

Abstract
The kinetic, thermodynamic and structural basis for fidelity of nucleic acid polymerases remains controversial. An understanding of viral RNA-dependent RNA polymerase (RdRp) fidelity has become a topic of considerable interest as a result of recent experiments that show that a two-fold increase in fidelity attenuates viral pathogenesis and a two-fold decrease in fidelity reduces viral fitness. Here we show that a conformational-change step preceding phosphoryl transfer is a key fidelity checkpoint for the poliovirus RdRp (3Dpol). We provide evidence that this conformational-change step is orientation of the triphosphate into a conformation suitable for catalysis, suggesting a kinetic and structural model for RdRp fidelity that can be extrapolated to other classes of nucleic acid polymerases. Finally, we show that a site remote from the catalytic center can control this checkpoint, which occurs at the active site. Importantly, similar connections between a remote site and the active site exist in a wide variety of viral RdRps. The capacity for sites remote from the catalytic center to alter fidelity suggests new possibilities for targeting the viral RdRp for antiviral drug development.
During each cycle of nucleotide incorporation, nucleic acid polymerases are presented with the challenge of having to select a nucleotide with the correct sugar configuration and the correct base. Fidelity of nucleotide selection is essential for maintenance of the information content and accurate expression of the genome. Importantly, some level of incorrect incorporation must exist in order to provide a mechanism for all organisms to survive challenges imposed by nature. Elucidation of the mechanisms employed by polymerases to maintain the appropriate balance between correct and incorrect nucleotide incorporation has been a primary goal of polymerase enzymology and structural biology.
The single-nucleotide-addition cycle of polymerases is likely conserved, consisting of at least five kinetically observable steps: (1) nucleotide binding; (2) a conformational change; (3) phosphodiester bond formation; (4) a second conformational change, perhaps translocation; and (5) pyrophosphate release (1–7). Theoretically, any of these steps could be employed as fidelity checkpoints. Empirically, the first three steps appear to be the primary steps governing fidelity of nucleotide addition (1–7). However, there is considerable debate regarding the capacity of the first conformational-change step to act as a fidelity checkpoint (8,9). Further complicating matters, is the absence of a clear structural explanation for the conformational change observed kinetically. Structural explanations range from inter- and intra-domain rearrangements (10–12) to triphosphate reorientation (1,7,13–17).
Studies of viral DNA-dependent DNA and RNA polymerases have been essential to developing the current state-of-the-art for polymerase structure, function and mechanism. However, until recently, very little structural and mechanistic information existed for the viral RNA-dependent RNA polymerase1 (RdRp) (7,17–22). Fidelity of RNA virus polymerases has become a topic of increased clinical importance because the capacity for RNA virus populations to escape neutralization by the immune system and to develop resistance to antiviral agents is a direct consequence of errors introduced by the polymerase during genome replication. Even more intriguing, however, is the recent realization that very subtle increases in RdRp fidelity can attenuate viral pathogenesis (23) and very subtle decreases in polymerase fidelity can force the virus into error catastrophe (lethal mutagenesis) (24).
The RdRp from poliovirus (PV), 3Dpol, has emerged as an important model system for understanding the structure, function and mechanism of this class of polymerases (7,17–22,25). However, given the structural and mechanistic similarities of 3Dpol to DNA polymerases, RNA polymerases and reverse transcriptases (7,21,22), it is likely that the structural and mechanistic basis for 3Dpol fidelity will be relevant to all classes of nucleic acid polymerases. Although it is often suggested that the RdRp has a high error rate, the RdRp error rate is actually equivalent to replicative DNA polymerases in the absence of the proofreading exonuclease (7).
In this article, we describe a PV mutant (G64S PV) that is less sensitive to an antiviral nucleoside yet more sensitive to other classes of antiviral compounds due to a mutation in 3Dpol-coding sequence that restricts genomic diversity. We show that this polymerase derivative, G64S 3Dpol, has a higher fidelity than the wild-type enzyme owing to a change in the equilibrium constant for the conformational-change step preceding the chemical step. Although position 64 is remote from the active site, the backbone of this residue is connected via hydrogen bonding to the conserved structural motif A that functions, in part, to hold the triphosphate of the incoming nucleotide in a conformation appropriate for catalysis. Importantly, similar connections between remote sites and motif A exist in polymerases from a wide variety of RNA viruses. Together, these data provide genetic evidence for a role of the first conformational-change step in polymerase fidelity, provide a link between the conformational change and the orientation of the triphosphate of the incoming nucleotide, and define sites remote from the polymerase active site as targets for development of antiviral agents capable of functioning by modulating nucleotide incorporation fidelity. The conclusions of this study regarding fidelity mechanisms employed by the RdRp are germane to all classes of nucleic acid polymerases.
Experimental Procedure
Materials
[γ-32P]ATP (> 7,000 Ci/mmol) was from ICN; [α-32P]UTP was from New England Nuclear; nucleoside 5′-triphosphates (all nucleotides were ultrapure solutions) and adenosine 5′-O-(1-thiotriphosphate) were from Amersham Pharmacia Biotech, Inc; dT15 was from Integrated DNA Technologies, Inc.; poly(rA) was from Sigma; RNA oligonucleotides were from Dharmacon Research, Inc. (Boulder, CO); T4 polynucleotide kinase was from New England Biolabs, Inc.; All other reagents were of the highest grade available from Sigma or Fisher.
Cells, viruses and plasmids
HeLa S3 cells (ATCC CCL 2.2) were grown in tissue culture flasks in Dulbecco’s modified Eagle medium-nutrient mixture F-12 (Ham) (1:1) (CellGro), supplemented with 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U of penicillin and streptomycin per ml, and 10 % newborn calf serum (Invitrogen). wild-type (WT) poliovirus type 1 Mahoney was used throughout this study. Plasmid prib(+)XpA (hereupon referred to as pXpA) contains the cDNA of poliovirus preceded by the hammerhead ribozyme and the T7 RNA polymerase promoter permitting the generation of infectious synthetic RNAs.
Construction of recombinant G64S PV clone
The codon encoding Gly-64 of the poliovirus RdRP was changed to Ser in the cDNA of WT poliovirus type 1 Mahoney in the plasmid pXpA. Specifically, nucleotides 6746-8 (GGT) were mutagenized to TCA using appropriate primers (available upon request) and the Quickchange Mutagenesis XLII kit (Stratagene). Positive clones were identified and confirmed by sequencing. The resulting plasmid pG64S, permit the transcription of infectious full-length genomes, expressing the mutant G64S polymerase.
In vitro transcription and RNA transfection
Poliovirus transcripts corresponding to full length genomic RNA (pXpA or pG64S) were obtained using a T7 Megascript transcription kit (Ambion) after linearization of cDNA plasmids. The integrity of the resulting RNAs was confirmed by gel electrophoresis. Transfection of HeLa S3 cells with these synthetic RNAs was performed with an Electro Cell Manipulator600 (BTX) following manufacturer’s directions.
Effect of ribavirin on virus titer
HeLa S3 cells were infected at an MOI of 0.1 with either passage 3 WT poliovirus (P3) or passage 3 G64S virus in the presence of 0–1000 μM ribavirin. Upon total cell lysis (CPE), virus was recovered and titered by plaque assay.
WIN compound escape mutants assay
WIN 51711 (disoxaril) compound and the assay protocol were obtained as a kind gift from Dan Pevear, ViroPharma, Inc.. Survival curves were established by plating serial 10-fold dilutions of 106 pfu/ml virus stocks on duplicate HeLa S3 cell monolayers in 6-well plates. After a 1 hour adsorption period the inoculum was removed and 3 ml of overlay medium was added containing 1.0 % agarose and different concentrations of disoxaril (10 μM, 1 μM, 0.1 μM, 0.01 μM or no drug). Percent inhibition was plotted as a function of virus titer obtained at each concentration of WIN compound over control titer obtained with mock treatment.
RNA isolation, cDNA synthesis and sequencing
Eighteen viral isolates belonging to each of the WT and G64S populations were obtained by plaque isolation. The viral isolates were then replicated on fresh HeLa cell monolayers (in 24 well plates) for 6 hours, and viral RNA was extracted and purified by Trizol extraction. RT-PCR was performed on the RNA using the Thermoscript kit (Invitrogen) and Pfu ultra fidelity polymerase (Stratagene). The RT reaction was performed using the oligo-dT primer. Two PCR products were obtained for each viral isolate, a product spanning the 5′-UTR and capsid coding region (nt 300–3300) and a product spanning 3CD and 3′-UTR (nt 5800–7441). Primers for the 5′-product were 5′-CAGAGTGTAGCTTAGGC-3′ (forward) and 5′-GGTGGACGCGGGCACC-3′ (reverse). Primers for the 3′-product were 5′-CAGGGATATCTAAATCTC-3′ (forward) and 5′-CTCCGAATTAAAGAAAAATTTACCC-3′ (reverse). The 36 PCR fragments from each population, representing 18 different viral clones of either WT or G64S, were purified using a Qiagen PCR purification kit and sequenced.
Virus competition experiment
HeLa cells were plated in 10 cm2 dishes and infected at an MOI of 10 with a virus mixture composed of passage 0 WT and G64S viruses at a ratio of 1:10. After adsorption, input virus was removed, and infection was allowed to proceed to cell lysis. Virus from the supernatants of infected cells was harvested by 3 freeze-thaw cycles and used to re-infect fresh HeLa monolayers (passages 1 to 3). At each passage, viral RNA was extracted by treatment with Trizol-HCl for RT-PCR. RT-PCR was performed as above, using the same primers that amplified the 3′-product. Direct sequencing of the PCR product was performed to determine the proportion of progeny virus belonging to the WT or G64S populations.
Construction of G64S expression vector
The G64S expression vector was constructed by introducing the G64S-encoding mutation into the WT 3Dpol gene by using overlap extension PCR with the appropriate primers (available upon request) and subcloning into the pET26-Ub-3D expression plasmid essentially as described previously (26). Mutations were confirmed by DNA sequencing (Nucleic Acid Facility, Pennsylvania State University).
Expression and purification of 3Dpol
Expression and purification of 3Dpol WT and G64S was performed as described previously (26).
Purification, 5′-32P labeling and annealing of sym/sub
RNA oligonucleotides were purified, labeled and annealed as described previously (18).
Poly(rU) polymerase specific activity assay
Reactions contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2, 0.05 μM 3Dpol, 500 μM UTP, 0.2 μCi/μL [α-32P]UTP, 5 mM MgCl2, 1.88 μM dT15 and poly(rA) (93.4 μM AMP). Reactions were initiated by 3Dpol and incubated at 30 °C for 5 min at which time the reactions were quenched by addition of EDTA to a final concentration of 50 mM. Products were analyzed by DE81 filter binding asp previously described (27).
Sym/sub assays
Reactions contained 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol, 5 mM MgCl2 or 5 mM MnCl2, 500 μM NTP, sym/sub and 3Dpol. Reactions were either quenched by addition of EDTA to a final concentration of 0.3 M or by addition of HCl to a final concentration of 1 M. Immediately after addition of HCl, the solution was neutralized by addition 1 M KOH and 300 mM Tris (final concentration). Specific concentrations of primer/template and 3Dpol, along with any deviations from the above, are indicated in the appropriate figure legend. 3Dpol was diluted immediately prior to use in: 50 mM HEPES, pH 7.5, 10 mM 2-mercaptoethanol and 20% glycerol. The volume of enzyme added to any reaction was always less than or equal to one-tenth the total volume. All reactions were performed at 30 °C.
Rapid chemical-quench-flow experiments
Rapid mixing/quenching experiments were performed by using a Model RQF-3 chemical-quench-flow apparatus (KinTek Corp., Austin, TX). Experiments were performed at 30 °C by using a circulating water bath. 3Dpol-sym/sub complexes were assembled by mixing 3Dpol and sym/sub for three minutes at room temperature and then rapidly mixed with the nucleotide substrate. After mixing, reactant concentrations were reduced by 50 %. Reactions were either quenched by addition of EDTA to a final concentration of 0.3 M or by addition of HCl to a final concentration of 1 M followed by immediate neutralization.
Determination of the kinetic parameters (kpol and Kd,app) for3Dpol-catalyzed nucleotide incorporation
3Dpol (2 μM) was incubated with sym/sub (1 μM duplex) and rapidly mixed with varying concentrations of nucleotide substrate (1/5 × Kd,app to 5 × Kd,app). At various times after mixing reactions were either quenched by addition of EDTA or HCl. Time courses at fixed nucleotide concentration were fit to (Eq.1):
where A is the amplitude of the burst, kobs is the observed first-order rate constant describing the burst, t is the time and C is a constant. The observed rate constants were plotted as a function of nucleotide concentration and the data fit to (Eq. 2):
where kpol is the maximal observed rate constant for nucleotide incorporation and Kd,app is the apparent dissociation constant. Data were fit by non-linear regression using the program, KaleidaGraph (Synergy Software, Reading, PA).
Kinetic simulation
Kinetic simulations were performed by using KinTekSim Version 2.03 (KinTek Corp., Austin, TX). Schemes employed are provided as figures. All rate constants were determined experimentally, except where noted. The agreement between the experimental data and kinetic simulations was determined by visual inspection.
RESULTS
G64S PV population has reduced genomic diversity
The genomic diversity of RNA virus populations permits rapid adaptation of the population to environmental stresses. Maximal adaptability requires maximal genomic diversity. However, the consequence of maximal genomic diversity is susceptibility to extinction under conditions in which a subtle increase in mutation frequency occurs. This mutation-induced extinction has been termed lethal mutagenesis, a promising broad-spectrum antiviral strategy (28–33). Studies with PV have shown that the mechanism of action of ribavirin, a clinically useful antiviral nucleoside, is lethal mutagenesis (24). The PV RdRp can incorporate ribavirin triphosphate ambiguously into the genome, forcing the virus population into error catastrophe (24). One mechanism of resistance to nucleoside-based mutagens is enhanced RdRp fidelity. Isolation of such a mutant would provide significant insight not only into the importance of genomic diversity for viral infectivity and pathogenesis but also into mechanisms of fidelity employed by polymerases.
Serial passage of PV in the presence of ribavirin yields a mutant with reduced sensitivity to ribavirin (23,34). This mutant contains a single nucleotide substitution in 3Dpol-coding sequence that converts Gly-64 to Ser. A recombinant G64S PV was constructed to preclude genetic reversion. As shown in Fig. 1A, in the absence of ribavirin, this virus grows to titers equivalent to WT PV, suggesting that this mutation does not alter the fitness of the virus in tissue culture. As the concentration of ribavirin is increased, G64S PV exhibits reduced sensitivity to the drug (Fig. 1A).
Figure 1. Biological analysis of G64S PV.
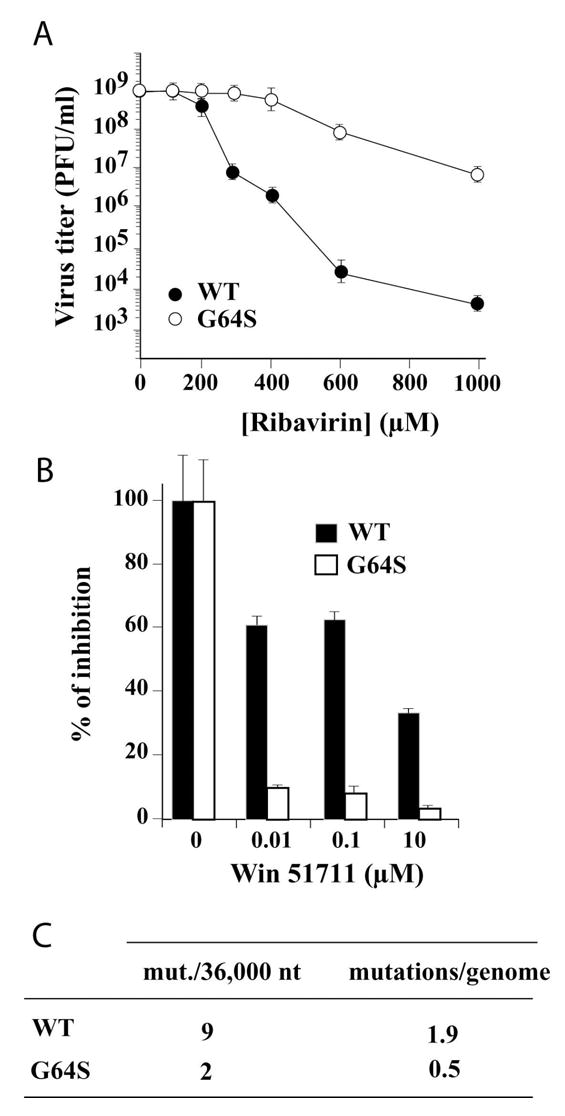
(A) The G64S substitution confers resistance to ribavirin. Titer of surviving virus (pfu/ml) at 20 hours post-infection for WT PV (•) and G64S PV (○) in the presence of increasing concentrations of ribavirin. (B) The G64S PV population contains a reduced number of drug-resistant variants. Percentage of inhibition for WT PV (▪) and G64S PV (□) in the presence of increasing concentrations of WIN 51711 (disoxaril). Error bars indicate variance between two separate experiments. (C) The G64S virus population exhibits a reduced mutation frequency. Total number of mutations observed per 36,000 nucleotides sequenced and the calculated average number of mutations per genome. The mutation frequencies of the virus populations were found to be statistically different (p<0.04799) by Mann-Whitney U test.
PV and other entero- and rhinoviruses in the Picornavirus family are sensitive to a class of inhibitors, commonly called WIN compounds, that inhibit virus uncoating by binding to the mature virus capsid and preventing conformational changes required for uncoating (35–37). Resistance to some of these compounds, for example WIN 51711 (disoxaril), rapidly emerges owing to the existence of drug-resistant variants in the virus population (36,37). WT PV resists challenge by WIN 51711 over a wide concentration range (Fig. 1B), likely reflecting drug-resistant mutants in the population. If G64S PV encodes a polymerase with enhanced fidelity, then fewer drug-resistant variants should be present in this population, conferring enhanced sensitivity of G64S PV to WIN 51711. As shown in Fig. 1B, G64S PV is more sensitive to WIN 51711. In order to confirm that the diversity of the G64S PV population was reduced relative to WT PV, we sequenced 36,000 nt of capsid-coding sequence. The G64S PV population contains 4-fold fewer mutations per genome than the WT PV population (Fig. 1C). These results are consistent with enhanced fidelity of G64S 3Dpol mediating the reduced sensitivity of G64S PV to ribavirin.
Biochemical defects associated with G64S 3Dpol
We expressed and purified G64S 3Dpol and compared its basic biochemical properties to WT 3Dpol (26). At temperatures greater than 4 °C, WT 3Dpol has a finite lifetime in solution in the absence of nucleotide and nucleic acid (Fig. 2A) (18). The observed rate constant for inactivation of G64S 3Dpol at 30 °C in the presence of 500 μM ATP is 9-fold faster than WT 3Dpol (Fig. 2A). The faster rate of inactivation of G64S 3Dpol leads to a 15-fold reduction in the specific activity of this derivative relative to WT enzyme as determined by using a poly(rU) polymerase activity assay (Fig. 2B). Product formed by using this assay reflects template-switching activity of the few complexes that assemble productively on this primer-template substrate (27). An increase in the rate constant for inactivation will cause a proportional reduction in complexes competent for primer extension on these large primer-template substrates (18).
Figure 2. G64S 3Dpol has a faster inactivation rate that leads to reduced activity on homopolymeric substrates and reduced assembly on a heteropolymeric primer-template substrate.
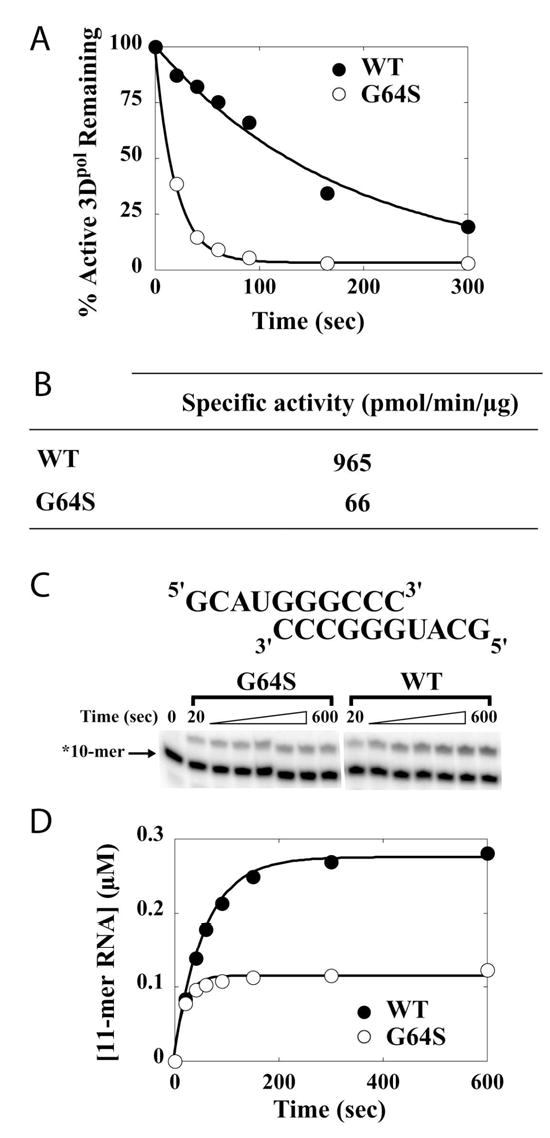
(A) Inactivation of 3Dpol. Percentage of active WT 3Dpol (•) and G64S 3Dpol (○) remaining plotted as a function of time. The solid lines represent the fit of the data to a single exponential with a kinact(obs) of 5.6 ± 0.7 × 10−3 s−1 for WT 3Dpol and 5.1 ± 0.2 × 10−2 s−1 for G64S 3Dpol. 3Dpol (2 μM) was incubated in 1X reaction buffer with ATP (500 μM) at 30 oC and at the indicated times the reaction was initiated by addition of sym/sub (0.5 μM duplex) and allowed to proceed for an additional 90 sec at which point the reaction was quenched by addition of EDTA. (B) Specific activity (pmol/min/ug) for WT 3Dpol and G64S 3Dpol on oligo(dT)-primed poly(rA). Reactions were performed as described in Experimental Procedures. (C) Primer/template (sym/sub-U) employed. Substrate (10-mer) can be resolved from product(s) by denaturing PAGE and visualized by phosphorimaging. (D) WT 3Dpol and G64S 3Dpol assemble on and utilize sym/sub-U. Kinetics of assembly on and utilization of sym/sub-U by WT 3Dpol (•) and G64S 3Dpol (○). Product (11-mer) formed in panel C plotted as a function of time. The solid lines represent the fit of the data to a single exponential with an observed rate constant of 0.017 ± 0.001 s−1 and an amplitude of 0.27 ± 0.01 μM for WT 3Dpol and an observed rate constant of 0.050 ± 0.007 s−1 and an amplitude of 0.11 ± 0.01 μM for G64S 3Dpol. Reactions contained 3Dpol (1 μM), sym/sub (0.5 μM duplex) and ATP (500 μM). Reactions were initiated by 3Dpol.
We have developed a synthetic primer-template substrate for evaluation of the kinetics and mechanism of 3Dpol-catalyzed nucleotide incorporation (18). This substrate, referred to as sym/sub-U (Fig. 2C), is a 10-nt self-complimentary RNA that forms a 6-bp duplex and two 4-nt 5′-overhangs capable of templating correct incorporation of each of the four ribonucleotides. The “U” designation identifies the first templating base of the substrate. Products can be resolved from substrate by electrophoresis through denaturing polyacrylamide gels, detected by phosphorimaging and quantitated (Fig. 2C). Assembly of 3Dpol onto sym/sub-U is slow, and the yield of assembled complex is modulated by thermal inactivation (18). The rate constant for assembly of G64S 3Dpol onto sym/sub-U is 3-fold faster than WT 3Dpol; however, the yield of assembled complexes is 2.5-fold lower than WT 3Dpol (Fig. 2D). These biochemical defects were not predicted by the biological data which showed that G64S PV was capable of growing to titers equivalent to WT PV (Fig. 1A).
G64S PV exhibits reduced fitness relative to WT PV
The observation that G64S 3Dpol was not as robust as WT 3Dpol in vitro suggested the possibility that fitness defects associated with G64S PV had been overlooked by evaluating this virus in isolation. Therefore, we created a mixed viral population consisting of WT PV and G64S PV at a ratio of 1:10. The mixed population was used to infect HeLa cells and serially passaged. The ratio of each virus present at each stage of the experiment was determined by sequencing. After the first passage, G64S PV represented less than 20% of the virus population (Fig 3). After the second passage, G64S PV is barely detectable (Fig. 3). The capacity of WT PV to outcompete G64S PV so efficiently shows that G64S PV has a substantially reduced fitness relative to WT PV.
Figure 3. WT PV outcompetes G64S PV.
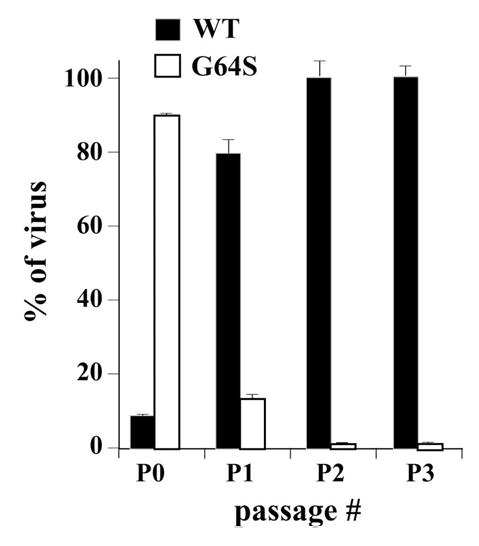
Percent of WT PV (▪) and G64S PV (□) remaining after 0–3 serial passages. The initial virus mixture contained a ratio of WT PV:G64S PV of 1:10.
G64S 3Dpol exhibits enhanced fidelity of nucleotide incorporation
In order to determine whether the incorporation fidelity of G64S 3Dpol is greater than WT 3Dpol, elongation complexes must be evaluated. Although the yield of assembled G64S-3Dpol-sym/sub-U complexes is reduced by 2.5-fold relative to WT 3Dpol (Fig. 2D), this value could be increased to within 80% of the value measured for WT 3Dpol by increasing the concentration of sym/sub-U employed (y-intercept in Fig. 4A). The rate-limiting step for single nucleotide incorporation under steady-state conditions is dissociation of polymerase from sym/sub-U product complex (18). WT 3Dpol dissociates faster from the end of template (use of all four NTPs) than from internal positions (use of ATP only) (Fig. 4A). G64S 3Dpol was competent for single and multiple cycles of nucleotide incorporation, and the corresponding steady-state rate constants for this enzyme were equivalent to WT 3Dpol (Fig. 4A).
Figure 4. Assembled G64S 3Dpol-sym/sub complexes are stable and competent for nucleotide incorporation.

(A) Steady-state incorporation of single (○) and multiple (•) nucleotides by WT 3Dpol and G64S 3Dpol. The solid lines represent the fit of the data to a line. The y-intercept is 0.88 ± 0.02 μM for AMP incorporation and 0.86 ± 0.06 μM for NMP incorporation by WT 3Dpol; The corresponding values for kcat (slope/y-int) are 1.4 ± 0.1 × 10−4 s−1 for AMP incorporation and 1.2 ± 0.1 × 10−3 s−1 for NMP incorporation by WT 3Dpol. The y-intercept is 0.68 ± 0.01 μM for AMP incorporation and 0.74 ± 0.04 μM for NMP incorporation by G64S 3Dpol; The corresponding values for kcat (slope/y-int) are 1.3 ± 0.1 × 10−4 s−1 for AMP incorporation and 7.7 ± 0.4 × 10−4 s−1 for NMP incorporation by G64S 3Dpol. Reactions contained sym/sub (10 μM duplex), 3Dpol (1 μM) and either ATP (500 μM) or ATP, CTP, GTP and UTP (500 μM each). Reactions were initiated by addition of 3Dpol and quenched at the indicated times by EDTA. (B) Stability of G64S 3Dpol-sym/sub complexes. Experimental design and kinetics of dissociation of 3Dpol-sym/sub complexes for WT 3Dpol (•) and G64S 3Dpol (○). The percentage of complex remaining competent for nucleotide incorporation is plotted as a function of time. The solid lines represent the fit of the data to a single exponential with a kdiss(obs) of 5.0 ± 0.6 × 10−4 s−1 for WT 3Dpol and 7.0 ± 0.8 10−4 s−1 for G64S 3Dpol. 3Dpol (1 μM) was incubated with sym/sub (0.5 μM duplex) for 90 sec at which point trap, unlabeled sym/sub (100 μM duplex), was added to the reaction. At fixed times after the addition of trap, ATP (500 μM) was added, and the reaction allowed to proceed for 30 sec and then quenched by addition of EDTA.
Because the steady-state analysis does not measure dissociation directly and provides information on the 3Dpol-sym/sub-UA product complex rather than the 3Dpol-sym/sub-U substrate complex, we performed the experiment shown in Fig. 4B. G64S or WT 3Dpol was mixed with labeled sym/sub-U for 90 sec to permit complexes to form. Excess unlabeled sym/sub-U was added as a trap for free and dissociating enzyme. ATP was added at various times after addition of trap, and the reaction was quenched 30 sec later. The amount of complex remaining was determined by determination of the amount of 11-mer product formed. The rate constant for dissociation was determined by fitting the plot of the percentage of complex remaining as a function of time to a single exponential decay. The rate constants for dissociation are equivalent. Consistent with previous results, the stability of the 3Dpol-sym/sub-U substrate complex is ~ 4-fold less stable than the 3Dpol-sym/sub-UA product complex (18).
We evaluated the ATP concentration dependence of AMP incorporation into sym/sub-U for the G64S and WT enzymes. The results of this analysis are shown in Table 1. The apparent dissociation constant (Kd,app) for ATP is the same for both enzymes. In contrast, there is an ~ 3-fold reduction in the maximal observed rate constant for nucleotide incorporation (kpol) measured for the G64S enzyme relative to WT 3Dpol. Importantly, the overall efficiency of RMP incorporation into sym/sub-U by G64S 3Dpol is reduced by 5-fold relative to WT 3Dpol (Table 1). Again, this difference is due to a reduction in the observed rate constant for RMP incorporation without any significant change in the value for the apparent dissociation constant for RTP. Similarly, the overall efficiency of GMP incorporation into sym/sub-U by G64S 3Dpol is reduced by 4-fold. We conclude that G64S 3Dpol has an increase in fidelity relative to WT 3Dpol when nucleotides lacking canonical basepairing capacity are employed.
Table 1.
G64S 3Dpol has a higher fidelity than WT 3Dpol in vitroa
| Nucleotide | Enzyme | Kd,app (μM) | kpol (s−1) | kpol/Kd,app (μM−1 s−1) | (kpol/Kd,app)WT/(kpol/Kd,app)G64S |
|---|---|---|---|---|---|
| ATP | WT | 130 ± 20 | 90 ± 5 | 0.65 | |
| G64S | 160 ± 10 | 30 ± 5 | 0.20 | 3 | |
| RTP | WT | 390 ± 40 | 1.0 ± 0.2 × 10−2 | 2.8 × 10−5 | |
| G64S | 370 ± 40 | 2.0 ± 0.2 × 10−3 | 5.7 × 10−6 | 5 | |
| GTP | WT | 330 ± 50 | 1.5 ± 0.2 × 10−2 | 4.5 × 10−5 | |
| G64S | 320 ± 50 | 3.5 ± 0.2 × 10−3 | 1.1 × 10−5 | 4 |
Experiments were performed as described in Experimental Procedures.
In order to rule out rate-limiting steps after incorporation of ribavirin as an additional contributor to the decreased sensitivity of G64S PV to ribavirin, we evaluated the kinetics of incorporation of UMP after RMP incorporation by G64S 3Dpol and compared these data to those obtained for WT 3Dpol (Fig. 5). UMP incorporation into sym/sub-U is undetectable in the absence of ribavirin, but the kinetics of UMP incorporation are identical to the kinetics of RMP incorporation when both RTP and UTP are present in the reaction, suggesting that translocation does not become a rate-limiting step after RMP incorporation by G64S or WT 3Dpol.
Figure 5. G64S 3Dpol is not blocked for elongation after incorporation of ribavirin.

(A) Primer/template (sym/sub-U) employed. (B) Experimental design. 3Dpol (2 μM) was mixed with sym/sub (1 μM duplex) for 3 min to assemble 3Dpol-sym/sub complexes after which RTP (1 mM) or RTP (1 mM) and UTP (10 μM) was added to the reaction. At fixed times after the addition of nucleotide the reaction was quenched by addition of EDTA. (C) Denaturing PAGE of the 32P-labeled products from WT 3Dpol-catalyzed RMP incorporation into sym/sub-U. (D) Denaturing PAGE of the 32P-labeled products from G64S 3Dpol-catalyzed RMP incorporation into sym/sub-U.
The final mechanism explored to explain the decreased sensitivity of G64S PV to ribavirin was that G64S 3Dpol exhibited biased incorporation opposite RMP in a template relative to wild-type 3Dpol. In order to test this possibility, we employed sym/sub-UR, which contains RMP as the second templating nucleotide (Fig. 6A). Elongation complexes were assembled by mixing 3Dpol, sym/sub-UR and ATP for 3 min to yield an 11-mer product complex. Next, CTP or UTP was added for incorporation opposite RMP, and GTP was also present in this mixture to evaluate elongation of a C:R or U:R basepair (Fig. 6B). When CTP and GTP are employed, a 14-mer product should accumulate; when UTP and GTP are employed, a 13-mer product should accumulate. The expected products accumulate in reactions catalyzed by either WT 3Dpol (Fig. 6C) or G64S 3Dpol (Fig. 6D). Relative to WT 3Dpol, G64S 3Dpol shows a 2-fold decrease in the observed rate of CMP (compare Fig. 6E to Fig. 6F) and UMP (compare Fig. 6G to 6h) incorporation opposite RMP as well as a 2-fold decrease in the observed rate of GMP incorporation after the C:R or U:R basepair (Figs. 6g–6h). We conclude that biased incorporation of CMP or UMP opposite RMP in the template does not contribute to the increased tolerance of ribavirin by G64S PV
Figure 6. G64S 3Dpol does not exhibit biased incorporation opposite ribavirin in the template.
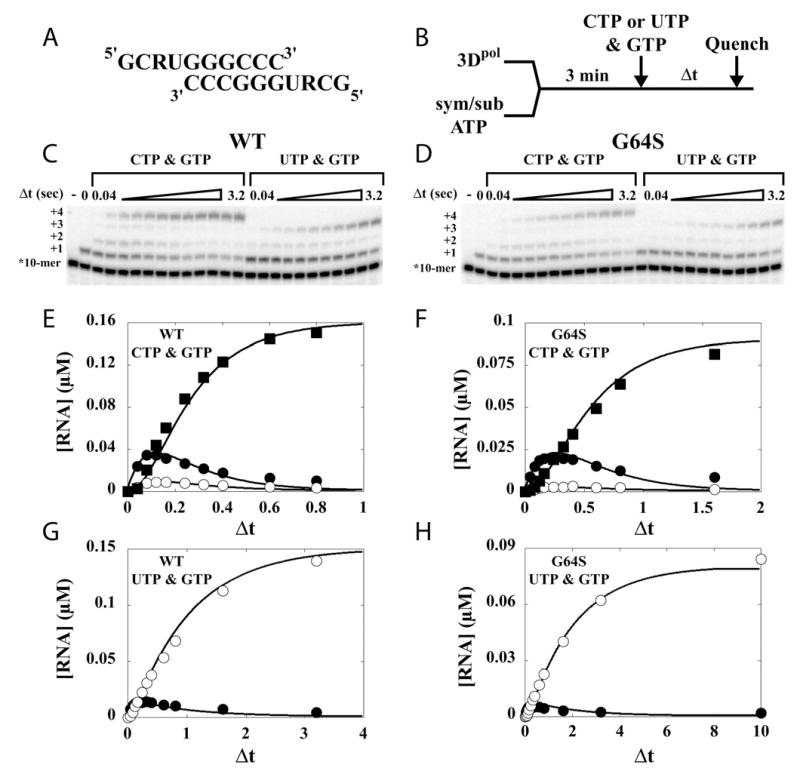
(A) Primer/template (sym/sub-UR) sequences. (B) Experimental design. 3Dpol (2 μM) was mixed with sym/sub (1 μM duplex) and ATP (5 μM) for 3 min to assemble 3Dpol-sym/sub product complexes after which CTP (1 mM) and GTP (500 μM) or UTP (1 mM) and GTP (500 μM) was added to the reaction. At fixed times after the addition of nucleotide the reaction was quenched by addition of EDTA. (C) Denaturing PAGE of the 32P-labeled products from WT 3Dpol-catalyzed nucleotide incorporation into sym/sub-UR. (D) Denaturing PAGE of the 32P-labeled products from G64S 3Dpol-catalyzed nucleotide incorporation into sym/sub-UR. (E) Kinetics of formation and disappearance of 12-mer (5 s−1), 13-mer (12 s−1) and 14-mer (50 s−1) by WT with CTP and GTP. (F) Kinetics of formation and disappearance of 12-mer (2.5 s−1), 13-mer (5 s−1) and 14-mer (50 s−1) by G64S with CTP and GTP. (G) Kinetics of formation and disappearance of 12-mer (1 s−1) and 13-mer (10 s−1) by WT with UTP and GTP. (H) Kinetics of formation and disappearance of 12-mer (0.5 s−1) and 13-mer (5 s−1) by G64S with UTP and GTP.
G64S 3Dpol exhibits a decrease in the equilibrium constant for the conformational-change step preceding phosphoryl transfer
The kinetics of AMP incorporation into sym/sub-U follows the five-step kinetic mechanism shown in Fig. 7A (7). ATP binds to the 3Dpol-sym/sub-U complex, followed by an isomerization step to form the catalytically competent complex. Catalysis occurs followed by a second isomerization step and pyrophosphate release. The observed reduction in the kpol values for G64S 3Dpol could therefore be a reflection of changes in either step 2 or step 3.
Figure 7. The conformational-change step preceding phosphoryl transfer is a fidelity checkpoint.

(A) Complete kinetic mechanism for WT 3Dpol-catalyzed nucleotide incorporation in the presence of Mg2+. ERn (3Dpol-sym/sub elongation complex); NTP (nucleotide); ERnNTP (ternary complex); *ERnNTP (activated ternary complex); *ERn+1PPi (activated product complex); ERn+1PPi (product complex); ERn+1 (translocated 3Dpol-sym/sub product complex); PPi (pyrophosphate). (B) AMP incorporation into sym/sub-U by WT 3Dpol (•) and G64S 3Dpol (○) in the presence of Mn2+. The solid lines represent the fit of the data to a single exponential with a kobs of 27 ± 2 s−1 for WT 3Dpol and 19 ± 1 s−1 for G64S 3Dpol. 3Dpol (2 μM) was incubated with sym/sub (1 μM duplex) and rapidly mixed with ATP (1 mM) as described in Experimental Procedures. (C) Partial kinetic mechanism employed for simulation of the data shown below in panel D. (D) Accumulation of the activated ternary complex prior to phosphoryl transfer. Kinetics of AMP incorporation by WT 3Dpol and G64S 3Dpol in the presence of Mn2+ quenched by using either EDTA (•) or HCl (○). The solid line represents the simulation of the data to the mechanism shown in panel C with values for Kconf and kchem shown in Table 2. EDTA-quench data reflects *ERnNTP and *ERn+1PPi; HCl-quench data reflects *ERn+1PPi only. 3Dpol (2 μM) was incubated with sym/sub (1 μM duplex) and rapidly mixed with ATP (1 mM) as described in Experimental Procedures.
In the presence of Mg2+, both steps 2 and 3 are partially rate limiting for nucleotide incorporation for WT 3Dpol (Fig. 7A); however, in Mn2+ step 3 is rate limiting (7,17). We evaluated the kinetics of AMP incorporation into sym/sub-U in the presence of Mn2+ (Fig. 7B). The values for the rate constant for AMP incorporation for both enzymes are now within 30% of each other (Fig. 7B). This observation points to step 2 as the primary determinant for the decrease in the kpol value for G64S in the presence of Mg2+.
In order to obtain additional support for the conclusion that step 2 is affected in the G64S derivative, we performed two additional experiments. First, we evaluated the phosphorothioate effect – that is, the efficiency of AMPαS incorporation into sym/sub-U relative to AMP incorporation into sym/sub-U. If the conformational change is more rate limiting for the G64S enzyme, then the value of kobs,ATP/kobs,ATPαS (Eobs) should decrease for this enzyme relative to wild-type 3Dpol (7). In the presence of Mg2+, the phosphorothioate effect for G64S is 3.2 ± 0.2 while that for WT is 4.2 ± 0.4. In the presence of Mn2+, the values for the phosphorothioate effect are equivalent (Table 2). The elevated value of the phosphorothioate effect in the presence of Mn2+ is consistent with chemistry as the rate-limiting step for nucleotide incorporation (17).
Table 2.
The chemical step is not affected by changing Gly-64 to Ser.
Kconf and kchem were determined by kinetic simulation of the data shown in Fig. 7D to the mechanism shown in Fig. 7C.
The observed phosphorothioate effect is calculated using (kobs)ATP/(kobs)ATPαS. The observed rate constant for nucleotide incorporation was determined at a concentration of ATP (1 mM) or ATPαS (1 mM) in the presence of Mn2+.
The second experiment performed exploited the observation that reactions performed in the presence of Mn2+ yield information on the concentration of the isomerized ternary complex and product complex when quenched by using EDTA and yield information only on the product complex when quenched by using HCl (17). These data yield results similar to isotope-trapping experiments and can yield values for the equilibrium constant across the conformational-change step preceding phosphoryl transfer (Kconf) and the rate constant for phosphoryl transfer (kchem) by using computer simulation to fit the data to the mechanism shown in Fig. 7C. As shown in Fig. 7D, the amplitude of the first phase of reactions quenched by using EDTA is greater for WT 3Dpol than for G64S 3Dpol, suggesting a decrease in the value of Kconf for the G64S enzyme. By simulating the data shown in Fig. 7D to the mechanism shown in Fig. 7C, we obtain a value of 1 for Kconf (Table 2) for G64S 3Dpol. This value is 3-fold lower than obtained for WT 3Dpol (Table 2) (17). Taken together, our kinetic analysis of correct nucleotide incorporation catalyzed by the G64S 3Dpol suggests that the 3-fold reduction in the observed rate constant for nucleotide incorporation for this derivative relative to WT 3Dpol is caused by a 3-fold reduction in the equilibrium constant across the conformational-change step preceding phosphoryl transfer.
DISCUSSION
PV exists as a quasispecies with each viral genome containing 1–2 transition mutations introduced by the viral RdRp, 3Dpol, during genome replication as a result of the intrinsic error rate of the polymerase (28). The quasispecies nature of poliovirus, and RNA viruses in general, likely prevents viral extinction when the viral population is challenged by antiviral mechanisms of the host. A consequence of the existence as a quasispecies is the enhanced sensitivity of the viral population to accumulation of additional mutations (28–33). Therefore, agents that increase the mutation frequency of the virus should be effective antivirals. This prediction was not only confirmed but also given clinical significance by studies of ribavirin that showed that the mechanism of action of this antiviral is lethal mutagenesis (24). Ribavirin triphosphate (RTP) is a substrate for 3Dpol and RMP is incorporated opposite both cytidine and uridine in RNA templates (24).
In our initial report describing the mutagenic activity of ribavirin, we suggested that resistance to lethal mutagens would require a more faithful polymerase, which would not likely emerge given the importance of a viral quasispecies for virus survival (24). Nevertheless, we endeavored to isolate such a mutant because of the insight this mutant could provide on the biological and biochemical consequences of enhanced RdRp fidelity and the kinetic and structural basis for polymerase fidelity. We isolated a PV mutant with a single missense mutation in 3Dpol-coding sequence that changed Gly-64 to Ser. G64S PV was also isolated and reported earlier by Pfeiffer and Kirkegaard (34). That study demonstrated reduced sensitivity of G64S PV to ribavirin and other antiviral nucleosides in tissue culture as well as a decrease in guanidine-resistant variants in the G64S virus population.
G64S PV is, indeed, less sensitive to ribavirin (Fig. 1A). However, this virus population is more sensitive to inhibitors of uncoating (Fig. 1B). The enhanced sensitivity of G64S PV to drugs that target the capsid is likely a reflection of the enhanced fidelity of G64S 3Dpol and the corresponding reduction of drug-resistant variants in the population. This conclusion is supported by direct sequencing of capsid-coding sequence, which reveals a four-fold reduction in mutation frequency (Fig. 1C). We have gone on to show that this reduced genomic diversity of G64S PV leads to fitness decreases, restricted tissue tropism and attenuated pathogenesis by using a mouse model for PV infection (23).
Our initial biochemical experiments revealed enhanced thermal inactivation of 3Dpol in vitro (Fig. 2A) that leads to reduced poly(rU) polymerase activity (Fig. 2B) and a reduced yield of stable elongation complexes on a synthetic primer-template substrate (Fig. 2C). Such substantial defects in the biochemical properties of G64S 3Dpol were unexpected because G64S PV grows to essentially the same titers as WT PV in the absence of selection (Fig. 1A). It is possible that the temperature sensitivity of the mature G64S 3Dpol is not present in the context of the precursor used to establish genome-replication complexes in cells. However, these biochemical differences prompted a direct evaluation of the fitness of G64S PV relative to WT PV by performing a competition experiment in tissue culture. Surprisingly, WT PV outcompeted G64S PV after the first passage under conditions in which the ratio of WT PV to G64S PV was 1:1000 (Fig. 3).
Emergence of drug-resistance to an antiviral therapy is a major obstacle in the clinic. Therefore, the finding that PV can develop resistance to ribavirin suggested that lethal mutagenesis may not be as effective as suggested by previous studies (24,28–30). However, the finding that two different groups isolated the same ribavirin-resistant mutant suggests that very few solutions exist to the problem of resistance to ribavirin and, perhaps, other lethal mutagens. Indeed, as indicated by the discussion above, G64S PV is one, not-so-good solution. This lethal-mutagen-resistant virus is less fit and more sensitive to other drugs than WT PV, suggesting that application of lethal mutagens in combination with other drugs should prevent rapid development of resistance in a clinical setting. We conclude that lethal mutagenesis remains a promising antiviral strategy with limited potential for development of resistance.
Once assembled, G64S 3Dpol-primer/template complexes are as stable as complexes formed with WT 3Dpol (Fig. 4). Defects to assembly can be overcome by increasing the concentration of primer-template substrate employed (Fig. 4A). Assembled complexes support fast rates of nucleotide incorporation (Table 1) and are competent for multiple cycles of nucleotide addition (Fig. 4A and Fig. 5). Nucleotide binding by G64S 3Dpol is equivalent to wild-type 3Dpol (Table 1). Therefore, the only significant defect to the elongation complex is a 3-fold reduction in the observed rate constant (kpol) for nucleotide incorporation (Table 1).
Two steps in the kinetic mechanism dictate the observed rate constant for nucleotide incorporation when Mg2+ is employed as the divalent cation cofactor: a conformational-change step preceding phosphoryl transfer (referred to as step 2) and phosphoryl transfer (referred to as step 3) (7). Because phosphoryl transfer is the sole rate-limiting step when Mn2+ is employed as the divalent cation cofactor (17), the observation that the observed rate constant for G64S 3Dpol-catalyzed nucleotide incorporation in Mn2+ was 70% of the value obtained for wild-type 3Dpol in Mn2+ (Fig. 7D) suggested that step 2 was affected most by the G64S substitution. More thorough evaluation of this step showed that the value for the equilibrium constant across the conformational-change step (Kconf) for G64S 3Dpol is, indeed, reduced by 3-fold relative to WT 3Dpol (Table 2 and Fig. 7D).
The destabilization of the catalytically competent ternary complex appeared to be exacerbated by employing nucleotide substrates incapable of forming a canonical basepairing geometry, for example R:U and G:U basepairs, based upon the reductions in the observed rate constants for RTP and GTP utilization (Table 1). The reduction in the observed rate constant for RMP incorporation into sym/sub-U is clearly related to Kconf for G64S 3Dpol as this defect could not be rescued by using Mn2+ as the divalent cation cofactor (data not shown). There is an ongoing debate regarding the importance of step 2 for polymerase fidelity (8,9). The ability to show that an amino acid substitution in 3Dpol that increases polymerase fidelity causes a change in the value for Kconf provides an indisputable link between step 2 and fidelity.
In addition to the controversy surrounding step 2 and fidelity, there has been some debate regarding the molecular basis of step 2 (15,16). Crystal structures of polymerase ternary complexes have been used to argue that step 2 reflects interdomain rearrangements that convert the catalytic site from an “open” to a “closed” conformation in response to nucleotide binding (10–12). More recently, additional rearrangements have been observed in DNA and RNA polymerase structures after formation of the "closed" conformation that permit the triphosphate moiety of the incoming nucleotide to achieve a catalytically-competent conformation (16,38,39). For the RdRp, we have argued that step 2 reflects reorientation of the triphosphate into a catalytically-competent conformation (7), a process that would occur after formation of the closed complex. This interpretation creates an inextricable link between step 2 and the observed rate constant for phosphoryl transfer. Destabilization of the active conformation of the triphosphate would reduce the frequency of molecules in an optimal position for attack by the 3′-OH of the primer terminus.
The active conformation of the triphosphate of all polymerases relies on interactions between the phosphate and two divalent cations, in most cases magnesium ions. The divalent cations are also coordinated by carboxylates originating from aspartic acid residues conserved in all polymerases. An established model of this interaction for 3Dpol is shown in Fig. 8A (19,20). Structural motif A consists of residues 233–241. Asp-233 is a ligand to the metal, Asp-238 interacts with the ribose of the incoming nucleotide and the backbone in between these residues participates in hydrogen bonding interactions that stabilize the active conformation of the triphosphate. Therefore, perturbations of motif A should alter the equilibrium position of the triphosphate. Based on the discussion above, a change in the equilibrium position of the triphosphate should be manifested as a change in Kconf for step 2.
Figure 8. A link between the fingers subdomain and conserved structural motif A of the palm subdomain of PV 3Dpol.
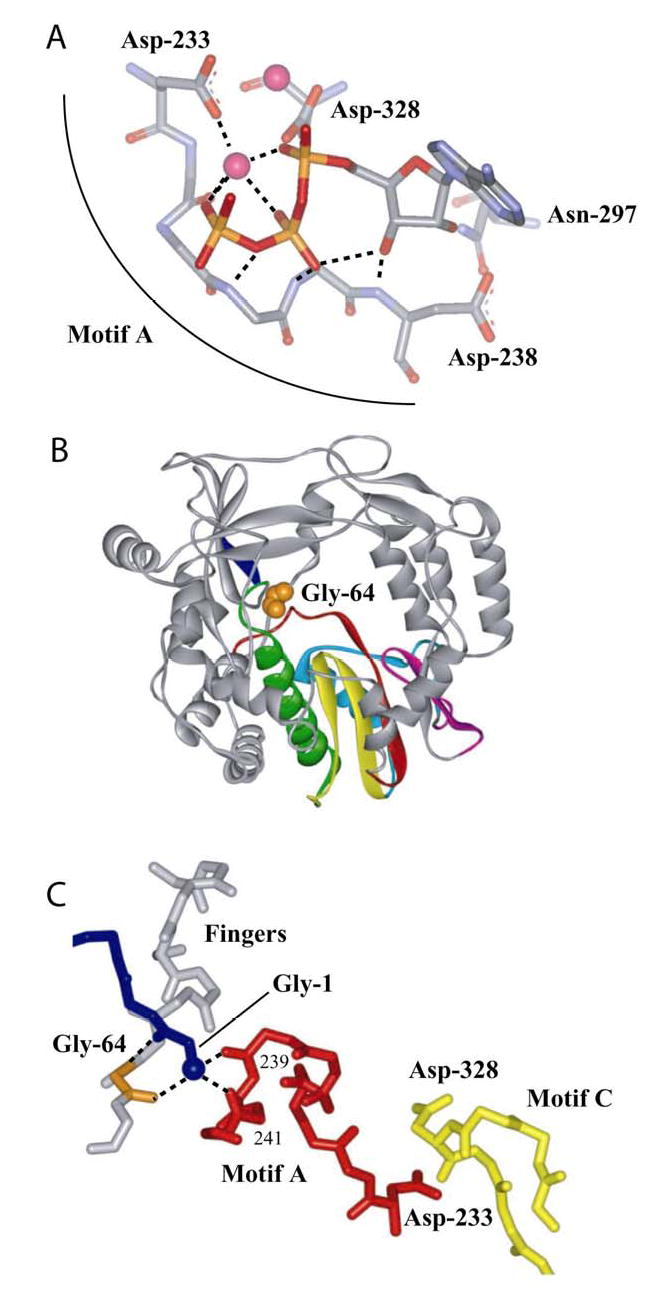
(A) Motif A functions, in part, to orient the triphosphate of the incoming nucleotide for catalysis. Model for interaction of 3Dpol with bound nucleotide (19,20). ATP and metal ions required for catalysis are labeled. In this model, the side chains for Asp-233 and Asp-238 have been rotated to permit interactions with ATP. (B) Gly-64 is located in the fingers subdomain. Complete structure for PV 3Dpol (22). The conserved structural motifs in the palm subdomain are colored as follows: motif A, red; motif B, green; motif C, yellow; motif D, blue; and motif E, purple. Van der Waal’s projection of Gly-64 (orange). (C) Gly-64 backbone orients the amino terminus, and the amino terminus interacts with motif A. The amino terminus of 3Dpol is in blue, Gly-64 is in orange, motif A is in red, motif C is in yellow and hydrogen bonds are shown as dashed lines. Misalignment of position 64 will cause defects to the orientation of the triphosphate as well as the efficiency of phosphoryl transfer owing to misalignment of motif A.
Gly-64 is located in the fingers domain (Fig. 8B). At first glance, it would appear that this residue is remote from the catalytic center. However, upon inspection of the structure for 3Dpol (22), it becomes clear that a direct link exists between position 64 and motif A. The backbone of Gly-64 is hydrogen bonded to the amino terminus, which, in turn, is hydrogen bonded to the backbone of residues Ala-239 and Leu-241 of motif A (Fig. 8C). In order for the backbone of Gly-64 to achieve interactions with the amino terminus, this residue has to assume a conformation that is disallowed in the Ramachandran plot (22). Therefore, Ser at this position would preclude an optimal interaction between the position-64 backbone and the amino terminus with negative consequences on the position of motif A and stabilization of the triphosphate. We conclude that the conformational change preceding chemistry is orientation (k+conf) and stabilization (k−conf) of the triphosphate in the active conformation and serves as the first fidelity checkpoint in the kinetic mechanism for RdRp-catalyzed nucleotide incorporation.
The use of triphosphate reorientation and stabilization of the isomerized triphosphate as a fidelity checkpoint can also be extrapolated to DNA-dependent DNA and RNA polymerases. Structural data available for DNA polymerase I from Bacillus stearothermophilus (BF) (Fig. 9A) and the RNA polymerase from bacteriophage T7 (T7 Rp) (Fig. 9B) show similar rearrangements of the triphosphate during a nucleotide addition cycle (16,38,39). In both cases, there is an initial interaction of the triphosphate with helix O that collapses into the active conformation. The active conformation is characterized by an interaction of the triphosphate with a divalent cation, which, in turn, is interacting with a conserved aspartic acid. This aspartic acid is equivalent to Asp-233 in 3Dpol (Asp-653 in BF and Asp-537 in T7 Rp). Binding of a nucleotide with an incorrect base or sugar configuration would prevent maximal stabilization of the isomerized triphosphate, causing a reduced efficiency of incorporation and shifting the overall equilibrium of the reaction in the direction of nucleotide release to provide an opportunity for binding of another nucleotide.
Figure 9. Triphosphate reorientation as a fidelity checkpoint in all polymerases.
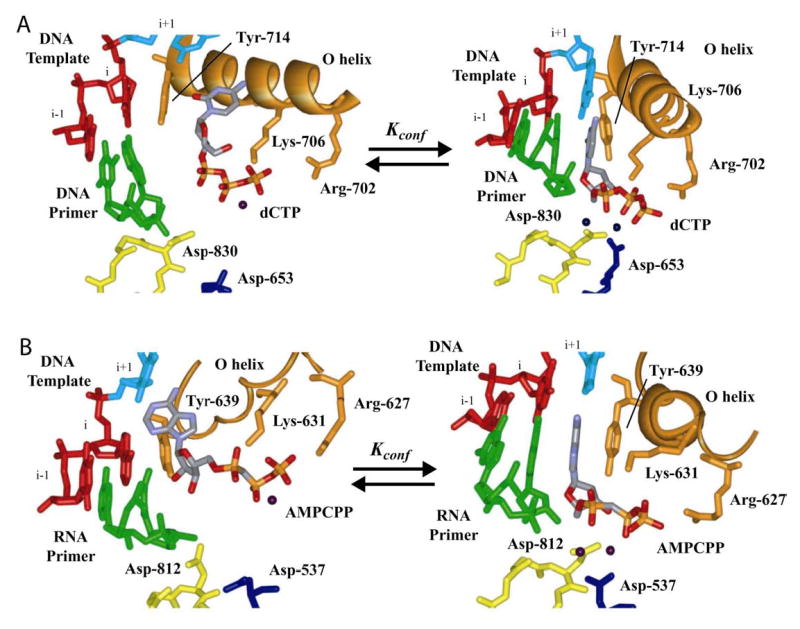
(A) Bacillus stearothermophilus DNA polymerase I fragment. The panel on the left was derived from 1L3U (16). We inserted a dCTP molecule to maintain interaction between the triphosphate and the basic amino acids of the O helix. The panel on the right was derived from 1LV5 (16). (B) T7 RNA polymerase. The panel on the left was derived from 1S0V (38). The panel on the right was derived from 1S76 (39). In both polymerases, two conformations of the incoming nucleotide can be observed. Complexes on the right are poised for catalysis. We propose that these two complexes represent ERnNTP and *ERnNTP observed as the first fidelity checkpoint in the kinetic mechanism for PV 3Dpol (Fig. 7A), suggesting that this checkpoint is conserved in all classes of nucleic acid polymerases with a canonical, palm-based active site.
Mutational analysis of residues in helix O has established a role for this helix in DNA polymerase fidelity; however, precise kinetic and structural explanations are lacking (40–42). Tyr-639 is located at the end of helix O in T7 Rp and modulates sugar specificity, again without a clear molecular explanation (43). We suggest that mutations in helix O change the equilibrium position of the triphosphate, leading to decreases in the efficiency of correct nucleotide incorporation without substantial changes for incorrect nucleotides, the combination of which accounts for the observed reduction in fidelity.
The connection between the fingers and motif A observed in PV 3Dpol is not only conserved in other picornavirus polymerases, for example human rhinovirus (Fig. 10A), but also in other supergroup I polymerases, for example Norwalk virus (Fig. 10B), and supergroup II polymerases, for example hepatitis C virus (Fig. 10C). We conclude that control of fidelity by sites remote from the catalytic center as demonstrated here for PV 3Dpol is a conserved feature in all RdRps. While the picornavirus polymerases employ the amino terminus for interactions with motif A, an arginine serves this purpose in the other enzymes (Fig. 10). In all cases, the region of the fingers in contact with motif A is solvent accessible. Small molecules that interact with this surface could cause changes in polymerase activity that range from increased or decreased fidelity to complete inactivation. Any of these outcomes should lead to a significant antiviral effect. The capacity of small molecules to target remote sites of a polymerase and interfere with incorporation at the active site has been shown recently by the design of a new class of inhibitors effective against Escherichia coli RNA polymerase (44).
Figure 10. A link between the fingers subdomain and the catalytic site for all RNA-dependent RNA polymerases.

Conserved interactions between the fingers and motif A in supergroups I and II RdRps. (A) Human rhinovirus type 14 (HRV-14) polymerase (45). (B) Norwalk virus (NV) polymerase (46). (C) Hepatitis C virus (HCV) polymerase (47). Labeling is the same as for PV 3Dpol shown in Fig. 8C. In the RdRps for NV and HCV, the connection to the fingers is made directly by an Arg sidechain instead of indirectly by the amino terminus. Perturbations in the conformation of the region of the fingers domain shown should be transmitted to the active site via perturbation in the conformation of motif A.
Footnotes
This work was supported, in part, by research grants AI45818 (C.E.C.) and AI40085 (R.A.) from the US National Institutes of Health. C.E.C. is the recipient of an Established Investigator Award (0340028N) from the American Heart Association.
The abbreviations used are: RdRp, RNA-dependent RNA polymerase; PV, poliovirus; WT, wild-type; RTP, ribavirin triphosphate; BF, Bacillus stearothermophilus; T7 Rp, bacteriophage T7 RNA polymerase; HRV-14, human rhinovirus type 14; NV, norwalk virus; HCV, hepatitis C virus.
References
- 1.Kuchta RD, Mizrahi V, Benkovic PA, Johnson KA, Benkovic SJ. Biochemistry. 1987;26:8410–8417. doi: 10.1021/bi00399a057. [DOI] [PubMed] [Google Scholar]
- 2.Dahlberg ME, Benkovic SJ. Biochemistry. 1991;30:4835–4845. doi: 10.1021/bi00234a002. [DOI] [PubMed] [Google Scholar]
- 3.Wong I, Patel SS, Johnson KA. Biochemistry. 1991;30:526–537. doi: 10.1021/bi00216a030. [DOI] [PubMed] [Google Scholar]
- 4.Patel SS, Wong I, Johnson KA. Biochemistry. 1991;30:511–525. doi: 10.1021/bi00216a029. [DOI] [PubMed] [Google Scholar]
- 5.Kati WM, Johnson KA, Jerva LF, Anderson KS. J Biol Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- 6.Reardon JE. J Biol Chem. 1993;268:8743–51. [PubMed] [Google Scholar]
- 7.Arnold JJ, Cameron CE. Biochemistry. 2004;43:5126–37. doi: 10.1021/bi035212y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joyce CM, Benkovic SJ. Biochemistry. 2004;43:14317–24. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- 9.Showalter AK, Tsai MD. Biochemistry. 2002;41:10571–10576. doi: 10.1021/bi026021i. [DOI] [PubMed] [Google Scholar]
- 10.Huang H, Chopra R, Verdine GL, Harrison SC. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 11.Ollis DL, Brick P, Hamlin R, Xuong NG, Steitz TA. Nature. 1985;313:762–766. doi: 10.1038/313762a0. [DOI] [PubMed] [Google Scholar]
- 12.Doublie S, Ellenberger T. Curr Opin Struct Biol. 1998;8:704–712. doi: 10.1016/s0959-440x(98)80089-4. [DOI] [PubMed] [Google Scholar]
- 13.Ferrin LJ, Mildvan AS. Biochemistry. 1985;24:6904–6912. doi: 10.1021/bi00345a024. [DOI] [PubMed] [Google Scholar]
- 14.Ferrin LJ, Mildvan AS. Biochemistry. 1986;25:5131–5145. doi: 10.1021/bi00366a023. [DOI] [PubMed] [Google Scholar]
- 15.Vande Berg BJ, Beard WA, Wilson SH. J Biol Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 16.Johnson SJ, Taylor JS, Beese LS. PNAS. 2003;100:3985–3900. doi: 10.1073/pnas.0630532100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arnold JJ, Gohara DW, Cameron CE. Biochemistry. 2004;43:5138–48. doi: 10.1021/bi035213q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnold JJ, Cameron CE. J Biol Chem. 2000;275:5329–5336. doi: 10.1074/jbc.275.8.5329. [DOI] [PubMed] [Google Scholar]
- 19.Gohara DW, Crotty S, Arnold JJ, Yoder JD, Andino R, Cameron CE. J Biol Chem. 2000;275:25523–25532. doi: 10.1074/jbc.M002671200. [DOI] [PubMed] [Google Scholar]
- 20.Gohara DW, Arnold JJ, Cameron CE. Biochemistry. 2004;43:5149–58. doi: 10.1021/bi035429s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen JL, Long AM, Schultz SC. Structure. 1997;5:1109–1122. doi: 10.1016/s0969-2126(97)00261-x. [DOI] [PubMed] [Google Scholar]
- 22.Thompson AA, Peersen OB. Embo J. 2004;23:3462–71. doi: 10.1038/sj.emboj.7600357. Epub 2004 Aug 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vignuzzi, M., Stone J.K., Arnold J.J., Cameron C.E. and Andino R. (2005) Nature, (Submitted).
- 24.Crotty S, Maag D, Arnold JJ, Zhong W, Lau JY, Hong Z, Andino R, Cameron CE. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 25.Castro C, Arnold JJ, Cameron CE. Virus Res. 2005;107:141–9. doi: 10.1016/j.virusres.2004.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gohara DW, Ha CS, Kumar S, Ghosh B, Arnold JJ, Wisniewski TJ, Cameron CE. Protein Expr Purif. 1999;17:128–138. doi: 10.1006/prep.1999.1100. [DOI] [PubMed] [Google Scholar]
- 27.Arnold JJ, Cameron CE. J Biol Chem. 1999;274:2706–2716. doi: 10.1074/jbc.274.5.2706. [DOI] [PubMed] [Google Scholar]
- 28.Crotty S, Cameron CE, Andino R. Proc Natl Acad Sci U S A. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Holland JJ, Domingo E, de la Torre JC, Steinhauer DA. J Virol. 1990;64:3960–3962. doi: 10.1128/jvi.64.8.3960-3962.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CH, Gilbertson DL, Novella IS, Huerta R, Domingo E, Holland JJ. J Virol. 1997;71:3636–3640. doi: 10.1128/jvi.71.5.3636-3640.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Domingo E. Virus Res. 2005;107:115–6. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Domingo E, Escarmis C, Lazaro E, Manrubia SC. Virus Res. 2005;107:129–139. doi: 10.1016/j.virusres.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 33.Vignuzzi M, Stone JK, Andino R. Virus Res. 2005;107:173–81. doi: 10.1016/j.virusres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 34.Pfeiffer JK, Kirkegaard K. Proc Natl Acad Sci U S A. 2003;100:7289–7294. doi: 10.1073/pnas.1232294100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinz BA, Rueckert RR, Shepard DA, Dutko FJ, McKinlay MA, Fancher M, Rossmann MG, Badger J, Smith TJ. J Virol. 1989;63:2476–85. doi: 10.1128/jvi.63.6.2476-2485.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mosser AG, Rueckert RR. J Virol. 1993;67:1246–54. doi: 10.1128/jvi.67.3.1246-1254.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mosser AG, Sgro JY, Rueckert RR. J Virol. 1994;68:8193–201. doi: 10.1128/jvi.68.12.8193-8201.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Temiakov D, Patlan V, Anikin M, McAllister WT, Yokoyama S, Vassylyev DG. Cell. 2004;116:381–91. doi: 10.1016/s0092-8674(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 39.Yin YW, Steitz TA. Cell. 2004;116:393–404. doi: 10.1016/s0092-8674(04)00120-5. [DOI] [PubMed] [Google Scholar]
- 40.Carroll SS, Cowart M, Benkovic SJ. Biochemistry. 1991;30:804–13. doi: 10.1021/bi00217a034. [DOI] [PubMed] [Google Scholar]
- 41.Bell JB, Eckert KA, Joyce CM, Kunkel TA. J Biol Chem. 1997;272:7345–51. doi: 10.1074/jbc.272.11.7345. [DOI] [PubMed] [Google Scholar]
- 42.Minnick DT, Bebenek K, Osheroff WP, Turner RM, Jr, Astatke M, Liu L, Kunkel TA, Joyce CM. J Biol Chem. 1999;274:3067–75. doi: 10.1074/jbc.274.5.3067. [DOI] [PubMed] [Google Scholar]
- 43.Sousa R, Padilla R. Embo J. 1995;14:4609–21. doi: 10.1002/j.1460-2075.1995.tb00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Artsimovitch I, Chu C, Lynch AS, Landick R. Science. 2003;302:650–4. doi: 10.1126/science.1087526. [DOI] [PubMed] [Google Scholar]
- 45.Love RA, Maegley KA, Yu X, Ferre RA, Lingardo LK, Diehl W, Parge HE, Dragovich PS, Fuhrman SA. Structure (Camb) 2004;12:1533–44. doi: 10.1016/j.str.2004.05.024. [DOI] [PubMed] [Google Scholar]
- 46.Ng KK, Pendas–Franco N, Rojo J, Boga JA, Machin A, Alonso JM, Parra F. J Biol Chem. 2004;279:16638–45. doi: 10.1074/jbc.M400584200. [DOI] [PubMed] [Google Scholar]
- 47.Lesburg CA, Cable MB, Ferrari E, Hong Z, Mannarino AF, Weber PC. Nat Struct Biol. 1999;6:937–43. doi: 10.1038/13305. [DOI] [PubMed] [Google Scholar]