

Abstract
Transforming growth factor beta (TGF-β) proteins play important roles in morphogenesis of many craniofacial tissues; however, detailed biological mechanisms of TGF-β action, particularly in vivo, are still poorly understood. Here, we deleted the TGF-β type I receptor gene Alk5 specifically in the embryonic ectodermal and neural crest cell lineages. Failure in signaling via this receptor, either in the epithelium or in the mesenchyme, caused severe craniofacial defects including cleft palate. Moreover, the facial phenotypes of neural crest-specific Alk5 mutants included devastating facial cleft and appeared significantly more severe than the defects seen in corresponding mutants lacking the TGF-β type II receptor (TGFβRII), a prototypical binding partner of ALK5. Our data indicate that ALK5 plays unique, non-redundant cell-autonomous roles during facial development. Remarkable divergence between Tgfbr2 and Alk5 phenotypes, together with our biochemical in vitro data, imply that (1) ALK5 mediates signaling of a diverse set of ligands not limited to the three isoforms of TGF-β, and (2) ALK5 acts also in conjunction with type II receptors other than TGFβRII.
Keywords: Alk5, Cleft face, Cleft palate, Cranial neural crest, Craniofacial malformation, Mandible, Palatal fusion
Introduction
Facial malformations including cleft lip and cleft palate are among the most common human birth defects, which often arise from a poorly understood failure in cell–cell and/or matrix–cell signaling. In this context, the key cell types are epithelial cells derived from the ectoderm and ectomesenchymal cells largely derived from the neural crest (Francis-West et al., 1998). Both these cell types express multiple components of the transforming growth factor beta (TGF-β) signaling pathway, which has been shown to play a critical role in many aspects of craniofacial development, including palatogenesis (Dudas and Kaartinen, 2005). TGF-βs typically signal via heterotetrameric receptor complexes composed of two type II (TGFβRII) and two type I (ALK5) receptors, which phosphorylate intracellular mediators known as receptor-regulated rSmads (Smad2, Smad3). Phosphorylated rSmads, in turn, complex with Smad4 and translocate into the nucleus to modulate transcription of target genes (Massague and Chen, 2000; Derynck and Feng, 1997). Although multiple alternative (non-canonical) downstream pathways have also been identified (Dudas and Kaartinen, 2005), their exact connection to TGF-β receptors is currently unknown.
The palate, i.e., the upper wall of the oral cavity, is formed from two embryonic palatal shelves, which fuse in the midline (Ferguson, 1988). The fusion process requires a tight mutual adhesion of the two apposing shelves and a subsequent degradation of the epithelial seam, resulting in mesenchymal confluence (Dudas and Kaartinen, 2005). Mice deficient in either TGF-β2 or TGF-β3 demonstrate defects in palatal fusion (Sanford et al., 1997; Kaartinen et al., 1995; Proetzel et al., 1995). Whereas Tgf-β2 is expressed in the palatal mesenchyme, Tgf-β3 expression is strictly limited to the pre-fusion palatal midline epithelium (Dudas et al., 2004a; Saika et al., 2001; Sanford et al., 1997; Dickson et al., 1993). Based on studies using misexpression and chemical inhibition, it has been suggested that the TGF-β/ALK5/Smad2 pathway has an important function in disintegration of the midline seam (Cui et al., 2005; Dudas et al., 2004a). Three major mechanisms for the MES (midline epithelial seam) disintegration have been proposed in recent literature: apoptosis, cell migration, and epithelial-to-mesenchymal transdifferentiation (Martinez-Alvarez et al., 2000). However, the exact epithelium-specific role of TGF-β signaling via ALK5 in palatal fusion in vivo is still to be defined.
The mesenchyme underlying the palatal epithelium is formed from the craniofacial neural crest (CNC), a pluripotent cell population originating from the dorsal ridges of the neural tube (Ito et al., 2003). Many mouse mutants lacking genes that control proliferation of palatal mesenchymal cells display defects in palatal fusion, thus confirming that controlled growth and patterning of the palatal mesenchyme is a prerequisite for successful palatal fusion in vivo (Dudas and Kaartinen, 2005; Martinez-Alvarez et al., 2004; Rice et al., 2004; Brewer et al., 2004; Dudas et al., 2004b; Darling et al., 2003; Zhang et al., 2002; Beverdam et al., 2001; Lavrin et al., 2001; Qu et al., 1999; Satokata and Maas, 1994). Several signaling processes have been shown to control neural crest cells (NCCs) in a cell-autonomous fashion (Sasaki et al., 2005; Brewer et al., 2004; Jeong et al., 2004; Dudas et al., 2004b; Ito et al., 2003; Tallquist and Soriano, 2003; Brault et al., 2001). Among these pathways, TGF-β/BMP superfamily signaling is of particular interest because many TGF-β-related ligands have been shown to play indispensable roles in facial development (Dudas et al., 2004b; Ito et al., 2003; Taya et al., 1999; Sanford et al., 1997; Kaartinen et al., 1995; Proetzel et al., 1995).
Studies on the roles of TGF-β/BMP receptors during facial development in vivo have been hampered by the fact that embryos lacking these genes die before the face is formed. Recently, tissue-specific gene knockout technologies utilizing Cre/loxP recombination have become powerful tools, which can be used to circumvent this problem. Here, we deleted the TGF-β type I receptor ALK5 specifically in the ectodermal epithelial and neural crest lineages by crossing mice carrying the conditional Alk5flox allele (Larsson et al., 2001) with K14-Cre mice (Andl et al., 2004), or with Wnt1-Cre mice (Danielian et al., 1998). In both cases, the resulting mutant mice display impaired palatal fusion. Moreover, neural crest-specific Alk5 mutants demonstrate severe nasal clefting with associated mandibular hypoplasia and several other craniofacial defects. These phenotypes differ remarkably from those seen in corresponding Tgfbr2 mutants (Ito et al., 2003), suggesting that during facial morphogenesis, ALK5 may also become activated by an additional mechanism that does not require a function of its typical binding partner, the TGF-β type II receptor.
Materials and methods
Mice and genotyping
Mice carrying Alk5flox and Alk5KO alleles were PCR genotyped as described (Larsson et al., 2001). Wnt1-Cre mice were a generous gift from A. McMahon and Rosa26R (R26R) Cre-reporter mice were purchased from the Jackson Labs (for detailed PCR-genotyping, see http://www.jax.org); K14-Cre mice were obtained from S. Millar (Andl et al., 2004). Tgf-β3−/ − mice were generated in our laboratory (Kaartinen et al., 1995). All mice were maintained on mixed genetic backgrounds. All studies were carried out at the Animal Care Facility of the Saban Research Institute in accordance with national and institutional guidelines.
Histology and stainings
Tissues were fixed with 4% formaldehyde for 12h, and paraffin sections were stained with hematoxylin–eosin. Embryos were stained for β-galactosidase activity as described (Hogan et al., 1994). Briefly, the specimens were fixed in 4% formaldehyde for 30min, washed in the detergent wash, and developed in the X-Gal solution. Skeletal staining with alcian blue and alizarin red dyes, and tissue clearing were performed as published (McLeod, 1980).
Electron microscopy
E14 heads with exposed pre-fusion palatal shelves were fixed in phosphate-buffered 4% formaldehyde with 0.2% glutaraldehyde for 24h and processed at DEI/USC/Norris Cancer Center Cell and Tissue Imaging Core. Pictures were recorded using the scanning electron microscope with computerized digital capture (Hitachi S-570). Cells were counted on both palatal shelves of each genotype, in areas of 20×40μm (approximately 20 cells), the longer side being parallel with the AP axis of palatal shelves.
Expression studies
Screening for expression changes was performed using expression arrays (GEArray® Q Series Mouse TGF-β/BMP Array, SuperArray Cat. # MM-023) according to manufacturer's instructions and accompanying software analysis tool and quantitative RT-PCR assays (Roche LightCycler® with Roche TaqMan® probes and primers designed online, http://www.roche-applied-science.com). Whole-mount and section in situ hybridization experiments were used to visualize the relevant expression patterns and carried out as described (Moorman et al., 2001; Hogan et al., 1994). We used probes specific for Msx1 (Furuta et al., 1997), Msx2 (Ishii et al., 2003), Tgfbr2 (Ito et al., 2003), Alk5 (Dudas et al., 2004a), Bmpr2 (nucleotides 1070–1566; accession number NM_007561), Acvr2A (nucleotides 1040–1277; accession number NM_007396) and Acvr2B (nucleotides 627–998; accession number NM_007397), Fgf8 (Tanaka et al., 1992), Tgif (nucleotides 343–755, accession number NM_009372), and Gdf11 (nucleotides 247–703, ID number ENSMUST00000026408, and Nakashima et al., 1999).
Apoptosis and cell proliferation
Apoptotic cells were detected in paraffin sections as a green fluorescence using DeadEnd Fluorometric TUNEL system (Promega). Cell proliferation was immunodetected using the phospho-histone H3 antibody (Cell Signaling) and Cy3-conjugated secondary antibody (Jackson ImmunoResearch Laboratories). Positively stained cells were counted manually in defined areas of tissues. Statistical analysis of cell counts in serial sections and comparison of mutant specimens with controls was performed using non-parametric Wilcoxon rank sum test.
Palatal organ culture and bead implantation
Mice were mated during the dark period of the controlled light cycle; presence of vaginal plugs was designated as day 0, hour 0. Females were euthanized by CO2, and E14 embryos were collected in Hanks' balanced salt solution on ice. Palatal shelves were microdissected, placed on Millipore filter discs, and cultured in BGJb medium (Gibco) supplemented with vitamin C (Kaartinen et al., 1997). Agarose beads (Affi-Gel Blue, Bio-Rad) were washed in PBS and incubated for 30min at 37°C in 0.1% BSA in PBS, with or without 5ng/ml TGF-β3 (Sigma). Beads were washed in a culture medium and placed between the edges of dissected palatal shelves during the arrangement of organ culture experiments, as described above. Tissues were harvested after 12h of culture at 37°C and 6% CO2 in humidified air, before the midline seam disintegrated.
Expression vectors, transfections, and Western blot analyses
Expression vectors for human Smad2 and rat Alk5 cDNAwere obtained from Rik Derynck. A segment of rat Alk5 cDNA encoding the intracellular portion of ALK5 (nucleotides 541–1543; accession number L26110) was PCR-amplified and subcloned in-frame into the SpeI site of the pC4-RHE plasmid. A fragment of cDNA encoding the entire kinase domain of Tgfbr2 (nucleotides 901–2027; accession number NM_009371) and Acvr2B (nucleotides 602–1651; accession number NM_007397) was reverse-transcribed and PCR-amplified from the mouse embryonal RNA (E10) and subcloned in-frame into the pC4M-F2E plasmid. pC4-RHE and pC4M-F2E plasmids were from Argent Heterodimerization kit (Ariad Pharmaceuticals, http://www.ariad.com/regulationkits. All constructs were sequenced to exclude mutations introduced by PCR and to verify in-frame ligation. DR26 cells were cultured in D-MEM/F12 medium (Invitrogen) and transfected on 24-well plates. For each transfection, 200ng of Smad2 vector, 400ng of type II receptor vector, and 400ng of Alk5 vector were mixed with 2.0μl of Lipofectamine™ 2000 in a total volume of 100μl of Opti-MEM (Invitrogen). The cells were incubated with the DNA/Lipofectamine mixture for 24h at 37°C, 5% CO2. Subsequently, cells were washed and incubated with 0nM, 50nM, and 500nM concentrations of the heterodimerizer (AP21967 from Ariad Pharmaceuticals). After 40min, cells were lysed in 2× Laemmli sample buffer and analyzed using Western blotting. Anti-phospho-Smad2 (Upstate Biotech.), anti-Smad2 (Santa Cruz Biotech.), anti-HA (USC Core facility), and anti-Flag (Sigma) antibodies were used.
Results
Deletion of Alk5 in the epithelium impairs palatal fusion
To dissect the function of ALK5 in the palatal epithelium during palatogenesis, we crossed mice carrying the floxed Alk5 allele (Alk5flox/flox) with K14-Cre transgenic mice (Andl et al., 2004), which were also heterozygous for the Alk5 knockout allele (i.e., Alk5KO/WT/K14-Cre+/−). Twenty-five percent of the resulting mutant embryos were compound heterozygotes for the Alk5flox and Alk5KO alleles, and carried K14-Cre, a transgene encoding the Cre recombinase under the control of the K14 promoter (herein termed Alk5/K14-Cre). K14-Cre is specifically expressed in ectodermal cells, including the palatal epithelium, as early as at E10 (data not shown), and has been shown to efficiently mediate the recombination of floxed alleles (Andl et al., 2004).
Despite widespread expression of Alk5 in many ectodermal locations, epithelial deletion of Alk5 did not result in any obvious macroscopic pathological features in E14 embryos or in newborns (data not shown). However, Alk5/K14-Cre mutant newborn mice died soon after birth, lacking milk in the stomach. Stereoscopic examination consistently revealed 100% penetrant cleft in the posterior part of the soft palate, affecting approximately 20% of the palatal length (Figs. 1G–J). Subsequent detailed histological analyses of serial sections (frontal orientation) revealed a persistent midline epithelial seam and fusion defects also in the remaining anterior portion of the secondary palate (Figs. 1H–I). Stereoscopically, this frontal area of the palate seemed to be well adhered at the time of inspection (Fig. 1G); the anterior cleft (Fig. 1H) was not visible and may have appeared during tissue fixation and dehydration, suggesting that the midline seam in Alk5/K14-Cre mutant is formed, but remains persistently weak throughout the embryonic development until birth. In addition, the anterior portions of palatal shelves consistently failed to fuse with the nasal septum (Fig. 1H).
Fig. 1.
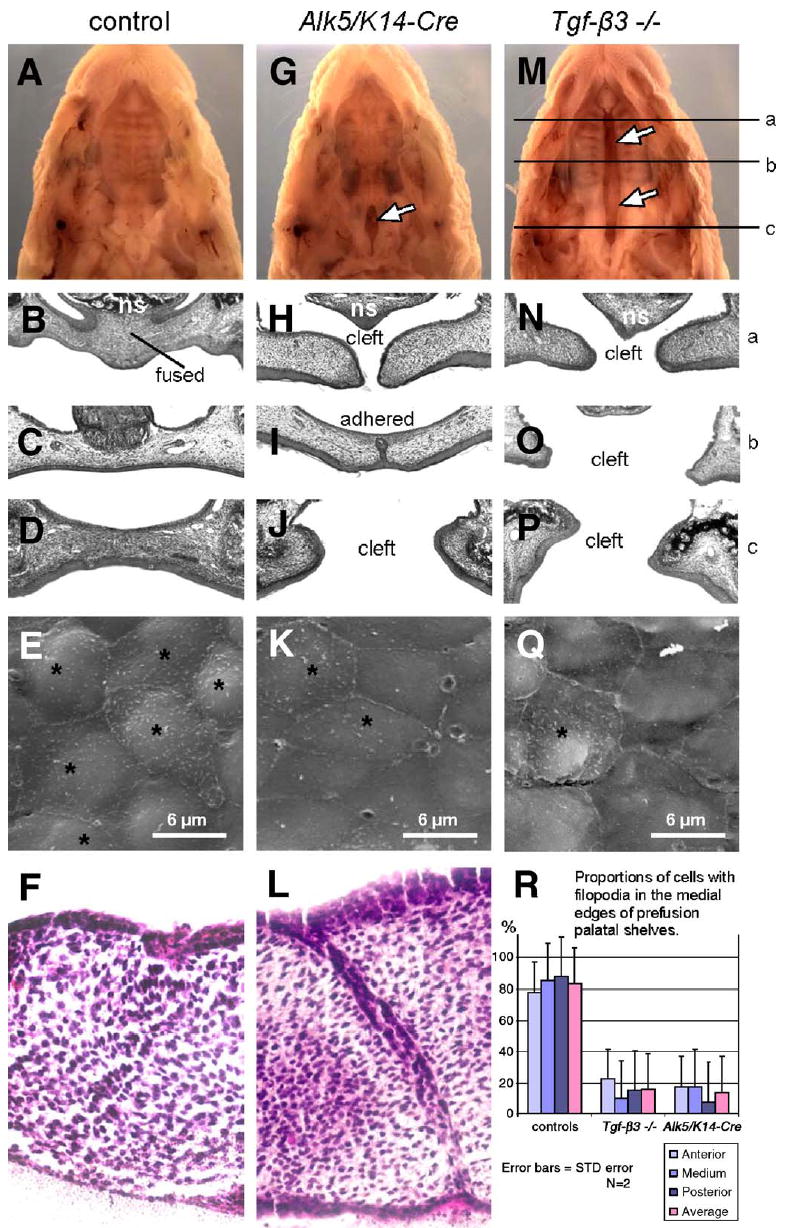
Epithelial deletion of Alk5 causes impaired fusion of palatal shelves and posterior palatal cleft. Alk5/K14-Cre pups (G–J) are born with a defect in the posterior part of the soft palate (arrow in panel G). In contrast, Tgf-β3 null newborns have a complete bilateral palatal cleft (M–P; arrows in panel M), whereas control newborns (A–D) have palates fully fused. The upper row shows stereoscopic images of formalin-fixed newborn heads after removal of the mandible (magnification 5×), the next three rows show histological sections through three different antero-posterior positions (a, b, c), as indicated in panel M (hematoxylin–eosin, magnification 20×). Alk5/ K14-Cre palatal shelves dissected on E14 fail to fuse in organ culture (L), whereas wild-type shelves (F) fuse normally (hematoxylineosin, magnification 40×). The fourth row (E, K, Q) demonstrates decreased proportion of cells with filopodia (asterisks) by E14 at the edges of pre-fusion palatal shelves in the Alk5/K14-Cre mutants (K) and Tgf-β3 null embryos (Q), in comparison with controls (E) (Scanning electron microscopy, magnification 5 000×). (R) Comparison of proportion of cells with filopodia in pre-fusion palatal shelves. Cells were counted in three areas (anterior, medium, posterior; 20×40μm) of the two palatal shelves for each genotype, and averaged within each group. f—the site of fusion of palatal shelves; ns—nasal septum; *—cells with filopodia.
Next we used serial sectioning to compare histological details of palatal fusion between mutants and controls at E14 (pre-fusion), at E14.5 (during adherence and fusion), and at E15 and E17 (post-fusion). Before and during palatal fusion, both mutant and control samples looked identical. At E14.5, palatal shelves of both genotypes formed the midline seam spanning about 90% of the total palate length, with small unadhered areas corresponding to the anterior and posterior regions. During later stages (E15 and E17), while control specimens demonstrated complete fusion, mutant palatal shelves were adherent but displayed a persistent midline seam, except in the areas of the posterior and anterior ends of the palate. The posterior regions demonstrated a cleft similar to the cleft in newborns shown in Fig. 1—albeit less extensive in size (less than 5% of the seam length in mutants on E15, and about 15% on E17). No differences in mesenchymal cell proliferation or apoptosis were found in Alk5/K14-Cre mutants, not even in the posterior or anterior ends of palatal shelves that failed to establish contact with the apposing shelf. Progressive increase in the relative size of the posterior cleft accounts for the final cleft reaching 20% of the palate length in Alk5/K14-Cre newborns. Moreover, in organ culture, Alk5/K14-Cre mutant shelves dissected from E14 embryos and placed within close contact displayed a persistent midline seam, confirming that the primary cause of the fusion defect is epithelial malfunction (Fig. 1L). These findings indicate that ALK5 plays an indispensable role in the disappearance of the palatal epithelial seam during palatal fusion, which in turn leads to clefting, particularly in the anterior and posterior aspects of the secondary palate.
Palatal edges of Alk5/K14-Cre mutants show a reduced number of cells with filopodia
It has been reported that MEE cells deficient in TGF-β3 fail to form filopodia on their apical surface (Taya et al., 1999), with implications to failed palatal fusion. Therefore, we have compared the MEE ultrastructure between controls, Tgf-β3 knockouts, and Alk5/K14-Cre mutants during the stage when approximately 5% of the MEEs are in contact. A vast majority (about 85%) of all MEE cells in controls displayed filopodia, whereas in both Tgf-β3−/ − and Alk5/K14-Cre samples, the number of filopodia-containing cells was markedly reduced (16.6 and 15%, respectively; Figs. 1E, K, Q, and R). MEE surfaces closely adjacent to the regions of adherence showed no differences in comparison with the remote (both anterior and posterior) parts of the medial edges. These results demonstrate that TGF-β3/ALK5 signaling plays a specific role in the morphological maturation of palatal midline epithelial cells.
Tgf-βsignaling and the MES disappearance
The exact contribution of cell death, migration, and epithelial-to-mesenchymal transformation to the removal of the MES from fusing palatal shelves has been discussed for decades (Dudas and Kaartinen, 2005; Cuervo and Covarrubias, 2004; Martinez-Alvarez et al., 2000; Shuler, 1995). To analyze, whether ALK5 signaling could mediate apoptosis in the MEE, we first compared the number of TUNEL-positive cells between controls, Tgf-β3 knockouts, and Alk5/K14-Cre mutants (Fig. 2). Controls demonstrated a large number of apoptotic cells, particularly in the nasal and oral epithelial triangles, whereas apposing Tgf-β3 knockout palatal shelves did not show any positively staining cells. Although palatal shelves in Alk5/K14-Cre mutants were adherent, they still failed to display apoptotic cells. Subsequently, we implanted beads soaked with recombinant TGF-β3 (1mg/ ml BSA; 5ng/ml TGF-β3) between the MEEs of Tgf-β3 knockout palatal explants. After 24-hour organ culture, control beads (BSA only) did not induce detectable apoptosis, whereas the explants incubated with TGF-β3 beads displayed a large number of apoptotic cells in the epithelial seam (Fig. 2E).
Fig. 2.

TGF-β-induced apoptosis in the midline epithelial seam is required for palatal fusion. Comparison of apoptotic cell death in control (A), Tgf-β3−/ − (B) and Alk5/ K14-Cre (C) palatal midline regions using TUNEL assay (green fluorescent signal) in embryonic heads fixed on E14.5. Intense clusters of apoptotic cells can be seen in controls (A), whereas positively staining cells cannot be seen in Tgf-β3 and Alk5/K14-Cre mutants. Arrow in panel A points to TUNEL-positive nuclei inside the disappearing midline seam. (D–F) Implantation of TGF-β3 beads (E) into the palatal midline region of E14 Tgf-β3−/ − explants leads to increased apoptosis of adjacent epithelial cells up to a level seen in control wild-type explants treated with BSA (bovine serum albumin) beads (F). DAPI counterstain, magnification 40×.
To follow the fate of palatal epithelial, we crossed Alk5flox/flox mice with R26R reporter mice, and subsequently the double homozygote Alk5flox/flox, R26R+/+ females were crossed with Alk5KO/WT/K14-Cre males (Jiang et al., 2000; Soriano, 1999). In resulting embryos, all cells expressing the active recombinase Cre and their descendants stained positively for β-galactosidase. At E17 (i.e., >2days after the palate has fused), palatal midlines in controls contained no positively staining mesenchymal cells (Fig. 3A), consistent with a report by others (Vaziri et al., 2005). In contrast, the mutant palates had either failed to adhere or demonstrated a persistent midline seam, with all epithelial regions sharply demarcated from the surrounding mesenchyme (Figs. 3B–F). No blue cells were invading nor joining the mesenchyme in this transitional region. The transitional region between the adhered and the cleft (posterior) region of the mutant palates displayed an elongated, thin epithelial bridge between the two palatal shelves (Fig. 3E).
Fig. 3.
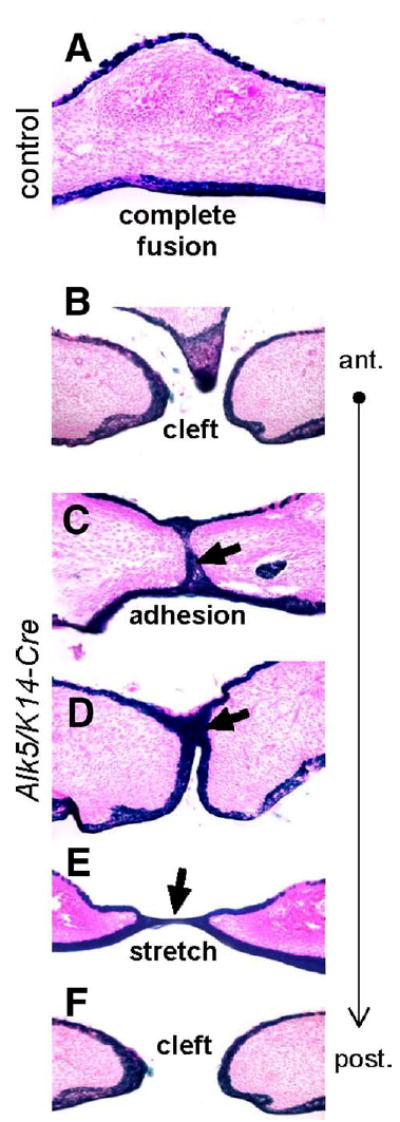
Fate mapping of midline epithelial cells in Alk5/K14-Cre mutants. Transverse palatal sections of E17 embryos expressing K14-Cre in the Rosa26 reporter background were stained for β-galactosidase activity (blue), which permanently marks epithelial cells and their descendants (eosin counterstain, magnification 20×). (A) Control palates have cells derived from the epithelium exclusively on the oral and nasal side of the palate, whereas the fully confluent mesenchyme (pink) along the entire anterior–posterior axis is devoid of positively staining cells. (B–F) Alk5/K14-Cre mutants show either no adhesion (cleft) or adhesion with a lack of fusion detected as persistent midline epithelial seam in the anterior palate (blue staining and arrows). Moreover, the nasal septum has failed to fuse with palatal shelves (B). The posterior part of the palate shows a stretched epithelial bridge (E), followed by palatal cleft in the most posterior region of the soft palate (F).
Taken together, these findings suggest that epithelial-to-mesenchymal transdifferentiation (EMT) may not be a major or a definitive TGF-β-driven mechanism to remove epithelial cells from the midline seam. Our experiments suggest that TGF-β signaling contributes to induction of programmed cell death in the palatal MES. Furthermore, the markedly stretched morphology of the epithelial bridge in the posterior palate of Alk5/ K14-Cre mutants suggests that a midline rupture, i.e., secondary cleft, may occur as an additional clefting mechanism.
ALK5 signaling in neural crest cells is required for normal craniofacial development
To analyze the role of TGF-β signaling via ALK5 in the palatal mesenchyme, we crossed the Alk5flox/flox mice with Alk5KO/WT mice carrying Wnt1-Cre knock-in (Danielian et al., 1998). In resulting Alk5/Wnt1-Cre embryos, Cre-mediated conversion of the Alk5flox allele to Alk5KO allele occurs only in cells that express Wnt1-Cre, i.e., in neural crest (NC) cells, and in the neural plate. This allows deletion of genes also in the palatal mesenchyme, which is composed largely of NC-derived cells. The Alk5/Wnt1-Cre embryos were born alive but were severely disfigured and died soon after birth. The most characteristic macroscopic external features included hypoplastic cranium, split snout, small mandible, cleft palate, and small tongue (Figs. 4A–C). Histological analyses verified the presence of a wide cleft between rudimentary palatal shelves (Fig. 4E). The region of the cranial vault was soft, and several newborns suffered from skin damage and external bleeding in that area. Because no such defect was seen if the fetuses were dissected from the uterus at E19, the forehead and parietal region were most likely injured during parturition. Additionally, no signs of exencephaly or neural closure defects were detected.
Fig. 4.
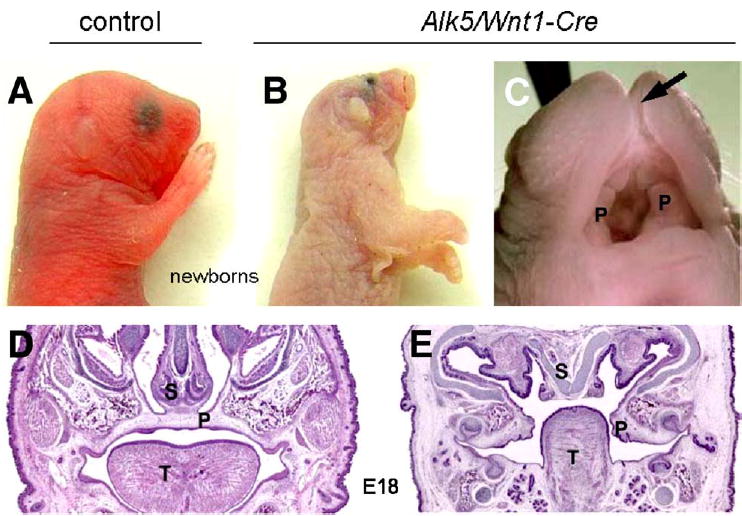
External facial phenotype of Alk5/Wnt1-Cre mutants. Lateral view of a newborn control (A) and a mutant (B) showing the malformed face and head. Mutant embryos are noticeably pale in comparison with controls. (C) Detailed view of the oral cavity of the newborn mutant. Arrow points to a cleft snout and upper lip; undersized palatal shelves are marked with P. Coronal sections through the palatal region of control (D) and Alk5/Wnt1-Cre mutant embryos (E) at E18 (hematoxylin and eosin, ×4). S—nasal septum; P—palatal shelves; T—tongue.
Missing and dysplastic craniofacial skeletal structures in Alk5/Wnt1-Cre mutants
To characterize the skeletal phenotype of Alk5/Wnt1-Cre embryos, we utilized the alizarin red and alcian blue staining, which stains calcified bone red and cartilage blue (Fig. 5). Calvaria phenotype was practically identical to Tgfbr2/Wnt1-Cre mutants (Ito et al., 2003), i.e., large portions of frontal bones were missing, and parietal bones were so severely developmentally retarded that they displayed only the most posterior border (Figs. 5A–D). In contrast, the more posterior structures, such as interparietal and supraoccipital bones, appeared to be well developed. Unlike the calvaria region, the face of the Alk5/Wnt1-Cre mice demonstrated a much more severe pathological phenotype when compared to the corresponding Tgfbr2 mutants (Figs. 4 and 5C–H). Almost all derivatives of the first pharyngeal arch were strongly affected. The nasal cartilage displayed a wide cleft, and both the squamosal zygomatic and maxillary zygomatic processes, as well as jugal bones, were absent. Moreover, the squamosal bones were missing, including the retrotympanic process. The premaxillary bones were hypoplastic, and both the palatal and maxillary bones were rudimentary and far apart, consistent with the presence of cleft palate. Meckel's cartilage was abnormally curved in the posterior part, and the tympanic ring was underdeveloped. The mandible was very small, and lacked identifiable coronoid, condylar, and angular processes. Moreover, the anterior cranial base, which is also derived from neural crest cells (Couly et al., 1993), lacked the presphenoid bone (Figs. 5E–F). Similarly, the middle ear ossicles were also abnormal; the manubrium of malleus was hypoplastic and the incus was missing. The stapes, derived from the second arch, was completely absent, and multiple additional structures derived from pharyngeal arches II–VI (Figs. 5I–J) showed severe and unique abnormalities, not found in corresponding Tgfbr2/Wnt1-Cre mutants (Ito et al., 2003).
Fig. 5.
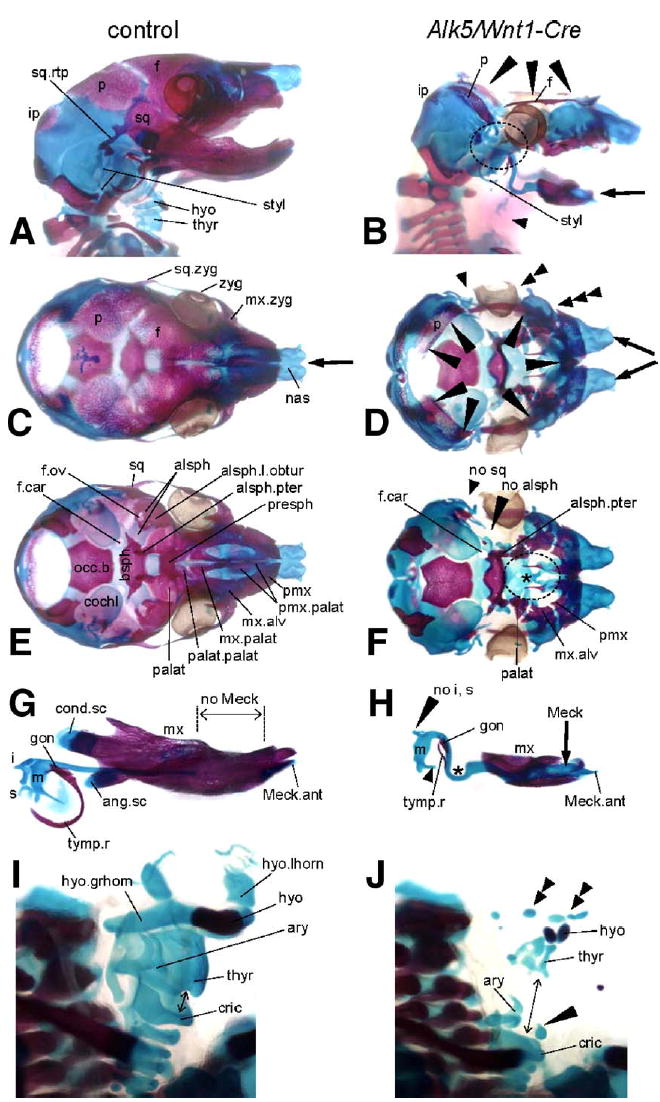
Bone and cartilage defects resulting from deletion of Alk5 in neural crest cells. Bone (red) and cartilage (blue) staining in E18 embryos; controls are on the left side, mutants on the right. Stereoscopic magnification ×5; magnification ×7 used for panels G and H, ×15 for panels I and J. (A–B) Lateral views demonstrate a large defect in calvaria, which is almost completely missing in Alk5/Wnt1-Cre mutants. Only small portions of the parietal and frontal bones remain and form a thin ring around the cranial base (long arrowheads). Shortened mandible (arrow) and multiple anomalies in the region of the absent temporomandibular joint (circled) are other prominent skeletal features of Alk5/Wnt1-Cre embryos, together with almost completely missing laryngeal cartilages and hyoid bone (short arrowhead). (C–D) Superior view shows a lack of all three components of the zygomatic arch (arrowheads), cleft in the region of nasal cartilages underlying the cleft snout (arrows), and missing calvaria as mentioned above (long arrowheads). (E–F) Inferior view after removal of the mandible with middle ear ossicles reveals a large defect in the bony palate (circled) due to rudimentary palatine, maxillary, and premaxillary palatal components. Arrowheads point to locations of missing squamosal and alisphenoid bones; asterisk marks missing presphenoid ossification. (G–H) Lateral view of the mandibular complexes with Meckel's cartilage and middle ear ossicles. Mutants show remarkable differences when compared to control littermates in the persistence and curvature of Meckel's cartilage (asterisk), defects in formation of the secondary cartilages, and impaired middle ear bones (missing incus and stapes, incomplete tympanic ring). (I–J) Semilateral view of the laryngeal region. In mutants, rudiments of the hyoid bone and thyroid cartilage appear as multiple small elements scattered in an anatomically correct location (double arrowheads). The cricoarytenoid complex is positioned abnormally low, at the level of the upper thoracic aperture, resulting in a gap in the laryngeal skeleton (arrow). The long arrowhead points to a midline cartilage of unknown origin. alsph—alisphenoid bone; alsph.l.obtur—lamina obturans; alsph.pter—pterygoid process of alisphenoid bone; ang.sc—secondary cartilage of the angular process; ary—arytenoid cartilage; bsph—basisphenoid bone; cochl—cochlear part of the temporal bone; cond.sc—secondary cartilage of the condylar process; cric—cricoid cartilage; f—frontal bone; hyo—hyoid bone; hyo.grhorn—greater horn of the hyoid bone; hyo.lhorn—lesser horn of the hyoid bone; i—incus; ip—intraparietal bone; f.car—foramen caroticum; f.ov—foramen ovale; gon—gonium; m—malleus; Meck.ant—anterior process of Meckel's cartilage; mx—maxilla; mx.alv—alveolar process of maxilla; mx.palat—palatal process of maxilla; mx.zyg—zygomatic process of maxilla; nas —nasal cartilages; no—used for missing structures; occ.b—basis of the occipital bone; p—parietal bone; palat—palatal bone; palat.palat—palatal process of palatal bone; pmx—premaxilla; pmx.palat—palatal process of premaxilla; s—stapes; sq—squamous bone; sq.rtp—retrotympanic process of the squamal bone; styl—styloid process; thyr—thyroid cartilage; tymp.r—tympanic ring; zyg—zygomatic (jugal) bone.
Fate mapping of neural crest cells in Alk5/Wnt1-Cre mutants
The fate of Alk5-deficient neural crest cells was followed during craniofacial development using the Rosa26 Cre reporter (R26R) β-galactosidase assay at E8.5, E11, and E15. Detailed analysis revealed that the staining pattern was indistinguishable between mutants and controls (Fig. 6). Intriguingly, this analysis also demonstrated that whereas the neural crest cells populate the pharyngeal region at E8.5–E11, the maxillary process of the first pharyngeal arch, as well as both nasal processes, fails to form properly and appear much smaller in mutants than in controls (Figs. 6E–H). In concordance with these findings, the mutant embryos display severe facial clefting at E15, although the facial processes are heavily populated with cells derived from the neural crest (Figs. 6K–L).
Fig. 6.
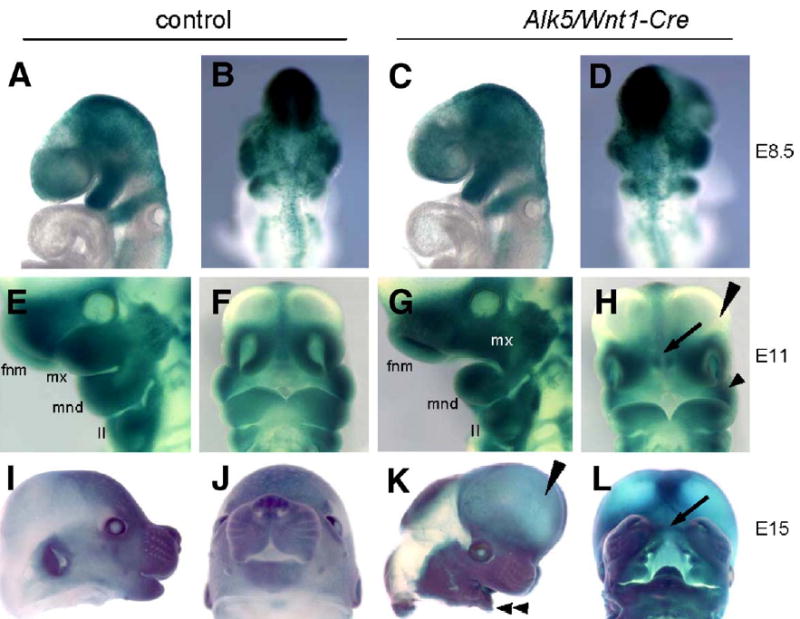
Normal neural crest cell migration but pronounced hypomorphism of mandibular, maxillary, and nasal processes in Alk5/Wnt1-Cre mutants. Embryos carrying the Rosa26 Cre reporter were stained for β-galactosidase activity. (A–D) Migration of neural crest cells (blue) in controls at E8.5 (A–B) is indistinguishable from mutants (C–D). (E–F) Control at E11. (G–H) Alk5/Wnt1-Cre mutant at E11. White mx in G marks an undersized maxillary process, arrow in panel H points to an enlarged gap between nasal pits, corresponding to the future clefting region (compare with panel L). Long arrowhead—forebrain vesicles protruding frontally (aggravated in panel K); short arrowhead—hypoplastic maxillary process. (I–J) Control littermate at E15. (K–L) Alk5/Wnt1-Cre mutant at E15. Long arrowhead points to frontally protruding forebrain region, double arrowhead points to a small mandible, and arrow indicates the facial cleft. II—2nd pharyngeal arch; fnm—frontonasal mass; mnd—mandibular process of the 1st pharyngeal arch; mx—maxillary process of the 1st pharyngeal arch.
Signaling via ALK5 in the neural crest is necessary for mesenchymal cell survival in the first pharyngeal arch
Analysis of Tgfbr2/Wnt1-Cre mutant embryos has shown that TGF-β signaling is required for cell proliferation in the palatal mesenchyme (Ito et al., 2003). We therefore hypothesized that the observed phenotypes in Alk5/Wnt1-Cre mutants could be caused by defective cell proliferation in the maxillary and mandibular processes of the first pharyngeal arch. Mutant and control embryos did not display significant differences in cell proliferation at E8.5, E9, E10, and E11 (Fig. 7). Subsequently, we considered the possibility that signaling via ALK5 would be necessary for cell survival. The rate of programmed cell death was determined using the TUNEL assay in the paraffin sections immediately adjacent to those used for the proliferation assays. Up until E9, identical size, shape, and apoptotic patterns were detected in pharyngeal arches of controls and mutants. The earliest detectable difference between them was observed at E10, when there was a slight change in the location (lateralization) of a typical patch of apoptotic cells apparent within the maxillary arch (Figs. 7A–D), whereas no differences were seen in the mandibular arch (Figs. 7E–H). By E11, Alk5/Wnt1-Cre mutants had a distinct increase in the number of apoptotic cells in the first pharyngeal arch (Figs. 7I–T) and forebrain (not shown). In summary, these results demonstrate that ALK5 signaling is critically involved in the regulation of cell death within the mesenchyme of the first pharyngeal arch.
Fig. 7.
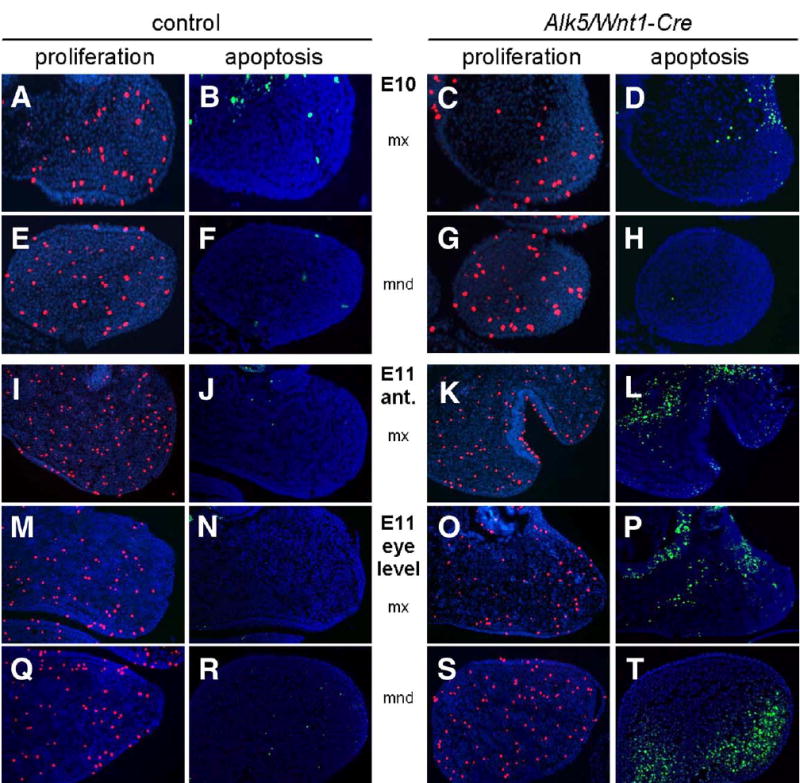
Increased apoptosis in pharyngeal arches of Alk5/Wnt1-Cre mutants. Frontal sections show a pronounced increase in number of TUNEL-positive apoptotic cells (green signal) in the maxillary (mx) and mandibular (mnd) processes of Alk5/Wnt1-Cre mutants on E11, but not on E10, when compared to controls. Cell proliferation (anti-phosphohistone H3 immunostaining, red signal) is comparable between mutants and controls (blue counterstaining with DAPI; magnification 20×).
Changes in proliferation and apoptosis in the palatal mesenchyme
To dissect the mechanism of palatal clefting in Alk5/Wnt1-Cre mutants on the cellular level, we analyzed growth, elevation, and fusion of palatal shelves between E12 and birth. Although mutant palatal shelves appeared symmetrically on the maxillary prominences, they were barely recognizable prior to E13 due to distinct hypoplasia. Between E14 and birth, these rudimentary shelves were located bilaterally in the horizontal orientation (Fig. 4E). Consistent with the findings of increased apoptosis in the mesenchyme of pharyngeal arches at E11, the palatal mesenchyme of Alk5/Wnt1-Cre mutants was affected with a dramatic increase in apoptosis (Fig. 8D). In addition, we detected more than 6-fold increase in the average number of proliferating mesenchymal cells per shelf cross-section (Figs. 8A–B, E). These results show that signaling via ALK5 is required for cell survival not only in the early facial mesenchyme at E10 and E11, but also in the palatal mesenchyme at E14.
Fig. 8.
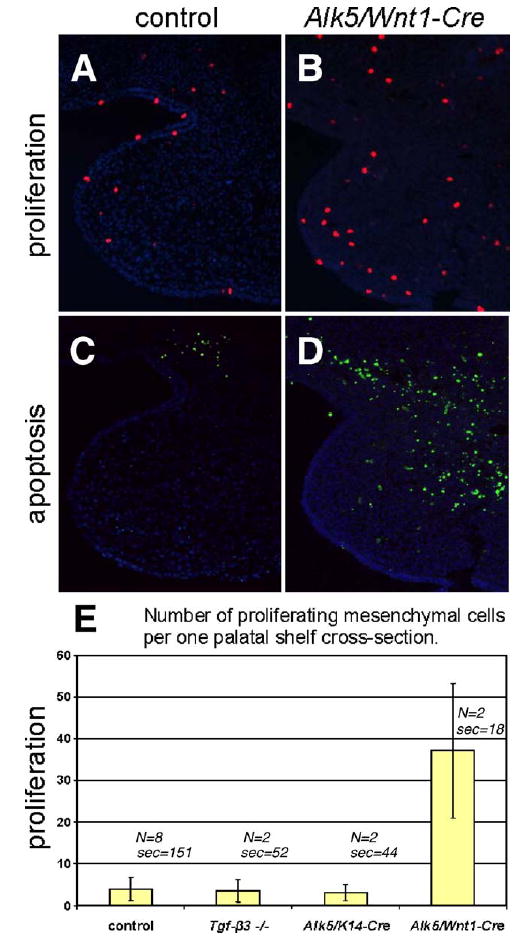
Increased apoptosis and proliferation rate in the palatal mesenchyme of Alk5/Wnt1-Cre mutants and comparison with Alk5/K14-Cre mutants and Tgf-β3 knockouts. (A–B) Proliferating cells labeled with anti-phospho-histone H3 antibody (red fluorescent signal) at E14. (C–D) Apoptotic cells labeled using a TUNEL assay at E14 (green fluorescent signal). Magnification 20×, DAPI counterstain. (E) Histogram demonstrating statistically significant increase in the absolute number of proliferating cells in the mesenchyme of Alk5/Wnt1-Cre mutants in comparison with wild-type controls, Alk5/K14-Cre mutants, and Tgf-β3 knockouts (Wilcoxon rank score test, P <0.05; error bars =standard deviations; N=number of samples; sec=total number of sections analyzed).
Changes in downstream gene expression in Alk5/Wnt1-Cre mutants
To identify signaling pathways that could link impaired signaling via ALK5 to the severe facial phenotype seen in Alk5/Wnt1-Cre mutants, we performed an extensive screen for changes in expression of potential target genes. We employed TGF-β/BMP signaling expression arrays, as well as individual qRT-PCR-based assays for rationally chosen genes involved in facial midline development. Significant changes in expression of Msx1, Fgf8, and Tgif were identified (not shown) and visualized by in situ hybridization (Fig. 9).
Fig. 9.
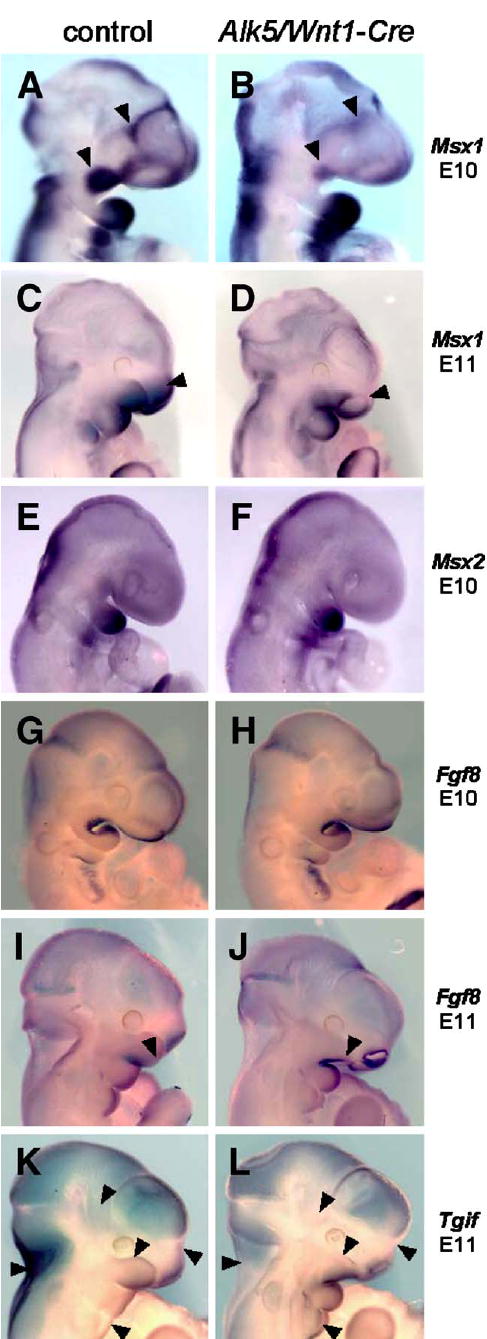
Attenuated expression of Msx1 and changes in expression patterns of Fgf8 and Tgif in Alk5/Wnt1-Cre mutants. Endogenous gene expression was visualized by in situ hybridization with biotinylated riboprobes (blue signal, magnification 5× for E10 and 4× for E11 samples). Whereas Msx2 did not show any change, Msx1 shows a remarkable decrease in expression in the maxillary process and frontonasal mass in mutants at E10 (B, arrowheads) and in the upper nasal pit region at E11 (D, arrowhead). Fgf8 expression in Alk5/Wnt1-Cre mutants spreads more anteriorly along the lower edge of the maxillary process at E11 (J, arrowhead), when compared with the control (I). Tgif expression shows a slight increase in the mandibular process and the 2nd pharyngeal arch in mutants at E11, whereas its expression in mutants of the same age is attenuated or absent in the upper portion of the maxillary process of the 1st pharyngeal arch, frontonasal process, temporoparietal region, and the nuchal area (K–L, arrowheads) The nasal pits and most of the calvaria show a comparable staining pattern and intensity in both mutants and controls.
Msx genes are well-documented effectors of TGF-β/BMP (Bei and Maas, 1998; Suzuki et al., 1997). At E10, Msx1 is normally expressed in the ectomesenchyme of both the maxillary and distal mandibular processes of the first pharyngeal arch (Tucker et al., 1998). In contrast, Msx1 expression was markedly reduced in the maxillary primordia of Alk5/Wnt1-Cre mutants, whereas the mandibular expression appeared unaffected (Fig. 9B). At E11, Msx1 continued to display attenuated expression in the maxillary and frontonasal processes (Fig. 9D), whereas the closely related Msx2 gene did not demonstrate noticeable differences between controls and mutants at any stage.
Fgf8, another gene important for facial midline development, displayed a normal characteristic expression pattern in both controls and mutants at E10. A notable difference in the shape of the maxillary processes between mutants and controls on E11 did not allow more direct comparison of Fgf8 expression, but Fgf8 was clearly more intensely expressed in the mutant anterior maxillary ectoderm at this stage (Fig. 9J), the location where physiological Fgf8 expression does not occur.
A homeodomain gene Tgif (Bertolino et al., 1995) was strongly expressed in controls at E11 in the maxillary and mandibular processes of the first pharyngeal arch, as well as in the frontonasal processes. In corresponding Alk5/Wnt1-Cre mutants, expression of Tgif was noticeably reduced in hypoplastic maxillary and frontonasal primordia, and also in normally shaped temporoparietal and neck regions (Fig. 9L).
Endogenous co-expression of Alk5 with Gdf11 and type II receptors in the facial primordia
As outlined above, the facial phenotypes of Alk5/Wnt1-Cre mutants were more severe than those of the corresponding Tgfbr2/Wnt1-Cre mutants (Ito et al., 2003). This raises the possibility that also other type II receptors, in conjunction with related TGF-β superfamily ligands, could activate ALK5 in the MEE and neural crest cells. Indeed, ligands such as GDFs 8, 9, and 11 have been shown to signal via ALK5 and activin type II receptors (Mazerbourg et al., 2004; Rebbapragada et al., 2003; Oh et al., 2002). Whereas GDF8 controls the growth of skeletal muscles (Hamrick et al., 2002; McPherron and Lee, 1997, 2002), and GDF9 is predominantly expressed in oocytes (Hanrahan et al., 2004; Carabatsos et al., 1998; Laitinen et al., 1998; McPherron and Lee, 1993), GDF11 is a likely candidate to function also in craniofacial morphogenesis. In addition to being expressed in the tail bud, as previously reported (Nakashima et al., 1999), GDF11 was strongly expressed in maxillary and mandibular primordia of the first pharyngeal arch, in lateral and medial nasal processes, in the second pharyngeal arch, and palatal shelves (Figs. 10A–C). We have also compared the expression patterns of Alk5 and type II receptors at the onset of craniofacial morphogenesis (Figs. 10D–H). Alk5 was uniformly expressed in the developing head, including the pharyngeal arches, with the most intense signal arising from the forebrain. Type II receptors displayed typical widespread expression with certain small areas staining stronger than the rest of the tissues, demonstrating a significant overlap with Alk5 expression.
Fig. 10.
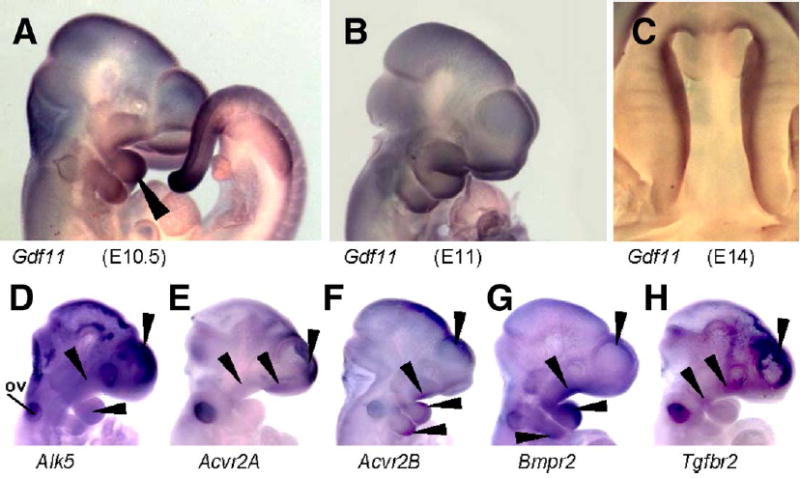
Endogenous co-expression of Alk5, type-II receptors, and Gdf11 in the facial primordia demonstrated by in situ hybridization. (A–C) Expression of Gdf11 in the tail and craniofacial primordia. Long arrowhead points to a strong signal in the mandibular arch at E10.5. (D–H) Whole-mount in situ hybridization with riboprobes for Alk5 (D), Acvr2A (E), Acvr2B (F), Bmpr2 (G), and Tgfbr2 (H). Arrowheads point to a positive signal in the nasal, maxillary and mandibular processes, second pharyngeal arch, and forebrain. ov—otic vesicle.
ALK5 can form functional signaling complexes with unconventional type II receptors in vitro
To further explore the possibility that ALK5 could be activated by several different type II receptor kinases in vivo, we made use of the rapamycin-based regulated heterodimerization system (Stockwell and Schreiber, 1998; Rivera et al., 1996). This methodology is based on a finding that the non-immunosuppressive rapamycin analogue AP21967 induces heterodimerization between an FKBP12 protein domain and a mutated version of a large PI3K homolog FRAP called FRB*, without interfering with the activity of endogenous FRAP (Pollock et al., 2000).
We constructed a mammalian expression vector encoding the FRB*-ALK5 cytoplasmic domain fusion protein (FRB*-ALK5cyt). In addition, we generated vectors containing a myristoylation sequence, two FKBP domains, and either the TGFβRII kinase domain (Myr-FKBP-TGFβRII-kin) or ACV-RIIB kinase domain (Myr-FKBP-ACVRIIB-kin) (Figs. 11A–B). We expected that these constructs could associate conditionally upon addition of the heterodimerizer AP21967, which would subsequently lead to the phosphorylation of a downstream signal transducer, Smad2, as shown in the schematic model (Fig. 11B). The constructs were transfected into Tgfbr2-deficient DR26 cells together with the Smad2 expression vector. After 24h, the cellular extracts were prepared and analyzed for Smad2 phosphorylation.
Fig. 11.
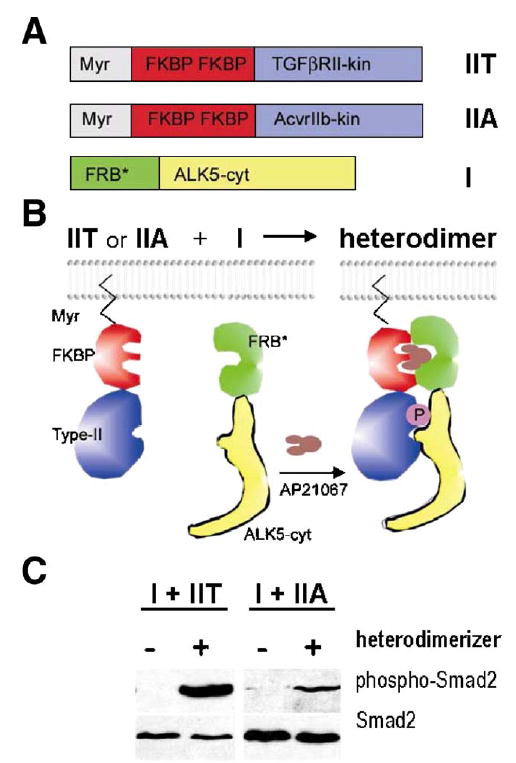
Activation of ALK5 by two different type II receptors in vitro. Regulated heterodimerization was used as a model system to test whether ALK5 may also be activated by ACVRIIB in addition to canonical TGFβRII. (A) Expression constructs for type II receptors contained N-terminal myristoylation signal (Myr), two copies of FK506 binding domains (FKBP), and regions coding for kinase domains of ACVRIIB (construct IIA in panel A) and TGFβRII (construct IIT in panel A), respectively. The ALK5 cytoplasmic domain was fused to FKBP-rapamycin binding domain (FRB*, see Materials and methods; construct I in panel A). (B) Heterodimerizing agent (AP21967, Ariad Pharmaceuticals; brown in the scheme) can be used to induce regulated heterodimerization of chimeric type I and type II receptor fusion proteins expressed in cell culture. (C) Western blot analysis shows that the kinase domain of ACVRIIB was able to activate the ALK5 fusion protein in Tgfbr2-deficient DR26 cells co-transfected with the Smad2 expression vector, albeit less efficiently than the corresponding TGFβRII domain, as demonstrated by phosphorylation of the effector protein Smad2. Left panel-exposure 5min; right panel—exposure 20min.
In the absence of the dimerizer, Smad2 phosphorylation was undetectable. Addition of the dimerizer to cells co-transfected with Myr-FKBP-TGFβRII-kin and FRB-ALK5cyt induced strong Smad2 phosphorylation, consistent with the earlier findings of Stockwell and Schreiber. They showed that this system can be used to investigate the mechanisms of Smad activation resulting from formation of a complex between cytoplasmic domains of TGFβRI and TGFβRII (Stockwell and Schreiber, 1998). Moreover, Myr-FKBP-ACVRIIB-kin and FRB-ALK5cyt fusion proteins, in the presence of the heterodimerizer, could also stimulate Smad2 phosphorylation, albeit more than 10-fold less efficiently than TGFβRII and ALK5 fusion proteins under identical conditions (Fig. 11C).
To conclude, these experiments provide direct biochemical evidence that ALK5 activation can be induced not only the kinase activity of TGFβRII, but also by the other type II receptor kinases such as ACVRIIB, provided that they are properly associated with each other.
Discussion
TGF-β3/ALK5 signaling and palatal fusion
The role of TGF-β signaling in palatogenesis is well established; Tgf-β3 expression is both spatially and temporally limited to the pre-fusion palatal epithelium (Fitzpatrick et al., 1990; Akhurst et al., 1990; Pelton et al., 1990a,b), and mice deficient in TGF-β3 demonstrate either a complete bilateral clefting of the secondary palate (50% of offspring), or posterior clefting and superficial anterior adherence (Kaartinen et al., 1995; Proetzel et al., 1995). TGF-β3, as a prototypical TGF-β ligand, signals via TGFβRII and ALK5, and therefore it was expected that mice lacking one of these receptors in the MEE would display a phenotype similar to that of Tgf-β3−/ − mice. In contrast, Alk5/K14-Cre consistently demonstrate posterior clefting and superficial adherence of the middle and anterior palate, but never a complete palatal cleft. These differences could be at least partly attributed to residual expression of Alk5 in the palatal epithelium as a result of incomplete K14-Cre-induced recombination in the Alk5flox locus. Alternatively, although no apparent changes in mesenchymal proliferation have been found in palatal shelves of these mutants, it is possible that epithelially expressed TGF-β3 enhances reshaping of the adjacent mesenchyme in the tips of Alk5/K14-Cre palatal shelves, thus locally improving the shelf midline contact-which cannot occur in mice completely lacking TGF-β3.
In agreement with the most recent observations (Vaziri et al., 2005), we show that epithelial–mesenchymal transdifferentiation is not a major TGF-β3-driven mechanism of palatal fusion. Furthermore, we demonstrate that addition of TGF-β3 beads into the Tgf-β3−/ − midline seam induces apoptosis, providing additional support for the critical role of programmed cell death as a primary mechanism of the MES breakdown (Martinez-Alvarez et al., 2000). As can be seen in Fig. 2, the increase in apoptosis is not only limited to the close proximity of the soaked beads, but also occurs in more remote parts of the MES. This may reflect the fact that the beads could not be firmly immobilized between the two slippery shelves and traveled either up or down before reaching their final position (thus releasing TGF-β3 in multiple locations), or this may have occurred through protein diffusion.
Anterior–posterior differences in palates of Alk5/K14-Cre mutants
As noted in Tgf-β3−/ − mice (Dudas et al., 2004a; Kaartinen et al., 1995), Alk5/K14-Cre mutants also show a difference in fusion of the anterior and posterior parts of the palate, a finding that arises as an intriguing biological problem in current developmental biology (Hilliard et al., 2005). A lack of palatal fusion in organ culture together with observations of establishing the palatal midline contact suggests that the posterior cleft in Alk5/K14-Cre mutants cannot be attributed to anterior–posterior differences in the fusion capability of the MEE. Normally, the very anterior and posterior parts of the secondary palate come in contact last and the shape of medial edges of palatal shelves is not straight but slightly rounded in the anterior and posterior aspects. As the shelves meet, the midline epithelial seam gradually disappears by programmed cell death, starting at the site where the contact was first established. This leads to a tighter contact between the apposing shelves, assisting other parts of the palate on the edges of the contact area to get closer to each other. Therefore, it is not surprising that anterior and posterior parts of the midline seam may be the first to break when appropriate epithelial fusion has not occurred, as seen in Alk5/K14-Cre mutants, and shelves become pulled apart by lateral growth of the head. Progressive elongation of the posterior cleft up to 20% of the palate length may result partly from the growth of the soft palate in length, and partly from post-adhesion seam rupture.
Filopodia and adherence of palatal shelves
The intercalation of cell membrane protrusions has been proposed as an important part of the machinery driving the fusion of epithelial edges in multiple organs during embryonic development across a wide range of animal species (Martin and Parkhurst, 2004). Furthermore, it has been suggested that a total failure of this process causes cleft palate in Tgf-β3 null mice (Taya et al., 1999). Our present data show that both Tgf-β3 null and Alk5/K14-Cre mutants suffer from a dramatic decrease in the proportion of MEE cells carrying filopodia. However, the morphological appearance of the rare cells with filopodia does not differ from those found abundantly in controls. Keeping in mind that a significant part of the palatal edges is tightly adhered in both mutants (Taya et al., 1999; Kaartinen et al., 1995), and that we did not find any anterior–posterior differences in the filopodia occurrence, we suggest that filopodia are not an absolute prerequisite for palatal adherence. The membrane protrusions in the MEE may play a role in navigation of apposing shelves towards each other in vivo, and/or may simply represent a phenotype of terminally differentiated MEE cells (Takigawa and Shiota, 2004).
Unique spectrum of craniofacial phenotypes in mice lacking ALK5 in neural crest cells
Several different signaling pathways have been suggested to function cell-autonomously in NC cells to regulate essential morphogenetic events during craniofacial development (Brewer et al., 2004; Dudas et al., 2004b; Ito et al., 2003; Tallquist and Soriano, 2003; Brault et al., 2001). As we report here, inactivation of the TGF-β type I receptor gene Alk5 in the neural crest severely impairs craniofacial development with 100% penetrance. Deletion of Alk5 in the neural crest affected also cardiovascular development, similarly as in the case of Alk2/Wnt1-Cre mutants (Kaartinen et al., 2004). Detailed investigation has revealed that craniofacial and cardiovascular phenotypes are not dependent on each other. Although the present study is focused on our findings within the craniofacial region, the complex cardiovascular defects will be analyzed elsewhere.
It is noteworthy that the observed spectrum of facial malformations in Alk5/Wnt1-Cre mutants is truly unique among neural crest cell phenotypes. Particularly, the nasal cleft is rarely seen in mouse mutants (Tallquist and Soriano, 2003; Beverdam et al., 2001; Lohnes et al., 1994). Similarly, severe midfacial cleft deformities, which occur either with or without associated holoprosencephaly, are also rare in humans (El Hawrani et al., 2006). Because our fate mapping studies showed that NC cells migrate normally to populate the craniofacial region in Alk5/Wnt1-Cre mutants, the severe phenotype appears to be caused, at least in part, by an early onset of a dramatic increase in mesenchymal apoptosis. This was detected in the first pharyngeal arch beginning at E10, i.e., around the time when the first visible phenotypic alterations appear. Although apoptotic cells often formed distinctive groups in multiple locations, it was not possible to match these patches with the primordia of individual anatomical structures, or to find any correlation of apoptotic regions with known expression patterns or pattern overlaps of any genes. From this stage onwards, the shape of facial prominences in mutants became morphologically altered to the extent that it was not possible to obtain sections matching controls. Later, palatal shelves of Alk5/Wnt1-Cre mutants at E14 demonstrated highly elevated mesenchymal apoptosis. Although we could not detect any differences in cell proliferation in the pharyngeal arch mesenchyme at E10 or E11, the palatal mesenchyme displayed a significant increase in cell proliferation. In contrast, corresponding Tgfbr2/Wnt1-Cre mutants displayed decreased levels of cell proliferation in the palatal mesenchyme (Ito et al., 2003), suggesting that ALK5 and TGFβRII mediate different responses in certain NC-derived tissues.
To conclude, the maxillary region is severely affected by the cell death in the first pharyngeal arch of Alk5/Wnt1-Cre mutants before the palatal shelves are even formed. The initially insufficient size of the palate is worsened by a persistently high rate of cell death within the palatal mesenchyme. This cannot be balanced by an increase in cell proliferation in later stages, which we think occurs as a compensative effect secondary to an apoptotic tissue loss.
Signaling via ALK5 differs from the TGFβRII-mediated signaling in vivo during craniofacial development
The defects in calvaria of the Alk5/Wnt1-Cre mice are analogous with those found in the corresponding Tgfbr2/Wnt1-Cre mutants (Ito et al., 2003). In both mutants, defects occurred not only in bones derived from the neural crest, but also in mesenchyme-derived structures. Because the role of TGF-β signaling in morphogenesis of cranial bones has already been studied in detail in Tgfbr2/Wnt1-Cre mutants (Sasaki et al., 2006), this study does not address calvarial development.
Interestingly, facial phenotypes of Alk5 mutant mice are far more severe than those seen in Tgfbr2/Wnt1-Cre embryos. To explain this phenotypic difference, we considered the possibility that during facial morphogenesis, ALK5 could become activated also by the other type II receptors. For example, GDF11, which is a ligand closely related to TGF-βs, has been shown in cell culture studies to bind ACVRIIA as well as ACVRIIB, and to activate Smad2 possibly via ALK5 (Mazerbourg et al., 2004; Oh et al., 2002). Here we show that Gdf11 is strongly expressed both in maxillary and mandibular primordia at E10–E11, and in the pre-fusion palatal MEE. Moreover, we show that its putative binding partner Acvr2B is strongly expressed in the developing facial prominences. These findings are in concordance with earlier studies demonstrating that both Gdf11 and Acvr2B null mutants exhibit cleft palate with variable penetrance (Ferguson et al., 2001; McPherron et al., 1999; Matzuk et al., 1995).
Consequently, we utilized the regulated heterodimerization system in a cell culture model to show that, at least in this simulated set-up, the kinase domain of ACVRIIB can activate the intracellular domain of ALK5, albeit less efficiently than that of TGFβRII. Therefore, GDF11 signaling via ACVRIIB and ALK5 in the neural crest derived facial mesenchyme, may account for different phenotypes we see in Alk5 mutants, when compared to corresponding Tgfbr2 mutants. Although not detected in our experiments, it is possible that ALK5 may also form signaling complexes with additional type II receptors, and/ or transmit signals mediated by more members of the TGF-β superfamily ligands than currently known. Thus, widespread and uniform deletion of Alk5 in the ectomesenchymal cells would be reflected in a complex phenotype with spatiotemporally complex pattern of localized expression changes of various genes, which is fully consistent with our findings in Alk5/Wnt1-Cre mutants.
Changes in gene expression in Alk5/Wnt1-Cre mutants
TGF-β superfamily signaling via Smads is linked to the etiology of craniofacial malformations with severe midline defects (Dudas and Kaartinen, 2005; Liu et al., 2004; Nomura and Li, 1998). Although the Alk5 gene in our Alk5/Wnt1-Cre mutants has been deleted in a large number of cells, including the entire craniofacial ectomesenchyme, increased apoptosis and changes in gene expression occurred in a regionalized and uneven manner, with no apparent overlap with expression patterns of genes encoding the TGF-β superfamily members, receptors, and downstream mediators. Seeing a large number of various structures affected in Alk5/Wnt1-Cre mutants, encompassing all pharyngeal arches, middle ear, frontonasal region, and skull, it is likely that indeed there is not a single universal TGF-β downstream gene globally and uniformly affected in all of these locations. Instead, it is more likely that expression patterns of multiple different genes are influenced by ALK5 signaling at different time points in small areas all over the head and neck region.
Our results show significantly reduced expression of the homeobox gene Msx1 in the maxillary primordia of Alk5/Wnt1-Cre mutants. The Msx genes, which are involved in craniofacial development both in mice (Satokata and Maas, 1994) and humans (Lidral et al., 1998), are required for NCC survival (Ishii et al., 2005). Therefore, a dramatic reduction in Msx1 expression could explain increased apoptosis and subsequent morphological changes localized in the maxillary derivatives. However, Msx1 expression was not affected in the mandibular process, indicating that ALK5-mediated signaling initiates different downstream responses in maxillary and mandibular primordia.
Expression of Tgif, a gene involved in craniofacial morphogenesis (Gripp et al., 2000; Wallis and Muenke, 1999), was altered in the upper portion of the maxillary arch, and in the supranasal region. TGIF is a member of the TALE superfamily of homeodomain proteins, which can recruit multiple transcriptional corepressors and repress a diverse set of genes (Wotton and Massague, 2001; Bertolino et al., 1995), including interactions with the TGF-β-activated Smads and repression of TGF-β target genes (Wotton and Massague, 2001; Wotton et al., 1999). Thus, our current results suggest that TGIF may represent a novel feedback mechanism to control the level of Smad activation in the developing face.
Alk5/Wnt1-Cre mutants also displayed subtle changes in the expression of Fgf8, which is another important player in craniofacial development (Gong et al., 2005; Albertson and Yelick, 2005; Mina, 2001; Tucker et al., 1999; Trumpp et al., 1999), with known interactions with TGF-β/BMP signaling (Shigetani et al., 2000), including anterior–posterior patterning of the first pharyngeal arch (Liu et al., 2005; Barlow et al., 1999). Altogether, our results suggest that TGF-β/ALK5 signaling is a critically important and widespread morphogenetic regulator of craniofacial development. In future, multiple detailed studies focusing on individual anatomical structures will be required to explain precisely how the ALK5 signaling is orchestrated with numerous other signaling pathways, and to elucidate its complex spatiotemporal mode of operation.
Acknowledgments
We thank A. McMahon and S. Millar for providing the Wnt1-Cre and K14-Cre mouse lines, S. Bellusci and A. Joyner for probes, J. Massague for DR26 cells, and R. Derynck for cDNAs. MD was supported by the CHLA RCDF Award, YC by grants from the NIH, and VK by grants from the Robert E. Schneider Foundation and the NIH (HL074862 and DE013085).
References
- Akhurst RJ, Fitzpatrick DR, Gatherer D, Lehnert SA, Millan FA. Transforming growth factor betas in mammalian embryogenesis. Prog Growth Factor Res. 1990;2:153–168. doi: 10.1016/0955-2235(90)90002-2. [DOI] [PubMed] [Google Scholar]
- Albertson RC, Yelick PC. Roles for fgf8 signaling in left-right patterning of the visceral organs and craniofacial skeleton. Dev Biol. 2005;283:310–321. doi: 10.1016/j.ydbio.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Andl T, Ahn K, Kairo A, Chu EY, Wine-Lee L, Reddy ST, Croft NJ, Cebra-Thomas JA, Metzger D, Chambon P, Lyons KM, Mishina Y, Seykora JT, Crenshaw EB, III, Millar SE. Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development. 2004;131:2257–2268. doi: 10.1242/dev.01125. [DOI] [PubMed] [Google Scholar]
- Barlow AJ, Bogardi JP, Ladher R, Francis-West PH. Expression of chick Barx-1 and its differential regulation by FGF-8 and BMP signaling in the maxillary primordia. Dev Dyn. 1999;214:291–302. doi: 10.1002/(SICI)1097-0177(199904)214:4<291::AID-AJA2>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Bei M, Maas R. FGFs and BMP4 induce both Msx1-independent and Msx1-dependent signaling pathways in early tooth development. Development. 1998;125:4325–4333. doi: 10.1242/dev.125.21.4325. [DOI] [PubMed] [Google Scholar]
- Bertolino E, Reimund B, Wildt-Perinic D, Clerc RG. A novel homeobox protein which recognizes a TGT core and functionally interferes with a retinoid-responsive motif. J Biol Chem. 1995;270:31178–31188. doi: 10.1074/jbc.270.52.31178. [DOI] [PubMed] [Google Scholar]
- Beverdam A, Brouwer A, Reijnen M, Korving J, Meijlink F. Severe nasal clefting and abnormal embryonic apoptosis in Alx3/Alx4 double mutant mice. Development. 2001;128:3975–3986. doi: 10.1242/dev.128.20.3975. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Brewer S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol. 2004;267:135–152. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Carabatsos MJ, Elvin J, Matzuk MM, Albertini DF. Characterization of oocyte and follicle development in growth differentiation factor-9-deficient mice. Dev Biol. 1998;204:373–384. doi: 10.1006/dbio.1998.9087. [DOI] [PubMed] [Google Scholar]
- Couly GF, Coltey PM, Le Douarin NM. The triple origin of skull in higher vertebrates: a study in quail-chick chimeras. Development. 1993;117:409–429. doi: 10.1242/dev.117.2.409. [DOI] [PubMed] [Google Scholar]
- Cuervo R, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- Cui XM, Shiomi N, Chen J, Saito T, Yamamoto T, Ito Y, Bringas P, Chai Y, Shuler CF. Overexpression of Smad2 in Tgf-beta3-null mutant mice rescues cleft palate. Dev Biol. 2005;278:193–202. doi: 10.1016/j.ydbio.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen- inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Darling DS, Stearman RP, Qi Y, Qiu MS, Feller JP. Expression of Zfhep/deltaEF1 protein in palate, neural progenitors, and differentiated neurons. Gene Expr Patterns. 2003;3:709–717. doi: 10.1016/s1567-133x(03)00147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Feng XH. TGF-beta receptor signaling. Biochim Biophys Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- Dickson MC, Slager HG, Duffie E, Mummery CL, Akhurst RJ. RNA and protein localisations of TGF beta 2 in the early mouse embryo suggest an involvement in cardiac development. Development. 1993;117:625–639. doi: 10.1242/dev.117.2.625. [DOI] [PubMed] [Google Scholar]
- Dudas M, Kaartinen V. Tgf-Beta superfamily and mouse craniofacial development: interplay of morphogenetic proteins and receptor signaling controls normal formation of the face. Curr Top Dev Biol. 2005;66:65–133. doi: 10.1016/S0070-2153(05)66003-6. [DOI] [PubMed] [Google Scholar]
- Dudas M, Nagy A, Laping NJ, Moustakas A, Kaartinen V. Tgf-beta3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev Biol. 2004a;266:96–108. doi: 10.1016/j.ydbio.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Dudas M, Sridurongrit S, Nagy A, Okazaki K, Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech Dev. 2004b;121:173–182. doi: 10.1016/j.mod.2003.12.003. [DOI] [PubMed] [Google Scholar]
- El Hawrani A, Sohn M, Noga M, El Hakim H. The face does predict the brain-Midline facial and forebrain defects uncovered during the investigation of nasal obstruction and rhinorrhea Case report and a review of holoprosencephaly and its classifications. Int J Pediatr Otorhinolaryngol. 2006;70:935–940. doi: 10.1016/j.ijporl.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Ferguson MW. Palate development. Development. 1988;103(Suppl):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Ferguson CA, Tucker AS, Heikinheimo K, Nomura M, Oh P, Li E, Sharpe PT. The role of effectors of the activin signalling pathway, activin receptors IIA and IIB, and Smad2, in patterning of tooth development. Development. 2001;128:4605–4613. doi: 10.1242/dev.128.22.4605. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Denhez F, Kondaiah P, Akhurst RJ. Differential expression of TGF beta isoforms in murine palatogenesis. Development. 1990;109:585–595. doi: 10.1242/dev.109.3.585. [DOI] [PubMed] [Google Scholar]
- Francis-West P, Ladher R, Barlow A, Graveson A. Signalling interactions during facial development. Mech Dev. 1998;75:3–28. doi: 10.1016/s0925-4773(98)00082-3. [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL. Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- Gong SG, Gong TW, Shum L. Identification of markers of the midface. J Dent Res. 2005;84:69–72. doi: 10.1177/154405910508400112. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Hamrick MW, McPherron AC, Lovejoy CO. Bone mineral content and density in the humerus of adult myostatin-deficient mice. Calcif Tissue Int. 2002;71:63–68. doi: 10.1007/s00223-001-1109-8. [DOI] [PubMed] [Google Scholar]
- Hanrahan JP, Gregan SM, Mulsant P, Mullen M, Davis GH, Powell R, Galloway SM. Mutations in the genes for oocyte-derived growth factors GDF9 and BMP15 are associated with both increased ovulation rate and sterility in Cambridge and Belclare sheep (Ovis aries) Biol Reprod. 2004;70:900–909. doi: 10.1095/biolreprod.103.023093. [DOI] [PubMed] [Google Scholar]
- Hilliard SA, Yu L, Gu S, Zhang Z, Chen YP. Regional regulation of palatal growth and patterning along the anterior–posterior axis in mice. J Anat. 2005;207:655–667. doi: 10.1111/j.1469-7580.2005.00474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan, B., Beddington, R., Costantini, F., Lacy, E., 1994. Manipulating the Mouse Embryo. A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York.
- Ishii M, Merrill AE, Chan YS, Gitelman I, Rice DP, Sucov HM, Maxson RE., Jr Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development. 2003;130:6131–6142. doi: 10.1242/dev.00793. [DOI] [PubMed] [Google Scholar]
- Ishii M, Han J, Yen HY, Sucov HM, Chai Y, Maxson RE., Jr Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development. 2005;132:4937–4950. doi: 10.1242/dev.02072. [DOI] [PubMed] [Google Scholar]
- Ito Y, Yeo JY, Chytil A, Han J, Bringas P, Jr, Nakajima A, Shuler CF, Moses HL, Chai Y. Conditional inactivation of Tgfbr2 in cranial neural crest causes cleft palate and calvaria defects. Development. 2003;130:5269–5280. doi: 10.1242/dev.00708. [DOI] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottmann AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Laitinen M, Vuojolainen K, Jaatinen R, Ketola I, Aaltonen J, Lehtonen E, Heikinheimo M, Ritvos O. A novel growth differentiation factor-9 (GDF-9) related factor is co-expressed with GDF-9 in mouse oocytes during folliculogenesis. Mech Dev. 1998;78:135–140. doi: 10.1016/s0925-4773(98)00161-0. [DOI] [PubMed] [Google Scholar]
- Larsson J, Goumans MJ, Sjostrand LJ, van Rooijen MA, Ward D, Leveen P, Xu X, ten Dijke P, Mummery CL, Karlsson S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001;20:1663–1673. doi: 10.1093/emboj/20.7.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavrin IO, McLean W, Seegmiller RE, Olsen BR, Hay ED. The mechanism of palatal clefting in the Col11a1 mutant mouse. Arch Oral Biol. 2001;46:865–869. doi: 10.1016/s0003-9969(01)00044-9. [DOI] [PubMed] [Google Scholar]
- Lidral AC, Romitti PA, Basart AM, Doetschman T, Leysens NJ, Daack-Hirsch S, Semina EV, Johnson LR, Machida J, Burds A, Parnell TJ, Rubenstein JL, Murray JC. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet. 1998;63:557–568. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Festing M, Thompson JC, Hester M, Rankin S, El Hodiri HM, Zorn AM, Weinstein M. Smad2 and Smad3 coordinately regulate craniofacial and endodermal development. Dev Biol. 2004;270:411–426. doi: 10.1016/j.ydbio.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Liu W, Selever J, Murali D, Sun X, Brugger SM, Ma L, Schwartz RJ, Maxson R, Furuta Y, Martin JF. Threshold-specific requirements for Bmp4 in mandibular development. Dev Biol. 2005;283:282–293. doi: 10.1016/j.ydbio.2005.04.019. [DOI] [PubMed] [Google Scholar]
- Lohnes D, Mark M, Mendelsohn C, Dolle P, Dierich A, Gorry P, Gansmuller A, Chambon P. Function of the retinoic acid receptors (RARs) during development: I. Craniofacial and skeletal abnormalities in RAR double mutants. Development. 1994;120:2723–2748. doi: 10.1242/dev.120.10.2723. [DOI] [PubMed] [Google Scholar]
- Martin P, Parkhurst SM. Parallels between tissue repair and embryo morphogenesis. Development. 2004;131:3021–3034. doi: 10.1242/dev.01253. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Tudela C, Perez-Miguelsanz J, O'Kane S, Puerta J, Ferguson MW. Medial edge epithelial cell fate during palatal fusion. Dev Biol. 2000;220:343–357. doi: 10.1006/dbio.2000.9644. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Blanco MJ, Perez R, Rabadan MA, Aparicio M, Resel E, Martinez T, Nieto MA. Snail family members and cell survival in physiological and pathological cleft palates. Dev Biol. 2004;265:207–218. doi: 10.1016/j.ydbio.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–360. doi: 10.1038/374356a0. [DOI] [PubMed] [Google Scholar]
- Mazerbourg S, Klein C, Roh J, Kaivo-Oja N, Mottershead DG, Korchynskyi O, Ritvos O, Hsueh AJ. Growth differentiation factor-9 signaling is mediated by the type I receptor, activin receptor-like kinase 5. Mol Endocrinol. 2004;18:653–665. doi: 10.1210/me.2003-0393. [DOI] [PubMed] [Google Scholar]
- McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. GDF-3 and GDF-9: two new members of the transforming growth factor-beta superfamily containing a novel pattern of cysteines. J Biol Chem. 1993;268:3444–3449. [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Suppression of body fat accumulation in myostatin-deficient mice. J Clin Invest. 2002;109:595–601. doi: 10.1172/JCI13562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of anterior/ posterior patterning of the axial skeleton by growth/differentiation factor 11. Nat Genet. 1999;22:260–264. doi: 10.1038/10320. [DOI] [PubMed] [Google Scholar]
- Mina M. Morphogenesis of the medial region of the developing mandible is regulated by multiple signaling pathways. Cells Tissues Organs. 2001;169:295–301. doi: 10.1159/000047894. [DOI] [PubMed] [Google Scholar]
- Moorman AF, Houweling AC, de Boer PA, Christoffels VM. Sensitive nonradioactive detection of mRNA in tissue sections: novel application of the whole-mount in situ hybridization protocol. J Histochem Cytochem. 2001;49:1–8. doi: 10.1177/002215540104900101. [DOI] [PubMed] [Google Scholar]
- Nakashima M, Toyono T, Akamine A, Joyner A. Expression of growth/differentiation factor 11, a new member of the BMP/TGFbeta superfamily during mouse embryogenesis. Mech Dev. 1999;80:185–189. doi: 10.1016/s0925-4773(98)00205-6. [DOI] [PubMed] [Google Scholar]
- Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M, Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes Dev. 2002;16:2749–2754. doi: 10.1101/gad.1021802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton RW, Dickinson ME, Moses HL, Hogan BL. In situ hybridization analysis of TGF beta 3 RNA expression during mouse development: comparative studies with TGF beta 1 and beta 2. Development. 1990a;110:609–620. doi: 10.1242/dev.110.2.609. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Hogan BL, Miller DA, Moses HL. Differential expression of genes encoding TGFs beta 1, beta 2, and beta 3 during murine palate formation. Dev Biol. 1990b;141:456–460. doi: 10.1016/0012-1606(90)90401-4. [DOI] [PubMed] [Google Scholar]
- Pollock R, Issner R, Zoller K, Natesan S, Rivera VM, Clackson T. Delivery of a stringent dimerizer-regulated gene expression system in a single retroviral vector. Proc Natl Acad Sci U S A. 2000;97:13221–13226. doi: 10.1073/pnas.230446297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Tucker SC, Zhao Q, deCrombrugghe B, Wisdom R. Physical and genetic interactions between Alx4 and Cart1. Development. 1999;126:359–369. doi: 10.1242/dev.126.2.359. [DOI] [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor beta-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial–mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, Magari SR, Phillips T, Courage NL, Cerasoli F, Jr, Holt DA, Gilman M. A humanized system for pharmacologic control of gene expression. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- Saika S, Saika S, Liu CY, Azhar M, Sanford LP, Doetschman T, Gendron RL, Kao CW, Kao WW. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Ito Y, Xu X, Han J, Bringas P, Jr, Maeda T, Slavkin HC, Grosschedl R, Chai Y. LEF1 is a critical epithelial survival factor during tooth morphogenesis. Dev Biol. 2005;278:130–143. doi: 10.1016/j.ydbio.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ito Y, Bringas P, Jr, Chou S, Urata MM, Slavkin H, Chai Y. TGF{beta}-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Development. 2006;133:371–381. doi: 10.1242/dev.02200. [DOI] [PubMed] [Google Scholar]
- Satokata I, Maas R. Msx1 deficient mice exhibit cleft palate and abnormalities of craniofacial and tooth development. Nat Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- Shigetani Y, Nobusada Y, Kuratani S. Ectodermally derived FGF8 defines the maxillomandibular region in the early chick embryo: epithelial-mesenchymal interactions in the specification of the craniofacial ectomesenchyme. Dev Biol. 2000;228:73–85. doi: 10.1006/dbio.2000.9932. [DOI] [PubMed] [Google Scholar]
- Shuler CF. Programmed cell death and cell transformation in craniofacial development. Crit Rev Oral Biol Med. 1995;6:202–217. doi: 10.1177/10454411950060030301. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stockwell BR, Schreiber SL. Probing the role of homomeric and heteromeric receptor interactions in TGF-beta signaling using small molecule dimerizers. Curr Biol. 1998;8:761–770. doi: 10.1016/s0960-9822(98)70299-4. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Ueno N, Hemmati-Brivanlou A. Xenopus msx1 mediates epidermal induction and neural inhibition by BMP4. Development. 1997;124:3037–3044. doi: 10.1242/dev.124.16.3037. [DOI] [PubMed] [Google Scholar]
- Takigawa T, Shiota K. Terminal differentiation of palatal medial edge epithelial cells in vitro is not necessarily dependent on palatal shelf contact and midline epithelial seam formation. Int J Dev Biol. 2004;48:307–317. doi: 10.1387/ijdb.041840tt. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- Tanaka A, Miyamoto K, Minamino N, Takeda M, Sato B, Matsuo H, Matsumoto K. Cloning and characterization of an androgen-induced growth factor essential for the androgen-dependent growth of mouse mammary carcinoma cells. Proc Natl Acad Sci U S A. 1992;89:8928–8932. doi: 10.1073/pnas.89.19.8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taya Y, O'Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Trumpp A, Depew MJ, Rubenstein JL, Bishop JM, Martin GR. Cre-mediated gene inactivation demonstrates that FGF8 is required for cell survival and patterning of the first branchial arch. Genes Dev. 1999;13:3136–3148. doi: 10.1101/gad.13.23.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AS, Al Khamis A, Sharpe PT. Interactions between Bmp-4 and Msx-1 act to restrict gene expression to odontogenic mesenchyme. Dev Dyn. 1998;212:533–539. doi: 10.1002/(SICI)1097-0177(199808)212:4<533::AID-AJA6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Tucker AS, Yamada G, Grigoriou M, Pachnis V, Sharpe PT. Fgf-8 determines rostral-caudal polarity in the first branchial arch. Development. 1999;126:51–61. doi: 10.1242/dev.126.1.51. [DOI] [PubMed] [Google Scholar]
- Vaziri SF, Hallberg K, Harfe BD, McMahon AP, Linde A, Gritli-Linde A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev Biol. 2005;285:490–495. doi: 10.1016/j.ydbio.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Wallis DE, Muenke M. Molecular mechanisms of holoprosencephaly. Mol Genet Metab. 1999;68:126–138. doi: 10.1006/mgme.1999.2895. [DOI] [PubMed] [Google Scholar]
- Wotton D, Massague J. Smad transcriptional corepressors in TGF beta family signaling. Curr Top Microbiol Immunol. 2001;254:145–164. [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massague J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]