody that recognized macrophages and neutrophils (CD11b) (Figure 4G–4L). Yptb pTTSS mutants were found in lymphocyte-rich areas whereas WT Yptb was occasionally observed adjacent to CD11b+ cells (Figure 4G–4L). To determine whether Yptb had a preference for either B or T cells, sections were stained with anti-CD4 and anti-CD8 or anti-B220 and antibody against Yptb. Both WT and pTTSS mutant strains were found in close proximity to both T and B lymphocytes (unpublished data), indicating no overt preference for either type of lymphocyte.
Figure 4. pTTSS Mutants Are Adjacent to B and T Lymphocytes Whereas WT Is Adjacent to B and T Lymphocytes and CD11b+ Cells.

BALB/c mice were intragastrically inoculated with 2 × 109 WT IP2666 or IP2666 pIB1−. At 4 d post-inoculation, MLN were harvested, sectioned, and examined by fluorescence microscopy. Staining with antibodies to Yptb (red) (A), (D), (G), and (J), to CD4-CD8-B220 (green) (B) and (E), or to CD11b (green) (H) and (K) was performed, and images merged (C), (F), (I), and (L). Pictures show the cortex–paracortex and are representative of multiple fields and samples for WT-infected mice and pIB1 mice stained with CD4-CD8-B220. Fewer fields had pIB1 bacteria and CD11b+ cells. Scale bars correspond to 22 μm for 900× magnification.
pTTSS Mutants Are Deficient in Colonization of Lymphoid Tissues in Rag1−/− Mice
Since pTTSS mutants survive in B and T lymphocyte areas of the MLN, we hypothesized that B and T cell–rich areas might confer a protective niche from neutrophils and macrophages. To determine whether the absence of B and T cells rendered the IP2666 pIB1− less capable of surviving in the MLN, BALB/c or congenic Rag1−/− mice were infected with WT IP2666 or IP2666 pIB1−. Rag1−/− mice lack mature B and T lymphocytes, although small numbers of immature lymphocytes are present [43]. Four days post-inoculation, the MLN and ileum colonization levels were compared in the BALB/c and Rag1−/− mice (Figure 5). Notably, the size and weight of the MLN in Rag1−/− mice varied depending on whether mice had been infected with WT IP2666 or pIB1− strains (Figure 5A). Rag1−/− mice infected with pTTSS mutants had larger MLN than Rag1−/− MLN infected with WT Yptb (Figure 5A), consistent with the idea that WT IP2666 can inhibit the pro-inflammatory response [22,23,25]. Therefore recovery of bacteria in the MLN was calculated both as cfu/g and cfu/MLN (Figure 5B). Total colonization levels in the MLN were similar in WT IP2666–infected BALB/c and Rag1−/− mice (Figure 5B). In contrast, colonization of the MLN by IP2666 pIB1− in Rag1−/− mice was 16-fold lower than colonization levels in BALB/c mice (Figure 5B). Levels of the IP2666 pIB1− in the ileum of BALB/c and Rag1−/− mice showed no differences (Figure 5C), indicating that the lack of recovery in the MLN of the pTTSS mutant in Rag1−/− mice was not secondary to a reduced ability to colonize the GI tract in Rag1−/− mice.
Figure 5. B and T Lymphocytes Are Important for pIB1− Colonization in the MLN.

BALB/c or Rag1−/− mice were intragastrically inoculated with 2 × 109 WT IP2666 or IP2666 pIB1−.
(A) Size and weight of MLN from BALB/c or Rag1−/− mice that were either uninfected or infected with WT IP2666 or IP2666 pIB1− were measured.
(B) and (C) Colonization of the MLN or luminal content of the ileum 4 d post-intragastric inoculation of BALB/c or isogenic Rag1−/− mice was determined. Each square indicates the cfu from one mouse calculated as log cfu/MLN (B) or the log cfu/g of luminal content of the ileum (C); bars represent the geometric mean. Each experiment was performed with two to three mice and repeated three times. Asterisks (*) and black circles (•) indicate statistically significant differences between the number of pIB1− recovered from BALB/c mice versus Rag1−/−, calculated by the t test (*p < 0.05) or by Mann-Whitney (•U < 0.05), respectively.
Since Rag1−/− mice lack PP, it was conceivable that the pIB1− mutants colonize the MLN poorly because they are unable to reach the MLN due to the lack of PP, rather than to survive in the MLN. Thus, Rag1−/− mice were infected intraperitoneally (i.p.) to bypass any requirement for trafficking from the GI tract through the PP to the MLN. Unexpectedly, after intraperitoneal inoculation with either WT or pTTSS mutants, the MLN were not detectable by eye in Rag1−/− mice. Our inability to detect MLN could have been a result of their initial small size and lack of inflammatory response after i.p. inoculation, or alternatively, the MLN in the Rag1−/− mice may have been destroyed by a robust immune response to infection prior to dissection.
pTTSS Mutants Localize Extracellularly in the MLN
Since it is well documented that the pTTSS allows Yersinia spp. to remain extracellular while docked to phagocytic cells [9], we investigated whether the pTTSS mutants were extracellular or intracellular in the MLN using an “ex vivo” gentamicin protection assay (see Materials and Methods). BALB/c mice were infected intragastrically with WT YPIII, WT IP2666, or pTTSS mutants, and 4- or 5-d post-inoculation cell suspensions of MLN were treated with gentamicin (Figure 6A). Although the differences were not significant, more pTTSS mutants were protected from gentamicin than the isogenic WT strains. However, only 1% of the YPIII yscBL and 3.4% of the IP2666 pIB1− mutant were protected, indicating that over 96% of the pTTSS mutants were extracellular. The observation that more WT IP2666 were protected from gentamicin than WT YPIII may reflect the ability of the IP2666 strain to survive and replicate within macrophages [38]. As a positive control, nearly 15 % of Salmonella typhimurium were intracellular in the MLN at 4 d post-inoculation. Overall, these results indicate that the majority of Yptb remain extracellular in the MLN, even in the absence of a functional pTTSS.
Figure 6. Yptb pTTSS Mutants Remain Extracellular in the MLN and Are Not Efficiently Internalized by B or T Lymphocytes in Culture.

(A) Single-cell suspensions of Yptb-infected MLN were assayed for the presence of intracellular bacteria using a gentamicin protection assay. BALB/c mice were intragastrically inoculated with WT YPIII (n = 6 mice), YPIII yscBL (n = 5 mice), WT IP2666 (n = 5 mice), IP2666 pIB1− (n = 7 mice), or S. typhimurium (n = 4 mice). Four days post-inoculation with IP2666 strains or 5 d post-inoculation with YPIII strains, the percentage of intracellular bacteria was assessed by generating a single-cell suspension of the MLN and treating half the suspension with gentamicin and half without gentamicin (see Materials and Methods).
(B) Murine macrophage RAW264.7 cells, human T cells SUP-T1, and B and T lymphocytes isolated from MLN (see Materials and Methods) were infected with WT YPIII, YPIII yscBL, WT IP2666, or IP2666 pIB1− strains at MOI of 10:1 for 30 min, and then treated with gentamicin for 90 min. The data are presented as 100 times the number of gentamicin-resistant bacteria divided by the number of input bacteria. Data from one representative experiment done in triplicate is shown. All experiments were performed at least three times.
The pTTSS is important for Yptb to inhibit phagocytosis by macrophages, neutrophils, and epithelial cells in cell culture assays [9]; thus, it was surprising that the vast majority of the pTTSS mutants remained localized extracellularly. To confirm that the pTTSS mutants were phagocytosed more efficiently than WT, gentamicin protection assays were performed using the murine macrophage-like cell line, RAW264.7. As expected, higher level of uptake by the macrophages of the pTTSS mutants was observed compared to WT YPIII or WT IP2666 (Figure 6B). WT IP2666 was phagocytosed more efficiently than YPIII, an observation consistent with our “ex vivo” gentamicin protection assays and previous results by others [38]. Yptb pTTSS mutants efficiently invade some types of non-professional phagocytes, such as HEp-2 cells, by binding to α4- or α5-β1 integrins using the bacterial protein, invasin [44,45]. Since the areas of the MLN infected by pTTSS mutants are composed primarily of lymphocytes [42], and lymphocytes express α4- and α5-β1 integrins [46,47], gentamicin protection assays were performed on two human T cell lines, SUP-T1 and H9, and primary B and T lymphocytes isolated from MLN after incubation with WT and pTTSS mutants. The pTTSS mutants were not efficiently internalized by either T cell lines or by primary B and T lymphocytes isolated from MLN (Figure 6B and unpublished data). These results are consistent with the “ex vivo” gentamicin protection assays, indicating that pTTSS mutants are extracellular in lymphocyte rich areas.
Invasin and the cTTSS Are Not Necessary for Survival in the MLN
Since chromosomal factors must play a role in Yptb survival in the MLN, two chromosomally encoded factors, invasin and cTTSS, were tested for growth in the MLN when the pTTSS was not present. Invasin-deficient strains (inv−) are less competent than WT in colonizing the PP following intragastric inoculation [30,48], indicating that invasin is crucial for Yptb to reach and/or survive in the PP. To evaluate the role of invasin in a pIB1− background, BALB/c mice were infected intragastrically and colonization levels were assessed 5 d post-inoculation. Notably, a pIB1−inv − strain was 100-fold more deficient in the colonization of the ileum compared to pIB1−, indicating that invasin plays a role in bacterial survival in the ileum that is independent of pTTSS (Figure 7A). In the PP and MLN, the pIB1−inv − strain was more deficient than pIB1− in colonization (Figure 7A); however, the lower levels in lymph tissues could reflect the lower levels of bacteria in the ileum. In order to bypass the requirement for invasin in the ileum, mice were infected intraperitoneally. No difference was observed between the pIB1− and the pIB1−inv − strains in MLN colonization after i.p. inoculation, indicating that although invasin is required for reaching the MLN after oral inoculation, it is not required for Yptb pIB1− survival in MLN (Figure 7B).
Figure 7. Invasin and cTTSS Are Not Essential for Yptb Survival in the MLN.

(A) BALB/c mice were intragastrically inoculated with 2 × 108 YPIII pIB1 or YPIII pIB1−inv−, and colonization levels of the ileum, PP, MLN, and spleen were determined 5 d post-inoculation.
(B) BALB/c mice were i.p. inoculated with 2 × 106 YPIII pIB1 or YPIII pIB1− inv−, and their colonization levels in the MLN and spleen were determined 3 d post-inoculation.
(C) BALB/c mice were inoculated intragastrically with 2 × 108 YPIII pIB1− or YPIII pIB1−cTTSS, and colonization levels of the ileum, PP, MLN, and spleen were determined. Data are from 6–11 mice from at least two different experiments. Each square represents log 10 cfu/g tissue recovered from one mouse. Bars represent the geometric mean. Open squares indicate than less than 10 cfu were recovered per tissue. Asterisks (*) and black circles (•) indicate statistically significant differences between the pIB1− and either pIB1− inv − or pIB1−cTTSS strains, calculated by the t test (*p < 0.01) or by Mann-Whitney (•U < 0.05), respectively.
To determine whether the cTTSS is important for MLN colonization, BALB/c mice were intragastrically infected with pIB1− and pIB1−cTTSS mutants. No differences were observed in the colonization of the lumen, PP, MLN, or spleen between pIB1− and pIB1−cTTSS at 5 d post-inoculation (Figure 7C). Thus, the cTTSS is not required for MLN colonization in the absence of pIB1.
Discussion
A prominent clinical feature of infection by pathogenic Yersinia is lymphadenitis, as large buboes [1] and mesenteric lymphadenitis [4] are frequent symptoms associated with infection by Y. pestis and enteric Yersinia pathogens, respectively. Little is known about the factors involved in Yersinia survival in the lymph nodes, although deletion of several Yops reduces the ability of Yersinia to colonize PP and MLN, which suggests that these Yops are required for reaching and/or replicating in lymph nodes [27,31]. Here, we show that Yptb replicates extracellularly in the cortex and paracortex of the MLN in the absence of pTTSS and Yops, indicating that the ability to reach and replicate in lymphocyte-rich areas is driven by chromosomally encoded factors. WT Yptb infection provoked necrotic and suppurative foci whereas pTTSS mutants induced mild lymphadenitis, demonstrating that the presence of pTTSS triggers a more acute host inflammatory response. Furthermore, B and T lymphocytes appear to provide a protective niche for pTTSS mutants since their absence results in significantly lower colonization levels of the pTTSS mutants.
Strikingly, a partial complement of Yops is more detrimental to the survival of Yptb in the MLN than the complete absence of pTTSS [24,27]. Strains lacking two or more Yops colonize the MLN very inefficiently [27], and strains lacking only yopH, yopJ, or yopE have a 5- to 10-fold defect in MLN colonization [24,27]. One explanation for these observations is based on the finding that the translocation apparatus damaged cultured cells and induced elevated levels of IL-8, which were decreased by YopH, YopE, or YopJ [25]. By analogy, during infection with WT or yop mutants, components of the pTTSS may damage lymphocytes and trigger pro-inflammatory signals in the MLN, so that in the absence of one or more Yops, the bacteria are killed by recruited macrophages and neutrophils. pTTSS mutants may survive because they do not damage host cells and therefore do not induce the same pro-inflammatory response. In addition, pTTSS mutants might replicate faster during infection than strains expressing the pTTSS, as strains lacking pTTSS have a modest growth advantage compared to WT Yptb in culture media (unpublished data). Nonetheless, the pTTSS mutants clearly have a growth advantage over mouse commensal E. coli in the MLN, indicating that Yptb chromosomal factors enable the bacteria to persist and replicate in the MLN. With this in mind, it is conceivable that a common ancestor of Yersinia may have been a commensal with low pathogenic capability but with the ability to persist in the MLN or host environments similar to lymph nodes of mammals. Acquisition of the pTTSS would have enabled the non-pathogenic species to spread to other tissue environments of mammals and survive within the GI tract, enhancing its transmissibility.
Although the Yptb pTTSS mutants are rapidly internalized by cells that are generally non-phagocytic, such as epithelial cells [30], pTTSS mutants establish an extracellular niche surrounded by B and T lymphocytes in lymphoid tissues. Non-phagocytic cells expressing α4- or α5-β1 integrins internalize pTTSS mutants in an invasin-dependent manner at frequencies often 100-fold greater than WT Yptb [30,49]. Although T lymphocytes express α4- and α5-β1 integrins [46,47], pTTSS mutants were not internalized significantly more than WT Yptb during infection, and did not invade T or B lymphocytes isolated from MLN. Although lymphocytes are not generally considered intracellular hosts for bacterial pathogens, S. enterica is found within late endosomal–lysosomal compartments in B lymphocytes of the spleen after intragastric inoculation [50]. Combined, these results indicate that the cellular mechanisms exploited by Yptb to prompt its invasion into non-phagocytic cells are not as active in B and T lymphocytes. In addition, invasin may not be expressed in the MLN [51].
It is well established that the pTTSS is an essential virulence organelle that enables Yptb to remain extracellular in the presence of phagocytes through the anti-phagocytic action of Yops [9,21,52]. WT Yptb, but not the pTTSS mutants, was occasionally found associated with macrophages and neutrophils, suggesting that our inability to detect pTTSS mutants adjacent to CD11b+ cells may be due to rapid internalization and killing of these mutants by phagocytic cells. Furthermore, in Rag1−/− mice, WT Yptb had a selective advantage compared to the pTTSS mutants, suggesting that pTTSS mutants are compromised for survival in lymph nodes that lack B and T lymphocytes. In the absence of lymphocytes, the protective niche may be lost and the bacteria may be more exposed to phagocytes in the MLN [53]. In the PP, Yptb first encounters dendritic cells, neutrophils, and macrophages immediately after transcytosing the M cells [48]. In contrast to the MLN, the pTTSS mutants colonize the PP poorly at day 2, suggesting that many pTTSS mutants are eliminated by these cells prior to reaching the follicular area rich in B and T cells.
The inability of pTTSS mutants to survive well in Rag1−/− mice compared to the isogenic BALB/c mice suggests that lymphocytes provide a protective niche for pTTSS. However, it is possible that the lack of PP hindered the pTTSS in reaching the MLN. If this were true, then the lack of PP was specifically detrimental to the pTTSS mutants, since the WT Yptb was found in the MLN of Rag1−/− mice. The latter result indicated that WT Yptb does not require PP for successful dissemination to the MLN. We attempted to investigate whether the low levels of pTTSS mutants observed in the MLN of Rag1−/− mice were replicating in the MLN or were the result of continual seeding of the MLN from the GI tract. To eliminate pTTSS mutants in the GI tract and therefore prevent reseeding, mice were given streptomycin orally as described in [54]; however, in control experiments we found that streptomycin delivered orogastrically killed Yptb colonizing the MLN after i.p. delivery, but not Yptb colonizing the spleen, which is consistent with results reported about colonization of the spleen in [54]. Thus, this experimental approach is unfeasible. Future experiments are directed at determining where the pTTSS mutants are in the MLN of Rag1−/− mice, and the immune response to infection with WT and pTTSS in lymph nodes.
Common host-driven pathways indiscriminately traffic bacteria to lymph nodes, in part by dendritic cells binding to bacteria in the intestinal lumen and then migrating to the MLN [40]. Consistent with these observations, we found non-pathogenic E. coli in the MLN at 6 h post-inoculation. Also, others have reported Yptb and Salmonella in the blood as early as 30 min after intragastric inoculation [40,55]. It is noteworthy that at 6 h post-inoculation E. coli colonizes the lumen at much higher levels than Yptb, but their levels in the MLN are comparable to Yptb. Thus, Yptb likely use several tactics to rapidly reach the MLN, including invasin-dependent [30,48,56] and host-driven mechanisms [40,55], whereas E. coli either fails to efficiently reach the MLN or is rapidly eliminated by innate immune defenses in the MLN.
The key to the persistence of Yptb pTTSS mutants in the cortex and paracortex may lie in adherence factors that facilitate the interaction of Yersinia with lymphocytes or the underlying extracellular matrix in these areas. We have ruled out the requirement of invasin, YadA, and potential cTTSS-secreted factors in MLN colonization. Other candidate adhesion factors include the pH6 antigen [57] and glycoproteins. In other organisms, glycoproteins play a key role in conferring cellular tropism [58,59], and intriguingly, in some cases, they have been shown to be sufficient to induce a state of proliferative unresponsiveness in lymphocytes [58]. One such example is the MV glycoprotein complex of measles virus, which interferes with the propagation of the IL-2 receptor signal by blocking activation [58]. One could envision that the presence of specific glycoproteins or specific sugars on the LPS determines Yptb tropism and/or moderates inflammatory cytokines produced by B and T cells. In addition, it is tempting to speculate that preferential interaction with certain subsets of lymphocytes, such as CD4+CD25+ T regulatory cells may further aid Yptb colonization. In fact, the presence of CD4+CD25+ T lymphocytes has been shown to favor the establishment of Helicobacter pylori in the gastric mucosa by moderating inflammation [60].
Despite the key roles that the pTTSS and Yops play in disease [27], there is a report of a serotype 4a Yptb pIB1− strain isolated from a 10-y-old girl diagnosed with acute mesenteric lymphadenitis [61]. In agreement with our data, characterization of that strain in mice as well as another serotype 4a pIB1− strain demonstrated that serotype 4a strains lacking the pIB1 plasmid colonized the MLN at levels comparable to WT Yptb for up to 7 d [61]. Interestingly, the fate of Ye pTTSS mutants in the MLN seems to be dependent on Ye serotype. Ye biotype 1A pYV− strains have been isolated from symptomatic patients [62]. On the other hand, a number of experiments with other Ye biotypes, Ye O3 pYV−, Ye O9 pYV−, and Ye8081 pYV−, indicate that these strains were eliminated from the MLN of rabbits, gnonobiotic piglets, or mice within 1 to 3 d ([63–65] and unpublished data). Taken together, these results indicate that both enteric Yersinia spp. reach the MLN in the absence of pTTSS, but only a subset of strains flourishes for longer periods, and suggest that Yptb has specific chromosomally encoded factors that enhance its survival. A comparative analysis of the chromosomal genomes of Yersinia strains that persist versus those that fail to persist in the MLN may reveal factors that enable replication in lymphoid tissues.
Numerous pathogens, including Mycobacterium spp., Salmonella spp., Brucella spp., viruses, and parasites such as Leishmania spp., are capable of producing lymphadenitis [39,66–69]. These pathogens use a variety of mechanisms to survive in the MLN. For example, S. typhimurium survives intracellularly within macrophages in the MLN, where it can be isolated for up to a year post-inoculation [39]. Brucella suis survives inside macrophages by inhibiting apoptosis [70], and Leishmania spp. survive inside granulomas in the MLN [69]. We have shown that Yptb replicates in B and T lymphocytes areas in a pTTSS-independent manner. The presence of the pTTSS provokes many necrotic lesions, and allows Yptb to survive in phagocyte-rich areas cells and invade other organs. Future directions are aimed at identifying both the bacterial and host factors that permit replication of Yptb in lymphocyte rich zones of the MLN to understand the mechanisms driving the lymphotropism of Yersinia.
Materials and Methods
Bacterial strains.
Two virulent serotype III Yptb strains were used: YPIII pIB1 [27], which has recently been shown to have a mutation in phoP [38], and IP2666 pIBI [38]. These strains are described as WT YPIII and WT IP2666. The YPIII pIBI−kan and IP2666 pIB1− were generated by growing WT YPIII kan [27], or WT IP2666 on Luria Agar supplemented with 20 mM of sodium oxalate, 20 mM MgCl2, and 0.05 % Congo Red Dye. Loss of a functional pTTSS results in white colonies. The absence of several genes located on different regions of the plasmid was confirmed by PCR, indicating that the whole plasmid was lost. The invasin-deficient strain JM494 [30] was cured of pIB1 as described above. The mouse commensal E. coli strain was isolated from the feces of BALB/c mice (Taconic, Hudson, New York, United States). This strain was transformed with either pBR322 or pBR322-expressing invasin, generating JMB44 or JMB45, respectively. Expression of invasin in this E. coli strain increased the uptake by 100-fold in gentamicin protection assays using HeLa cells (unpublished data); however, no difference in colonization of the GI tract, PP, or MLN was observed when either E. coli strains were used to infect mice (Figure 1 and unpublished data). S. typhimurium, SL1344, was used [39].
Additional mutant strains generated in this study were constructed in WT YPIII or WT IP2666 by allelic exchange using the suicide vector pCVD442, and methods described in [27]. Isogenic pTTSS mutants were constructed by deleting the regions between yscB to yscL, yscN to yscU, and yscB to yscU. The primer sequences used to generate the appropriate suicide plasmids were as follows: yscBL deletion, yscB1-GTGTGAGTCGACGCGGTGGCGCAA GAAAC, yscB2-GACAGTGCATGCAGTTGGCCAACGCTTG TTGCATTAG; yscL3-CCAACTGCATGCACTGTCCAGGAGCTATTCGTGAAC; and yscL4-CTGTCAGAGCTCGTCGCTATAATTGTCTCTAC; yscNU deletion, yscN1-GTGTGAGTCGACCTACTCCCTGAGATGAAC, yscN2-GACAGTGCATGC AGTTGGCCAAGTGGAATAAGTAATGC, yscU3-CCAACTGCATGCACTGTCTCA TCAGTGGTGGTAGCT, and yscU4-CTGTCAGAGCTCCCAATAGCCGGTGTTAATC; and yscBU deletion, yscB1, yscB2 and yscU1, yscU2. Colonies were screened for their ability to bind to Congo Red. Deletions were confirmed by PCR and then Southern blot.
The cTTSS deletion was constructed by deleting the putative structural genes spiA-ssaV. Primers used to generate the suicide plasmid were: spiA1-GTGTGAGTC GACCTGTGCCATAAAAGCGATCC; spiA2-GACAGTGCATGCAGTTGGTAG CGGAATATCGC; ssaV3-CCAACTGCATGCACTGTCCACATTTTGCCGTT; and ssaV4-CTGTCAGAGCTCCCTGTGCAATGCGGATAG. Deletions were confirmed by PCR and then Southern blot. In addition, the pTTSS mutants were grown at 26 °C and 37 °C, using Yop-inducing, i.e., calcium-depleted. medium [71], or non-inducing conditions, i.e., 2×YT supplemented with 5 mM CaCl2. At 26 °C, the growth of the mutants was comparable to WT. At 37 °C, the pTTSS mutants had a growth advantage compared to WT in Yop-inducing conditions, but they grew similarly when grown in non-inducing conditions. Yops were not secreted when Yptb were grown at 37 °C under Yop-inducing conditions [27].
The resulting mutants and strains used were given the following strain numbers: JMB35, YPIII yscBL; JMB31, YPIII yscNU; JMB38, YPIII yscBU; JMB5, YPIII pIB1 kan; JMB111, IP2666 pIB1−; JMB141, IP2666 yscNU; JMB100, pIB1−inv − ; and JMB84, YPIII pIB1−cTTSS.
Mouse infection.
Seven– to 8-w-old female BALB/c (Taconic, or Jackson Laboratories, Bar Harbor, Maine, United States), C.129S7 (B6)-Rag1tm1Mom/J, Rag1−/− (Jackson Laboratories), or C57BL6/J mice (Jackson Laboratories) were used. Yptb were grown and infections were performed as described in [27]. S. typhimurium and E. coli were grown in LB at 37 °C overnight with aeration. Mice were infected intragastrically with 200-μl PBS containing 2 × 108 or 2 × 109 Yptb, as indicated in the figure legends, 5 × 108 S. typhimurium, or 2 × 108 E. coli. Mice were infected i.p. with 200-μl PBS containing 1 × 106 Yptb. The 6-h, 2-d, 4-d, or 5-d post-intragastric inoculation or 3-d post-intraperitoneal inoculation mice were sacrificed, and tissues were dissected and processed as described in [27]; p-values were determined by two-tailed, unpaired Student t test by comparing the log cfu/g tissue or log cfu/MLN of WT Yptb to each mutant.
The Institutional Animal Care and Use Committee of Tufts University approved all animal procedures.
Histology and immunohistochemistry.
MLN from uninfected mice or mice infected intragastrically with 2 × 109 Yptb were dissected, fixed in formalin, embedded in paraffin, and cut in 10-μm sections. Sections were treated with xylene to remove paraffin prior to staining with hematoxylin-eosin or staining for immunohistochemistry. Lauren Richey, DVM, PhD, Diplomat ACVP, and members of our lab scored MLN histology sections blindly.
Immunohistochemistry was performed by treating the sections with 3% hydrogen peroxide (DAKO, Glostrup, Denmark) for 5 min, washing with 0.05% Tween-20 in PBS (PBS-Tw) for 5 min, incubating with 1:100 rabbit anti-Yptb for 30 min, washing with PBS-Tw, incubating with 1:300 polyclonal goat α-rabbit Ig (DAKO) for 30 min (gift from R. Isberg), washing with PBS-Tw, incubating with streptavidin-HRP (DAKO) for 15 min, and then incubating with substrate-chromatogen solution (DAKO). MLN were counterstained with 0.5% hematoxylin and examined with a Nikon Eclipse TE2000-U microscope (Melville, New York, United States).
Immunofluorescence.
MLN from uninfected mice or mice infected intragastrically with 2 × 109 with Yptb were harvested and snap frozen in Optimal Cutting Temperature Compound (Tissue-Tek, Fort Washington, Pennsylvania, United States). The 10-μm sections were cut, fixed with cold 100% acetone, and stored at −80 °C. For staining, sections were washed with PBS and blocked with 2% BSA for 30 min. Antibodies used were rat anti-mouse CD8 (BD Biosciences, San Jose, California, United States) and rat anti-mouse CD4 (BD Biosciences) for T lymphocytes, rat anti-mouse B220 (BD Biosciences) for B lymphocytes, rat anti-mouse CD11b (BD Biosciences) for neutrophils and macrophages, and rabbit anti-Yptb. Primary antibodies were diluted 1:200 in 2% BSA and incubated with the samples for 3 h at room temperature. Slides were washed three times with PBS-Tw for 15 min per wash and then incubated for 1 h with FITC conjugated to donkey anti-rat (Jackson ImmunoResearch, West Grove, Pennsylvania, United States) and Texas-Red–conjugated donkey anti-rabbit antibodies (Jackson ImmunoResearch). Slides were washed three times with PBS-Tw for 15 min per wash, and a cover slip was placed over the tissue before visualization using 20×, 40× and 100× objectives with a Nikon Eclipse TE2000-U microscope with fluorescence. Pictures were taken using a Roper Scientific camera (Trenton, New Jersey, United States) controlled by Open Lab software.
In vitro gentamicin protection assay.
The macrophage cell line RAW264.7 (ATCC# CRL-2278), the T cell line SUP-T1 (ATCC# CRL-1942), and the T cell line H9 (ATCC# HTB-176) were grown as recommended by ATCC (Manassas, Virginia, United States). Primary B and T lymphocytes were isolated from MLN using enrichment columns (Stem cells; R&D System, Minneapolis, Minnesota, United States). Yptb strains were grown under Yop-inducing conditions [71]. A total of 5 × 104 or 2 × 105 cells were infected at a multiplicity of infection (MOI) of 10:1 for 30 min at 37 °C in the presence of 5% CO2. Gentamicin was added to a final concentration of 100 μg/ml for 90 min, and then cells were washed three times with PBS. T and B cells were centrifuged at 200 g for 5 min between each wash. Cells were lysed with 100 μl of 1% Triton-X-100 for 5 min followed by addition of 900-μl LB, and lysates were diluted and plated to determine the number of intracellular bacteria. Results are presented as percent uptake equal to 100 times the recovered cfu divided by the input cfu.
Ex vivo gentamicin protection assay.
MLN were dissected from 7–8-wk female BALB/c mice infected with IP2666 or S. typhimurium. MLN were placed in 1-ml RPMI on ice and then pressed through a 70-μm filter (BD Labware, Palo Alto, California, United States) with the rubber end of a 1-ml syringe plunger. The sample was divided into two 0.5-ml aliquots: one aliquot was treated with 100-μg/ml gentamicin for 2 h at 37 °C to kill extracellular bacteria, and the other aliquot was untreated for 2 h at 37 °C to calculate the total cfu recovered. Cells were washed three times with PBS and lysed with 100 μl of 1% Triton-X-100 for 5 min followed by addition of 900-μl LB, and lysates were diluted and plated to determine the number of intracellular bacteria. Results are presented as number of gentamicin resistant bacteria (recovered after 2 h incubation) divided by the input bacteria times 100.
Acknowledgments
We thank Lauren Logsdon and Susan Butler for strains; Lauren Richey DVM, PhD, Diplomat ACVP, for histological analysis of tissues; Cynthia Castillo for advice about microscopy, staining of slides, and histological analysis; and members of the Mecsas lab, Mireia Guerau-de-Arellano, Molly Bergman, Jenifer Coburn, and Ralph Isberg, for useful discussion, suggestions, and critical review of the manuscript.
Abbreviations
- cfu
colony forming units
- CLN
cecal lymph node
- cTTSS
chromosomally encoded type III secretion system
- GI
gastrointestinal
- i.p.
intraperitoneal/ly
- MLN
mesenteric lymph nodes
- PP
Peyer's patches
- pTTSS
pIB1-encoded type III secretion system
- TTS
type III secretion system
- WT
wild-type
- Ye
Yersinia enterocolitica
- Yptb
Yersinia pseudotuberculosis
Footnotes
Competing interests. The authors have declared that no competing interests exist.
Author contributions. JMBL and JM conceived and designed the experiments, performed the experiments, analyzed the data, contributed reagents/materials/analysis tools, and wrote the paper.
Funding. This work was funded by the National Institutes of Health's National Institute of Allergy and Infectious Diseases grant R01AI056068, to JM, and Center for Gastroenterology Research on Absorptive and Secretory Processes (GRASP) P30DK-34928.
References
- Putzker M, Sauer H, Sobe D. Plague and other human infections caused by Yersinia species. Clin Lab. 2001;47:453–466. [PubMed] [Google Scholar]
- Achtman M, Zurth K, Morelli G, Torrea G, Guiyoule A, et al. Yersinia pestis, the cause of plague, is a recently emerged clone of Yersinia pseudotuberculosis . Proc Natl Acad Sci U S A. 1999;96:14043–14048. doi: 10.1073/pnas.96.24.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker RR. Factors promoting acute and chronic diseases caused by yersiniae. Clin Microbiol Rev. 1991;4:309–324. doi: 10.1128/cmr.4.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smego RA, Frean J, Koornhof HJ. Yersiniosis I: Microbiological and clinicoepidemiological aspects of plague and non-plague Yersinia infections. Eur J Clin Microbiol Infect Dis. 1999;18:1–15. doi: 10.1007/s100960050219. [DOI] [PubMed] [Google Scholar]
- Ibrahim A, Goebel BM, Liesack W, Griffiths M, Stackebrandt E. The phylogeny of the genus Yersinia based on 16S rDNA sequences. FEMS Microbiol Lett. 1993;114:173–177. doi: 10.1111/j.1574-6968.1993.tb06569.x. [DOI] [PubMed] [Google Scholar]
- Gemski P, Lazere JR, Casey T, Wohlhieter JA. Presence of a virulence-associated plasmid in Yersinia pseudotuberculosis . Infect Immun. 1980;28:1044–1047. doi: 10.1128/iai.28.3.1044-1047.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Moseley SL, Falkow S. Characterization of plasmids and plasmid-associated determinants of Yersinia enterocolitica pathogenesis. Infect Immun. 1981;31:775–782. doi: 10.1128/iai.31.2.775-782.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy DA, Falkow S. Virulence-associated plasmids from Yersinia enterocolitica and Yersinia pestis . J Bacteriol. 1981;148:877–883. doi: 10.1128/jb.148.3.877-883.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis GR. The Yersinia Ysc-Yop ‘type III' weaponry. Nat Rev Mol Cell Biol. 2002;3:742–752. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- Ghosh P. Process of protein transport by the type III secretion system. Microbiol Mol Biol Rev. 2004;68:771–795. doi: 10.1128/MMBR.68.4.771-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaoui A, Scheen R, Lambert de Rouvroit C, Cornelis GR. VirG, a Yersinia enterocolitica lipoprotein involved in Ca2+ dependency, is related to exsB of Pseudomonas aeruginosa . J Bacteriol. 1995;177:4230–4237. doi: 10.1128/jb.177.15.4230-4237.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik M, Toivanen P. LcrF is the temperature-regulated activator of the yadA gene of Yersinia enterocolitica and Yersinia pseudotuberculosis . J Bacteriol. 1992;174:2047–2051. doi: 10.1128/jb.174.6.2047-2051.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe NP, Minion FC, Goguen JD. Temperature sensing in Yersinia pestis: Regulation of yopE transcription by lcrF. J Bacteriol. 1992;174:4275–4286. doi: 10.1128/jb.174.13.4275-4286.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juris SJ, Shao F, Dixon JE. 2002. Yersinia effectors target mammalian signalling pathways. Cell Microbiol. 4:201–211. doi: 10.1046/j.1462-5822.2002.00182.x. [DOI] [PubMed] [Google Scholar]
- Monack DM, Mecsas J, Ghori N, Falkow S. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci U S A. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills SD, Boland A, Sory MP, van der Smissen P, Kerbourch C, et al. Yersinia enterocolitica induces apoptosis in macrophages by a process requiring functional type III secretion and translocation mechanisms and involving YopP, presumably acting as an effector protein. Proc Natl Acad Sci U S A. 1997;94:12638–12643. doi: 10.1073/pnas.94.23.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schesser K, Spiik AK, Dukuzumuremyi JM, Neurath MF, Pettersson S, et al. The yopJ locus is required for Yersinia-mediated inhibition of NF-kappaB activation and cytokine expression: YopJ contains a eukaryotic SH2-like domain that is essential for its repressive activity. Mol Microbiol. 1998;28:1067–1079. doi: 10.1046/j.1365-2958.1998.00851.x. [DOI] [PubMed] [Google Scholar]
- Fallman M, Andersson K, Hakansson S, Magnusson KE, Stendahl O, et al. 1995. Yersinia pseudotuberculosis inhibits Fc receptor-mediated phagocytosis in J774 cells. Infect Immun. 63:3117–3124. doi: 10.1128/iai.63.8.3117-3124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson C, Carballeira N, Wolf-Watz H, Fallman M. The PTPase YopH inhibits uptake of Yersinia, tyrosine phosphorylation of p130Cas and FAK, and the associated accumulation of these proteins in peripheral focal adhesions. EMBO J. 1997;16:2307–2318. doi: 10.1093/emboj/16.9.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosqvist R, Forsberg A, Rimpilainen M, Bergman T, Wolf-Watz H. The cytotoxic protein YopE of Yersinia obstructs the primary host defence. Mol Microbiol. 1990;4:657–667. doi: 10.1111/j.1365-2958.1990.tb00635.x. [DOI] [PubMed] [Google Scholar]
- Grosdent N, Maridonneau-Parini I, Sory MP, Cornelis GR. Role of Yops and adhesins in resistance of Yersinia enterocolitica to phagocytosis. Infect Immun. 2002;70:4165–4176. doi: 10.1128/IAI.70.8.4165-4176.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte R, Wattiau P, Hartland EL, Robins-Browne RM, Cornelis GR. Differential secretion of interleukin-8 by human epithelial cell lines upon entry of virulent or nonvirulent Yersinia enterocolitica . Infect Immun. 1996;64:2106–2113. doi: 10.1128/iai.64.6.2106-2113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LE, Hobbie S, Galan JE, Bliska JB. YopJ of Yersinia pseudotuberculosis is required for the inhibition of macrophage TNF-alpha production and downregulation of the MAP kinases p38 and JNK. Mol Microbiol. 1998;27:953–965. doi: 10.1046/j.1365-2958.1998.00740.x. [DOI] [PubMed] [Google Scholar]
- Monack DM, Mecsas J, Bouley D, Falkow S. Yersinia-induced apoptosis in vivo aids in the establishment of a systemic infection of mice. J Exp Med. 1998;188:2127–2137. doi: 10.1084/jem.188.11.2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viboud GI, So SS, Ryndak MB, Bliska JB. Proinflammatory signalling stimulated by the type III translocation factor YopB is counteracted by multiple effectors in epithelial cells infected with Yersinia pseudotuberculosis . Mol Microbiol. 2003;47:1305–1315. doi: 10.1046/j.1365-2958.2003.03350.x. [DOI] [PubMed] [Google Scholar]
- Straley SC, Cibull ML. Differential clearance and host-pathogen interactions of YopE− and YopK− YopL− Yersinia pestis in BALB/c mice. Infect Immun. 1989;57:1200–1210. doi: 10.1128/iai.57.4.1200-1210.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logsdon LK, Mecsas J. Requirement of the Yersinia pseudotuberculosis effectors YopH and YopE in colonization and persistence in intestinal and lymph tissues. Infect Immun. 2003;71:4595–4607. doi: 10.1128/IAI.71.8.4595-4607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KY, Reisner BS, Straley SC. YopM inhibits platelet aggregation and is necessary for virulence of Yersinia pestis in mice. Infect Immun. 1990;58:3262–3271. doi: 10.1128/iai.58.10.3262-3271.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin I, Wolf-Watz H. The plasmid-encoded Yop2b protein of Yersinia pseudotuberculosis is a virulence determinant regulated by calcium and temperature at the level of transcription. Mol Microbiol. 1988;2:237–245. doi: 10.1111/j.1365-2958.1988.tb00025.x. [DOI] [PubMed] [Google Scholar]
- Mecsas J, Bilis I, Falkow S. Identification of attenuated Yersinia pseudotuberculosis strains and characterization of an orogastric infection in BALB/c mice on day 5 postinfection by signature-tagged mutagenesis. Infect Immun. 2001;69:2779–2787. doi: 10.1128/IAI.67.5.2779-2787.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trulzsch K, Sporleder T, Igwe EI, Russmann H, Heesemann J. Contribution of the major secreted yops of Yersinia enterocolitica O:8 to pathogenicity in the mouse infection model. Infect Immun. 2004;72:5227–5234. doi: 10.1128/IAI.72.9.5227-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galyov EE, Hakansson S, Wolf-Watz H. Characterization of the operon encoding the YpkA Ser/Thr protein kinase and the YopJ protein of Yersinia pseudotuberculosis . J Bacteriol. 1994;176:4543–4548. doi: 10.1128/jb.176.15.4543-4548.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MT, et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- Young BM, Young GM. Evidence for targeting of Yop effectors by the chromosomally encoded Ysa type III secretion system of Yersinia enterocolitica . J Bacteriol. 2002;184:5563–5571. doi: 10.1128/JB.184.20.5563-5571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller JC, Carlson S, Pederson KJ, Pierson DE. A chromosomally encoded type III secretion pathway in Yersinia enterocolitica is important in virulence. Mol Microbiol. 2000;36:1436–1446. doi: 10.1046/j.1365-2958.2000.01964.x. [DOI] [PubMed] [Google Scholar]
- Venecia K, Young GM. Environmental regulation and virulence attributes of the Ysa type III secretion system of Yersinia enterocolitica biovar 1B. Infect Immun. 2005;73:5961–5977. doi: 10.1128/IAI.73.9.5961-5977.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto H, Young GM. Proteomic and functional analysis of the suite of Ysp proteins exported by the Ysa type III secretion system of Yersinia enterocolitica Biovar 1B. Mol Microbiol. 2006;59:689–706. doi: 10.1111/j.1365-2958.2005.04973.x. [DOI] [PubMed] [Google Scholar]
- Pujol C, Bliska JB. The ability to replicate in macrophages is conserved between Yersinia pestis and Yersinia pseudotuberculosis . Infect Immun. 2003;71:5892–5899. doi: 10.1128/IAI.71.10.5892-5899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Bouley DM, Falkow S. Salmonella typhimurium persists within macrophages in the mesenteric lymph nodes of chronically infected Nramp1+/+ mice and can be reactivated by IFN(gamma} neutralization. J Exp Med. 2004;199:231–241. doi: 10.1084/jem.20031319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson AJ, Uhr T. Induction of protective IgA by intestinal dendritic cells carrying commensal bacteria. Science. 2004;303:1662–1665. doi: 10.1126/science.1091334. [DOI] [PubMed] [Google Scholar]
- Crivellato E, Vacca A, Ribatti D. Setting the stage: An anatomist's view of the immune system. Trends Immunol. 2004;25:210–217. doi: 10.1016/j.it.2004.02.008. [DOI] [PubMed] [Google Scholar]
- Dumont F. Physical properties of mouse peripheral lymph node cells: Changes during development. Ann Immunol (Paris) 1977;128:1053–1063. [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- Isberg RR, Voorhis DL, Falkow S. Identification of invasin: A protein that allows enteric bacteria to penetrate cultured mammalian cells. Cell. 1987;50:769–778. doi: 10.1016/0092-8674(87)90335-7. [DOI] [PubMed] [Google Scholar]
- Isberg RR, Leong JM. Multiple beta 1 chain integrins are receptors for invasin, a protein that promotes bacterial penetration into mammalian cells. Cell. 1990;60:861–871. doi: 10.1016/0092-8674(90)90099-z. [DOI] [PubMed] [Google Scholar]
- Finkelstein LD, Reynolds PJ, Hunt SW, Shimizu Y. Structural requirements for beta1 integrin-mediated tyrosine phosphorylation in human T cells. J Immunol. 1997;159:5355–5363. [PubMed] [Google Scholar]
- Hauzenberger D, Sundqvist KG. Fibronectin at the lymphocyte surface. Evidence for activation-dependent binding to VLA4 and VLA5 integrins. Scand J Immunol. 1993;37:87–95. doi: 10.1111/j.1365-3083.1993.tb01669.x. [DOI] [PubMed] [Google Scholar]
- Marra A, Isberg RR. Invasin-dependent and invasin-independent pathways for translocation of Yersinia pseudotuberculosis across the Peyer's patch intestinal epithelium. Infect Immun. 1997;65:3412–3421. doi: 10.1128/iai.65.8.3412-3421.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krukonis ES, Dersch P, Eble JA, Isberg RR. Differential effects of integrin alpha chain mutations on invasin and natural ligand interaction. J Biol Chem. 1998;273:31837–31843. doi: 10.1074/jbc.273.48.31837. [DOI] [PubMed] [Google Scholar]
- Rosales-Reyes R, Alpuche-Aranda C, Ramirez-Aguilar Mde L, Castro-Eguiluz AD, Ortiz-Navarrete V. Survival of Salmonella enterica serovar Typhimurium within late endosomal-lysosomal compartments of B lymphocytes is associated with the inability to use the vacuolar alternative major histocompatibility complex class I antigen-processing pathway. Infect Immun. 2005;73:3937–3944. doi: 10.1128/IAI.73.7.3937-3944.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Lahrz A, Dersch P. Environmental control of invasin expression in Yersinia pseudotuberculosis is mediated by regulation of RovA, a transcriptional activator of the SlyA/Hor family. Mol Microbiol. 2001;41:1249–1269. doi: 10.1046/j.1365-2958.2001.02522.x. [DOI] [PubMed] [Google Scholar]
- Simonet M, Richard S, Berche P. Electron microscopic evidence for in vivo extracellular localization of Yersinia pseudotuberculosis harboring the pYV plasmid. Infect Immun. 1990;58:841–845. doi: 10.1128/iai.58.3.841-845.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izadjoo MJ, Polotsky Y, Mense MG, Bhattacharjee AK, Paranavitana CM, et al. Impaired control of Brucella melitensis infection in Rag1-deficient mice. Infect Immun. 2000;68:5314–5320. doi: 10.1128/iai.68.9.5314-5320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PD, Bergman MA, Mecsas J, Isberg RR. Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med. 2006;203:1591–601. doi: 10.1084/jem.20060905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Torres A, Jones-Carson J, Baumler AJ, Falkow S, Valdivia R, et al. Extraintestinal dissemination of Salmonella by CD18-expressing phagocytes. Nature. 1999;401:804–808. doi: 10.1038/44593. [DOI] [PubMed] [Google Scholar]
- Handley SA, Newberry RD, Miller VL. Yersinia enterocolitica invasin-dependent and invasin-independent mechanisms of systemic dissemination. Infect Immun. 2005;73:8453–8455. doi: 10.1128/IAI.73.12.8453-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Isberg RR. Transcriptional regulation of the Yersinia pseudotuberculosis pH6 antigen adhesin by two envelope-associated components. Mol Microbiol. 1997;24:499–510. doi: 10.1046/j.1365-2958.1997.3511719.x. [DOI] [PubMed] [Google Scholar]
- Schneider-Schaulies S, Schneider-Schaulies J, Niewiesk S, Ter Meulen V. Measles virus: Immunomodulation and cell tropism as pathogenicity determinants. Med Microbiol Immunol (Berl) 2002;191:83–87. doi: 10.1007/s00430-002-0121-6. [DOI] [PubMed] [Google Scholar]
- Van de Bovenkamp JH, Korteland-Van Male AM, Buller HA, Einerhand AW, Dekker J. The MUC5AC glycoprotein is the primary receptor for Helicobacter pylori in the human stomach. Helicobacter. 2003;8:521–532. doi: 10.1046/j.1523-5378.2003.00173.x. [DOI] [PubMed] [Google Scholar]
- Raghavan S, Holmgren J. CD4+CD25+ suppressor T cells regulate pathogen induced inflammation and disease. FEMS Immunol Med Microbiol. 2005;44:121–127. doi: 10.1016/j.femsim.2004.10.017. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Sato T, Nagasako R, Takeda I. Acute mesenteric lymphadenitis due to Yersinia pseudotuberculosis lacking a virulence plasmid. J Clin Microbiol. 1991;29:1271–1275. doi: 10.1128/jcm.29.6.1271-1275.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant T, Bennett-Wood V, Robins-Browne RM. Identification of virulence-associated characteristics in clinical isolates of Yersinia enterocolitica lacking classical virulence markers. Infect Immun. 1998;66:1113–1120. doi: 10.1128/iai.66.3.1113-1120.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian CJ, Hwang WS, Kelly JK, Pai CH. Invasiveness of Yersinia enterocolitica lacking the virulence plasmid: An in-vivo study. J Med Microbiol. 1987;24:219–226. doi: 10.1099/00222615-24-3-219. [DOI] [PubMed] [Google Scholar]
- Robins-Browne RM, Tzipori S, Gonis G, Hayes J, Withers M, et al. The pathogenesis of Yersinia enterocolitica infection in gnotobiotic piglets. J Med Microbiol. 1985;19:297–308. doi: 10.1099/00222615-19-3-297. [DOI] [PubMed] [Google Scholar]
- Bakour R, Balligand G, Laroche Y, Cornelis G, Wauters G. A simple adult-mouse test for tissue invasiveness in Yersinia enterocolitica strains of low experimental virulence. J Med Microbiol. 1985;19:237–246. doi: 10.1099/00222615-19-2-237. [DOI] [PubMed] [Google Scholar]
- Lee JH, Rhee PL, Lee JK, Lee KT, Son HJ, et al. The etiology and clinical characteristics of mesenteric adenitis in Korean adults. J Korean Med Sci. 1997;12:105–110. doi: 10.3346/jkms.1997.12.2.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler H, Dolan C, Forgacs P, George H. Brucella suis: An unusual cause of suppurative lymphadenitis in an outpatient. J Clin Microbiol. 1982;16:575–576. doi: 10.1128/jcm.16.3.575-576.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adcock PM, Nagaraj HS, Marshall GS. Pseudoappendicitis preceding infectious mononucleosis. Pediatr Emerg Care. 1998;14:345–346. [PubMed] [Google Scholar]
- Azadeh B, Sells PG, Ejeckam GC, Rampling D. Localized Leishmania lymphadenitis. Immunohistochemical studies. Am J Clin Pathol. 1994;102:11–15. doi: 10.1093/ajcp/102.1.11. [DOI] [PubMed] [Google Scholar]
- Dornand J, Gross A, Lafont V, Liautard J, Oliaro J, et al. The innate immune response against Brucella in humans. Vet Microbiol. 2002;90:383–394. doi: 10.1016/s0378-1135(02)00223-7. [DOI] [PubMed] [Google Scholar]
- Forsberg A, Wolf-Watz H. The virulence protein Yop5 of Yersinia pseudotuberculosis is regulated at transcriptional level by plasmid-plB1-encoded trans-acting elements controlled by temperature and calcium. Mol Microbiol. 1988;2:121–133. doi: 10.1111/j.1365-2958.1988.tb00013.x. [DOI] [PubMed] [Google Scholar]