

Summary
Glanzmann thrombasthenia (GT) is a rare autosomal recessive bleeding disorder caused by lack or dysfunction of αIIbβ3 in platelets. GT is relatively frequent in highly inbred populations. We previously identified a 13-bp deletion in the αIIb gene that causes in-frame deletion of six amino acids in three Palestinian GT patients. In this study, we determined the molecular basis of GT in all known Palestinian patients, examined whether Jordanian patients harbor the same mutations, analyzed whether there is a founder effect for the 13-bp deletion, and determined the mechanism by which the 13-bp deletion abolishes αIIbβ3 surface expression. Of 11 unrelated Palestinian patients, eight were homozygous for the 13-bp deletion that displayed common ancestry by haplotype analysis, and was estimated to have occurred 300–600 years ago. Expression studies in baby hamster kidney cells showed that substitution of Cys107 or Trp110 located within the deletion caused defective αIIbβ3 maturation. Substitution of Trp110, but not of Cys107, prevented fibrinogen binding. The other Palestinian patients harbored three novel mutations: G2374 deletion in αIIb gene, TT1616-7 deletion in β3 gene, and IVS14:−3C → G in β3 gene. The latter mutation caused cryptic splicing predicting an extended cytoplasmic tail of β3 and was expressed as dysfunctional αIIbβ3. None of 15 unrelated Jordanian patients carried any of the described mutations.
Keywords: founder effect, integrin αIIbβ3, variant Glanzmann thrombasthenia
Introduction
Glanzmann thrombasthenia (GT) is characterized by a moderate to severe mucocutaneous bleeding tendency, a normal platelet count, absence or reduced clot retraction, and absence or severely reduced platelet aggregation in response to all agonists except for ristocetin. The disease is inherited in an autosomal recessive fashion and is caused by lack or dysfunction of platelet αIIbβ3 that normally functions as a receptor for fibrinogen and von Willebrand factor and is essential for platelet aggregation. GT is rare worldwide but relatively frequent in inbred populations such as Iraqi-Jews [1], Indians [2], French Gypsies [3], Arabs in Jordan [4], Israel [5] and Saudi Arabia [6]. Many mutations in the αIIb or β3 genes have been identified in patients with GT including minor or major deletions, insertions, inversions and mostly point mutations located throughout both genes (for further information see database http://sinaicentral.mssm.edu/intranet/research/glanzmann/menu). Founder effects were discerned for mutations identified in Iraqi-Jews [7] and French Gypsies [8]. We have previously identified a 13-bp deletion at the intron 3–exon 4 junction of the αIIb gene in three Palestinian-Arab kindreds with members affected by GT. The deletion was shown to give rise to alternative splicing predicting an in-frame deletion of six amino acids [5]. The six deleted residues (Ala106-Gln111) are located in the second blade of the β-propeller domain, which interfaces with the βA domain of β3 through interaction of Trp110 of αIIb with Arg261 of β3 [9,10]. Among the deleted residues, Cys 107 was suspected to be critical for normal folding of αIIb and αIIbβ3 complex formation [5].
In this study, we examined whether the 13-bp deletion is also present in other unrelated Palestinian-Arab patients residing in Israel and the Palestinian territories or Jordanian-Arabs, and whether the mutation stems from a common founder. We also studied the mechanism by which the 13-bp deletion abolishes αIIbβ3 surface expression, as well as the molecular basis of GT in Palestinian patients not bearing the 13-bp deletion.
Methods
Patients
Eight Palestinian-Arab patients (four residing in Israel and four in the Palestinian territories) who did not know of any family connection to each other and who had lifelong mucocutaneous bleeding were referred to the Chaim Sheba Medical Center in Israel for evaluation between 1991 and 2004. Six were males and two were females whose age ranged between 3–29 years. Three previously described probands bearing the 13-bp deletion in the αIIb gene were also studied [5]. Ten of the 11 Palestinian probands were offspring of related parents. The diagnosis of GT patients was based on a normal platelet count, absent or minimal clot retraction, absent platelet aggregation in response to adenosine diphosphate (ADP), epinephrine, and collagen, and normal ristocetin-induced platelet aggregation. DNA samples of Jordanian GT patients belonging to 15 families were sent for analysis from the Rawhi Medical Center in Amman or from Children’s Mercy Hospital in Kansas City, MI by Dr Michael L. Begleiter. Control Palestinian-Arabs were patients consecutively admitted to the Chaim Sheba Medical Center in Israel. Approval of the study was obtained from the Institutional Review Board of the Chaim Sheba Medical Center.
Blood sampling and processing
Blood was taken in citrated buffer or in ethylenediaminetetra-acetic acid (EDTA) from the patients and healthy Arab controls. Citrated platelet-rich plasma (PRP) was used for flow cytometry and for immunoblot analysis of lysed platelets. DNA extraction from white blood cells was carried out in blood samples containing EDTA. RNA was extracted from washed platelets prepared from PRP using QuickPrep mRNA purification kit (Pharmacia Biotech, Piscataway, NJ, USA) and first strand cDNA was reverse transcribed using the reverse transcription system (Promega, Madison, WI, USA).
Flow cytometric analysis of platelets
A platelet suspension [20 μL of 200 000 cells/μL in phosphate buffered saline (PBS)] was incubated with fluorescein-isothiocyanate (FITC)-conjugated monoclonal antibodies against glycoprotein (GP) Ibα (CD42b; Dako, Glostrup, Denmark), αIIbβ3 (CD41/P2; Immunotech, Marseille, France), or αvβ3 (LM609; Chemicon, Temecula, CA, USA). Following incubation for 30 min at room temperature, platelets were washed with PBS and analyzed for cell-bound fluorescence in a flow cytometer (Coulter; EPICS, Luton, UK). The background was measured using FITC conjugated anti-mouse IgG.
Fibrinogen binding to resting and activated platelets
Diluted PRP (4 μL in 100 μL HEPES-buffered saline) was incubated for 2 min with 1.25 μM ADP (Dia-Med, Cressier sur Mora, Switzerland), 1.25 μg mL−1 collagen (Helena BioScience, Sunderland, UK), or with no agonist, and then 10 μL FITC-conjugated rabbit anti-human fibrinogen (Dako) was added. After 20 min, platelets were washed with HEPES-buffered saline and analyzed for cell-bound fluorescence in the flow cytometer. Non-specific binding was measured using FITC-conjugated anti mouse IgG.
Immunoblot analysis of platelet lysates
PRP was centrifuged at 14 000 g for 10 min and platelet pellets were lysed at 100 °C with loading buffer [40 mm Tris–HCl, pH 6.8, 1.5% sodium dodecyl sulphate (SDS), 8% glycerol, 1 mm 1,4-dithio-threitol (DTT), 0.01% bromophenol blue]. Samples were electrophoresed on NuPAGE Novex 3–8% Tris–Acetate gels (Invitrogen, Paisley, UK) and transferred to a polyvinyl-idenedifluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The membrane was immersed in Tris-buffered saline with 0.05% tween-20 (TBST) containing 1:1000 dilution of polyclonal antibody against αIIb [11] and polyclonal antibody against β3 (Chemicon), and then washed with TBST and immersed in TBST containing 1:2000 horseradish peroxidase conjugated-anti-rabbit IgG (Jackson Immuno Research, West Grove, PA, USA). Immunoreactive bands on the membrane were detected using an ECL immunoluminescence kit (Amersham, Buckinghamshire, UK) and X-ray film exposure.
Immunoprecipitation of the αIIbβ3 complex in platelet lysates was carried out by incubation of 10 μg 10E5 monoclonal antibody against the αIIbβ3 in lysis buffer [20 mm Tris–HCl, pH 7.5, 150 mm NaCl, 1% Nonident P40, 1% deoxycholic acid, and complete TM protease inhibitor mix (Roche Diagnosis, Mannheim, Germany)], followed by goat anti-mouse IgG-Agarose (Sigma-Aldrich, St Louis, MO, USA) as previously described [12]. The samples were electrophoresed in NuPAGE Novex 3–8% Tris–Acetate gels (Invitrogen) under non-reducing conditions; after transfer to PVDF membrane, proteins were detected by immunoblot analysis using monoclonal antibodies against αIIb (SZ22; Immunotech) and β3 (AP3, GTI, Brookfield, WI, USA) followed by horseradish peroxidase conjugate-anti mouse IgG (Jackson).
Mutation identification and analysis of polymorphisms
DNA samples of each patient were examined for the previously described 13-bp deletion in the αIIb gene [5] using direct polymerized chain reaction (PCR) amplification [13]. DNA fragments containing exons and flanking intronic regions of the αIIb and β3 genes were prepared by PCR using primers previously described [14,15]. The amplified fragments were directly sequenced by an automatic sequencer. Alternatively, the PCR products were first subjected to single-strand conformation polymorphism (SSCP) analysis, and when the SSCP revealed an abnormal fragment it was sequenced using the fmol DNA sequencing system (Promega).
For determination of a possible founder effect, haplotype analysis was performed using polymorphisms in the region of the αIIb and β3 genes on chromosome 17. These polymorphisms included microsatelite markers of the THRA1, BRCA1, and D17S579 loci that are adjacent to αIIb and β3 genes, and polymorphic sites within the αIIb (T2621G; HPA3) and β3 (CT repeats in IVS6 and A1545G; SmaI) genes [16].
Construction of expression vectors for mutant cDNAs
cDNAs of αIIb or β3 in pcDNA3 vector were generously provided by Dr Peter Newman (The Blood Center of Southeastern Wisconsin, Milwaukee, USA). A PCR fragment containing exons 1–7 of αIIb cDNA (nucleotides 4–800) was amplified from the cDNA of an Arab patient who carried the known 13-bp deletion by using a forward primer: 5′-GCCAGAGCTTTGTGTCCACTGC-3′ and a reverse primer: 5′-CCCAGTAGCCGTCGAAGTAC-3′. The PCR product containing the mutation was digested with NotI and SacII restriction enzymes and cloned into pcDNA3/IIb digested with the same restriction enzymes replacing the corresponding wild-type (WT) cDNA fragment. Cys107 or Trp110, located within the six-amino acid deletion, were substituted by Ser and Ala, respectively with QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) using pcDNA3/IIb as a template. Two overlapping oligonucleotide primers containing one or two base pair substitutions (underlined) were used: forward primer 5′-GTCATTGTGGCCTCCGCCCCCTGGCAG-3′ and reverse primer 5′-GCTGCCAGGGGGCGGAGGCCACAATGAC-3′ for the Cys107Ser substitution, or forward primer 5′-CCTGCGCCCCCGCGCAGCACTGGAACG-3′ and reverse primer 5′-CGTTCCAGTGCTGCGCGGGGGCGCAGG-3′ for the Trp110Ala substitution. WT β3 cDNA was excised from pcDNA3/β3 by EcoRI, filled in to create blunt ends, and subcloned into the PvuII site of pCEP4 mammalian expression vector carrying the hygromycin resistance gene as a selection marker (Invitrogen).
Co-transfection of αIIb and β3 cDNAs
Baby hamster kidney (BHK) cells were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 2 mg mL−1 l-glutamine and 5% fetal calf serum (Biological Industries, Beit-Haemek, Israel). The cells were co-transfected with 1 μg of WT or the mutated form of pcDNA3/αIIb and 1 μg of WT pCEP4/β3 using lipofectamine reagent (Gibco BRL, Paisley, UK). Mock cells were produced by transfecting BHK cells with 1 μg of pCEP4 and 1 μg of pcDNA3 and then subjecting them to selection in medium containing 0.7 mg mL−1 G418 (Gibco BRL) and 0.5 mg mL−1 hygromycin (Bohringer Gmbh, Mannheim, Germany) for at least 3 weeks before use.
Analysis of surface expression of αIIbβ3 and fibrinogen binding by flow cytometry
Transfected BHK cells were harvested with PBS/1 mm EDTA, pelleted and incubated in DMEM medium for 30 min at room temperature. Cells were pelleted again, resuspended in PBS, 1 mm CaCl2 and 1 mm MgCl2 (106 cells/100 μL) and incubated for 30 min at room temperature with a FITC-conjugated monoclonal antibody to the αIIbβ3 complex (CD41/P2 Immunotech). Subsequently, cells were diluted to 106 cells mL−1 and analyzed for surface fluorescence by flow cytometery (Coulter).
For fibrinogen binding, the cells were resuspended in TBS, 0.1% BSA (Sigma), 0.25 mm MgCl2 (106 cells 100 μL−1) and incubated for 1 h at room temperature with 10 μg/100 μL human fibrinogen (Sigma) and/or 1 μL anti LIBS6 (ligand induced binding site) antibody (kindly provided by Dr Mark Ginsberg, University of California, San Diego). Subsequently, cells were pelleted and resuspended in the same buffer and incubated for 20 min at room temperature with 10 μL FITC-conjugated rabbit anti-human fibrinogen (Dako) diluted to 106 cells mL−1 and analyzed by flow cytometery (Coulter). Background fluorescence was measured using the same antibodies in mock BHK cells.
Immunoblot analysis of BHK cells
Transfected BHK cells were lysed with lysis buffer and the lysate was electrophoresed on NuPAGE Novex 3–8% Tris–Acetate gels (Invitrogen) with 1 mm DTT. Seperated proteins were then transferred to a PVDF membrane (Millipore). The membrane was immersed in TBST containing a 1:1000 dilution of a monoclonal antibody to αIIb (SZ22; Immunotech), washed with TBST and then immersed in TBST containing 1:2000 horseradish peroxidase conjugated anti-mouse IgG. Immunoreactive bands on the membrane were detected using Super Signal West Pico Chemiluminescent substrate; (Pierce Biotechnology, Rockford, IL, USA) and X-ray film exposure.
Results
Platelet surface expression of αIIbβ3 and αvβ3
Flow cytometric analysis revealed that all 11 Palestinian-Arab probands expressed normal amount of GPIb/IX complex on their platelet surface (data not shown) and 10 patients had less than 5% of normal αIIbβ3 surface expression. One patient (proband 11 in Table 1) expressed approximately 50% of normal αIIbβ3 on his platelets (Fig. 1A), and consequently he was defined as bearing a variant mutation. Integrin αvβ3 was expressed on the platelets of probands 1–9 (data not shown), which was consistent with a mutation in the αIIb gene [12,17]. In one patient (proband 10 in Table 1), αvβ3 was not expressed on the platelet surface suggesting that the mutation was in the β3 gene.
Table 1.
Mutations identified in 11 Palestinian-Arab probands with Glanzmann thrombasthenia
| Unrelated probands | Mutated gene | DNA and mRNA alterations† | Predicted changes in protein† | Polymerized chain reaction primers and restriction enzyme for detection |
|---|---|---|---|---|
| 1–8* | αIIb | IVS4: −3, 13-bp deletion with cryptic splicing of exon 5 | In-frame deletion of Ala106-Gln111 | F: 5′-AGGAGGAGCCCAAGTCTCGCGC-3′
R: 5′-TCCCCATCGTGGCCCTTTC-3′ No restriction enzyme |
| 9 | αIIb | Exon 24: deletion of G2374 | 761–763 altered amino acids with a 764 stop | F: 5′-CCTTTCCAGCCTCCATGGTG-3′‡
R: 5′-CCAGGGACGCGAGGCTCC-3′ BstXI |
| 10 | β3 | Exon 10: deletion of TT1616–7 | 513–522 altered amino acids with a 523 stop | F: 5′-GGGCCCAACTGTGTCTAAAT-3′
R: 5′-TATATGAGGGGGTGTGGGTT-3′ BseNI |
| 11 | β3 | IVS14: −3C → G with cryptic splicing of exon 15 | 742–802 altered and extended amino acids with a 803 stop | F: 5′-AGGAAGTCACTGTAAGATGCT-3′
R: 5′-GGCACAGGCTGATTATGATCT-3′ BpmI |
Including three probands previously described [5].
Nucleotide numbering starts with the first ATG of the cDNA, amino acids numbering starts with the first amino acid of the mature protein.
One nucleotide mismatched in forward primer (underlined) was designed to create the recognition site in normal allele.
Fig. 1.
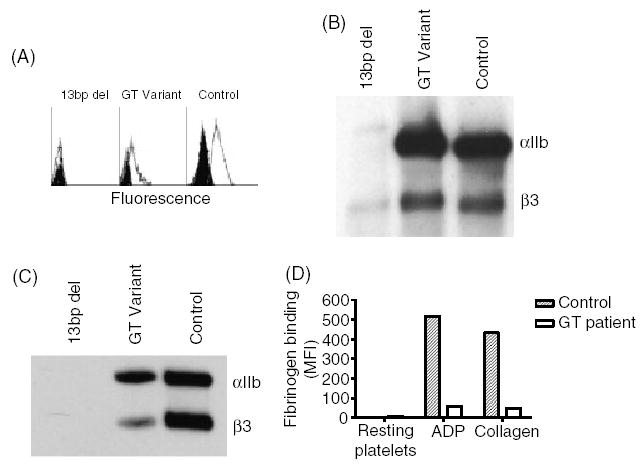
Platelet abnormalities in patients with Glanzmann thrombasthenia (GT). (A) Flow cytometric analysis of αIIbβ3 in platelets of a GT patient carrying the 13-bp deletion in the αIIb gene, a patient with a −3C → G mutation in intron 14–exon 15 junction of the β3 gene which causes variant GT, and a healthy control. fluorescein-isothiocyanate (FITC)-conjugated P2 monoclonal antibody against the αIIbβ3 complex was used. Background staining with FITC-conjugated mouse IgG is shown in black. (B) Immunoblotting under reduced conditions of sodium dodecyl sulphate-solubilized platelets using polyclonal antibodies against αIIb and β3. Notable is the trace amount of pro-αIIb and β3 in solubilized platelets of a patient with the 13-bp deletion and the normal amount of αIIb and β3 detectable in the patient with the variant mutation. (C) Immunoblot analysis under non-reducing conditions of αIIbβ3 immuno-precipitated by 10E5 monoclonal antibody, using SZ22 and AP3 monoclonal antibodies against αIIb and β3, respectively. Notable is the absence of αIIb and β3 in the patient with 13-bp deletion, and the reduced amounts of αIIb and β3 in the patient with the variant mutation compared with control. (D) Fibrinogen binding to resting platelets and platelets activated by 1.25 μm adenosine diphosphate or 1.25 μg mL−1 collagen. Note the impaired binding of fibrinogen to platelets of the patient with the variant mutation compared with control platelets.
Mutation identification
All eight newly examined Palestinian patients, as well as all 15 Jordanian patients, were tested for the presence of the 13-bp deletion in the αIIb gene[13]. Five patients, three residing in Israel and two residing in the Palestinian territories were homozygous for the 13-bp deletion. Thus, of 11 probands identified in this and our previous study[5], eight were homozygous for the 13-bp deletion. Of 158 Palestinian-Arabs controls (316 alleles), none was found to carry the 13-bp deletion in the αIIb gene consistent with a very low allele frequency (97.5%, CI; 0.0000–0.0116). None of the 15 Jordanian-Arab patients carried this mutation.
In each of the three Palestinian probands not bearing the 13-bp deletion, a homozygous novel mutation was identified (Table 1). In proband 9, a one nucleotide deletion (G2374) in exon 24 was found in the αIIb gene. This mutation predicts a shift in the reading frame leading to a stop at codon 764. In proband 10, a dinucleotides deletion (TT1616–7) in exon 10 of the β3 gene was identified, which also predicts a shift of the reading frame, resulting in a stop at codon 523. Both mutations occurred in homonucleotide sequences, (GG and TTT, respectively). In proband 11, whose platelet expressed approximately 50% of the normal level of αIIbβ3 (Fig. 1A), a −3C → G point mutation was identified. This mutation is one nucleotide upstream of the invariant AG of the intron14/exon 15 acceptor splice site of the β3 gene. This alteration gives rise to a new AG acceptor sequence flanking the natural AG acceptor.
For all three novel mutations, PCR-based assays combined with restriction enzyme digestion were designed for detection of carriers among the respective family members. None of the 15 Jordanian GT patients carried any of these three mutations.
Characterization of the novel variant GT mutation
Immunoblot analysis of the platelet lysates from patient 11 revealed apparently normal amounts of αIIb and β3 (Fig. 1B), and antibody 10E5 immunoprecipitated the αIIbβ3 complex from the patient’s platelet lysate (Fig. 1C).
The possible effects of the − 3C → G substitution at the intron 14/exon 15 junction on the mRNA splicing was analyzed using the Splice Site Prediction Program (http://www.fruitfly.org). In contrast to the score of 0.99 for the normal AG acceptor splice site at intron 14, the score for the − 3C → G mutant was <0.4. The newly created AG in the mutant sequence had a score of 0.93, suggesting that the mutation may result in alternative splicing. This was confirmed by sequencing the cDNA prepared from the patient’s platelets, which revealed that the normal AG acceptor was inserted into exon 15 (Fig. 2A, B). This insertion predicts a shift in the reading frame from residue 742 onward, creating a stop at codon 803 instead of codon 763, thereby extending the cytoplasmic tail of β3 by 40 amino acids (Fig. 2C).
Fig. 2.
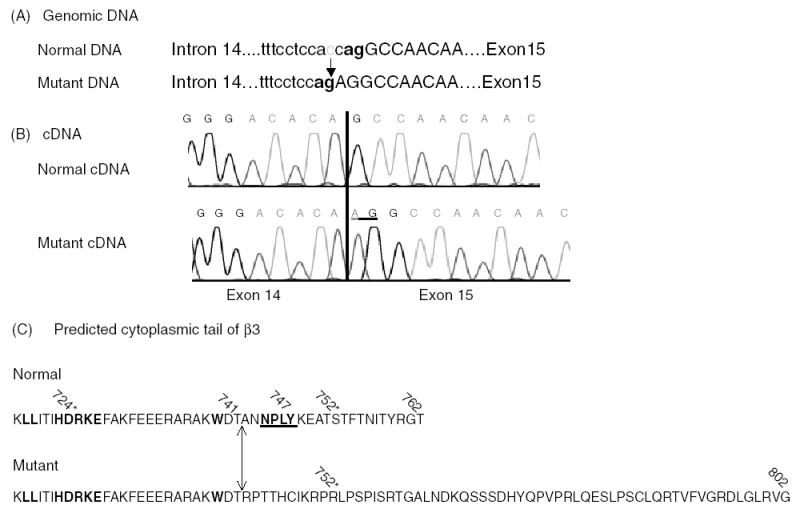
Alterations in DNA, cDNA and β3 protein in the variant −3C → G mutation (A) In the mutated gene, the depicted −3C → G alteration creates a new AG splice site for the intron 14/exon 15 junction, predicting a change in the reading frame. (B) Sequence of the cDNA from the normal and the patient’s platelets. Note the insertion of AG into exon 15 in the mutant. (C) The normal and the mutated β3 cytoplasmic tail. Because of the AG insertion into exon 15, the reading frame in the mutant protein is changed after residue 741, resulting in a shift of the stop codon from 763 to 803. The β3 tail is therefore extended by 40 amino acids. The conserved sequence LLxxxHDRKE (in bold), which has been implicated in the interaction with the αIIb tail is not changed in the mutated protein, whereas the NPxY motif (in bold and underlined), which interacts with talin, is missing in the mutant tail. Naturally accruing mutations causing variant GT patients are depicted by an asterisk.
The patient’s platelets bound only trace amounts of fibrinogen in response to stimulation by ADP or collagen, in spite of an αIIbβ3 surface expression of 50% (Fig. 1D).
Characterization of the 13-bp deletion
The 13-bp deletion causes an in-frame deletion of six amino acids (Ala106-Gln111), of which Cys107 was suspected to be critical for normal αIIbβ3 complex formation [5]. Analysis of the crystal structure of the αIIbβ-propeller in complex with the βA domain of β3 (PDB: 1TXV) revealed that Trp110 interacts directly with Arg261 of β3. We evaluated the effects of Cys107Ser substitution (in which the Cys107-Cys130 bond is abolished) and a Trp110Ala substitution (in which its interaction with Arg261 of the β3 subunit is abrogated). These mutated cDNAs were expressed in BHK cells with WT β3 cDNA. Flow cytometric analysis with the anti-αIIbβ3 monoclonal antibody P2 demonstrated lack of surface expression of αIIbβ3 in cells transfected with the natural 13-bp deletion, and about 30% of WT expression in cells transfected with Cys107Ser or Trp110Ala substitutions (Fig. 3A).
Fig. 3.
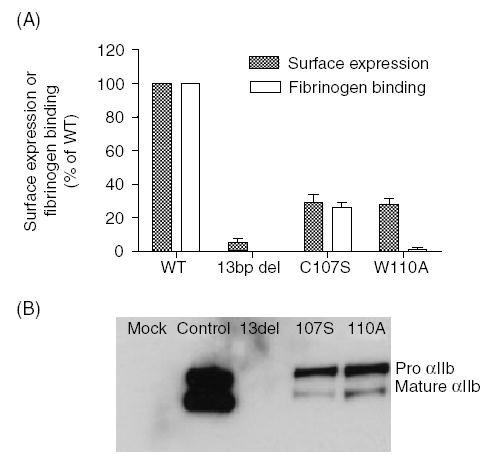
Expression of the natural 13-bp deletion and Cys107Ser or Trp110Ala artificial mutations. (A) Flow cytometric analysis of αIIbβ3 surface expression and fibrinogen binding to transfected baby hamster kidney (BHK) cells. BHK cells were transfected with normal β3 cDNA and either normal αIIb cDNA or mutated cDNAs (Cys107Ser, Trp110Ala or the 13-bp deletion). Cells were incubated with fluorescein-isothiocyanate (FITC)-conjugated P2 monoclonal antibody against the αIIbβ3 complex or with FITC-conjugated rabbit anti-human fibrinogen antibody following activation by anti LIBS6 antibody in the presence of human fibrinogen. Surface expression of αIIbβ3 or fibrinogen binding were normalized to 100% for control αIIbβ3 expression or fibrinogen binding, respectively, and is presented as mean ± SE of three experiments. Note the substantially reduced amount of αIIbβ3 expression in the Cys107Ser and Trp110Ala mutants and the lack of fibrinogen binding to cells containing Trp110Ala substitution. (B) Immunoblots of αIIbβ3 expressed in transfected BHK cells. BHK cells transfected with normal β3 and normal or mutated αIIb or pcDNA3 and pCEP4 plasmids without the inserts (mock) were lysed and subjected to immunoblot analysis under reducing conditions. In cells expressing the 13-bp deletion, no αIIb was detected, and in cells expressing Cys107Ser or Trp110Ala, most αIIb appeared as pro-αIIb.
Activation of αIIbβ3 by anti LIBS6 antibody yielded in cells containing Cys107Ser approximately 50% fibrinogen binding compared with WT cells, but no fibrinogen binding to Trp110Ala containing cells (Fig. 3A).
Immunoblot analysis using an antibody against αIIb (SZ22) showed no αIIb in cells containing the 13-bp deletion and markedly reduced amounts of αIIb in cells expressing either Cys107Ser or Trp110Ala (Fig. 3B). In cells expressing Cys107Ser or Trp110Ala mutations most αIIb appeared as pro-IIb suggesting impaired transport of the pro-αIIbβ3 complex to the Golgi (Fig. 3B).
Founder effect for the 13-bp deletion and an estimate of the mutation’s age
As the 13-bp deletion in αIIb was identified in eight unrelated families residing in different regions of Israel and the Palestinian territories, we assessed whether this mutation resulted from a founder effect or stemmed from several separate mutational events. Haplotype analysis in the eight probands and 50 Palestinian-Arab controls was performed using two polymorphic markers within the β3 gene, one polymorphic marker in the αIIb gene and three polymorphic markers (THRA1, BRCA1 and D17S579) located on chromosome 17 in the vicinity of αIIb and β3 genes. The analysis revealed that all GT patients carrying the 13-bp deletion had the same haplotype for the polymorphic marker within the αIIb gene, and for the D17S579 marker, which is located approximately 350 kb away from the αIIb gene (Table 2). At larger distances from the αIIb gene, homozygosities for polymorphic markers were frequent but some presumed recombination events were apparent. The frequency distribution of each polymorphism examined in control chromosomes and chromosomes bearing the 13-bp deletion differed significantly (Table 3). Collectively, these data are consistent with a founder effect for the mutation.
Table 2.
Haplotypes of eight unrelated probands carrying the 13-bp deletion in the αIIb gene*
| Probands
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Distance from αIIb gene (kb)† | Polymorphic sites‡ | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 |
| 4324 | THRA1 | 6 6 | 4 6 | 4 4 | 4 4 | 4 6 | 4 4 | 3 3 | 2 2 |
| 1234 | BRCA1 | 3 3 | 3 3 | 3 3 | 3 3 | 2 1 | 3 3 | 2 2 | 3 3 |
| 0 | αIIb-HPA3 | 2 2 | 2 2 | 2 2 | 2 2 | 2 2 | 2 2 | 2 2 | 2 2 |
| c349 | D17S579 | 5 5 | 5 5 | 5 5 | 5 5 | 5 5 | 5 5 | 5 5 | 5 5 |
| 3233 | β3: (CT)n | 8 4 | 8 8 | 8 8 | 8 8 | 4 4 | 6 8 | 4 5 | 8 8 |
| SmaI | 1 2 | 1 1 | 1 1 | 1 1 | 1 1 | 2 1 | 1 1 | 1 1 | |
The founder haplotype is boxed.
The distance from αIIb gene was calculated according to http://www.ncbi.nlm.nih.gov/mapview.
The allele designation is according to French et al. [16].
Table 3.
Comparison of allele frequencies of polymorphic markers in chromosome 17 in unrelated probands with Glanzmann thrombasthenia (GT) bearing the 13-bp deletion in the αIIb gene and Palestinian-Arab controls
| Polymorphic site | Allele | Controls (n = 100) | GT probands (n = 16) | χ2 analysis (P-value) |
|---|---|---|---|---|
| THRA1 | 2 | 0.03 | 0.125 | <0.0001 |
| 3 | 0.14 | 0.125 | ||
| 4 | 0.30 | 0.50 | ||
| 5 | 0.28 | 0 | ||
| 6 | 0.01 | 0.25 | ||
| Others | 0.24 | 0 | ||
| BRCA1 | 1 | 0.13 | 0.06 | <0.0001 |
| 2 | 0.15 | 0.19 | ||
| 3 | 0.19 | 0.75 | ||
| 5 | 0.10 | 0 | ||
| Others | 0.43 | 0 | ||
| αIIb-HPA3 | 1 | 0.67 | 0 | <0.0001 |
| 2 | 0.33 | 1.00 | ||
| D17S579 | 5 | 0.18 | 0 | <0.0001 |
| 8 | 0.04 | 1.00 | ||
| Others | 0.78 | 0 | ||
| β3-(CT) n | 3 | 0.43 | 0 | <0.0001 |
| 4 | 0.01 | 0.25 | ||
| 5 | 0.03 | 0.06 | ||
| 6 | 0.07 | 0.06 | ||
| 8 | 0.20 | 0.63 | ||
| Others | 0.26 | 0 | ||
| β3- smaI | 1 | 0.63 | 0.13 | 0.0002 |
| 2 | 0.37 | 0.87 |
n, number of alleles.
We estimated the time when the Palestinian 13-bp deletion occurred by analysis of BRCA1 and β3-CT polymorphisms, which are the closest loci on both sides of the mutation displaying recombination with the mutation. The age estimate was calculated by two methods. One method devised by Risch et al. [18] is based on the recombination fraction between the mutation and the closest marker related to the frequency of the marker on normal chromosomes. By this method, the range of age estimates was 19–31 generations, which translates into 380–620 years, assuming that one generation equals 20 years. The second method [19] was based the age estimate on linkage disequilibrium of multiple genetic markers in normal and affected individuals, and incorporates information about gene location, mutation frequency, and growth rate of the population examined, using a Markov chain Monte Carlo method [20]. Simulations using this program resulted in an estimate of 17 generations (340 years), with 95% credible sets of 14–22 generations, (280–440 years). Thus, the age estimate for the 13-bp deletion ranges between approximately 300 and 600 years.
Discussion
The data presented in this study, establish the predominance of the 13-bp deletion in the αIIb gene among Palestinian patients with GT. The mutation was detected in 8 of 11 unrelated probands (73%) and in none of 15 unrelated Jordanian-Arab patients. Haplotype analysis was consistent with a founder effect (Table 2, 3), and an age estimate based on two methods revealed that the mutation occurred probably 300–600 years ago. This ‘Palestinian mutation’ is thus the fourth mutation described that has produced a cluster of affected GT patients stemming from a founder effect. The other founder mutations are an 11 bp deletion in β3 exon 12 and an 11.2 kb β3 deletion in Iraqi Jews [7], and a splice site mutation IVS15(+1)G > A within the αIIb gene in French Gypsies [8]. The 13 bp Palestinian mutation involves deletion of 3 nt of intron 3 and 10 nt of exon 4, that lead to alternative splicing to a downstream AG acceptor producing an in-frame deletion of six amino acids, Ala106 – Gln111. The deleted residues are located in the second blade of the β-propeller domain of αIIb, which interfaces with the βA domain of β3. In our previous study we speculated that the absence of Cys107 within the deleted segment might have a critical effect on the folding of αIIb and its ability to form a normal complex with β3 [5]. Cys107 is completely conserved in 40 integrin β-propellers analyzed [21] and normally forms a disulfide bond with Cys130, maintaining a rigid structure for β-strands 2–3 of blade-2 within the β-propeller (Fig. 4A). Disruption of the Cys107–Cys130 bond by a Cys130Trp substitution was demonstrated to cause GT [22], and here we showed that disruption of the same Cys107-Cys130 bond by a Cys107Ser substitution resulted in impaired αIIb maturation and surface expression of αIIbβ3 (Fig. 3), but not in impaired fibrinogen binding (Fig. 3A). Another important residue within the Ala106-Gln111 deletion is Trp110 that interacts with Arg261 of the βA domain of β3 [10] and is also important for fibrinogen binding to αIIbβ3 as shown by others [23] and in the present study (Fig. 3A). Substitution of Trp110 by the much smaller residue Ala is predicted to disrupt the interaction with β3-Arg261 as shown in the model (compare Fig. 4A, B). BHK cells transfected with WT β3 cDNA, and αIIb cDNA in which Cys107 was substituted by Ser or Trp110 was substituted by Ala displayed in both instances only 30% of normal surface expression of αIIbβ3 (Fig. 3A). In each of these substitutions, most αIIb appeared as pro-αIIb (Fig. 3B), which suggests that pro-αIIbβ3 complexes form, but do not mature properly. In contrast to the complete 13-bp deletion, neither Cys107Ser nor Trp110Ala mutations resulted in complete loss of detectable αIIb (Fig. 3A, B). Interestingly, substitution of another residue in the deleted segment, Gln111Ala, did not affect surface expression of αIIbβ3 in Chinese hamster ovary cells but did reduce fibrinogen binding [24]. Thus, at least three of the six amino acids deleted as a result of the Palestinian mutation affect αIIbβ3 biogenesis and/or function.
Fig. 4.
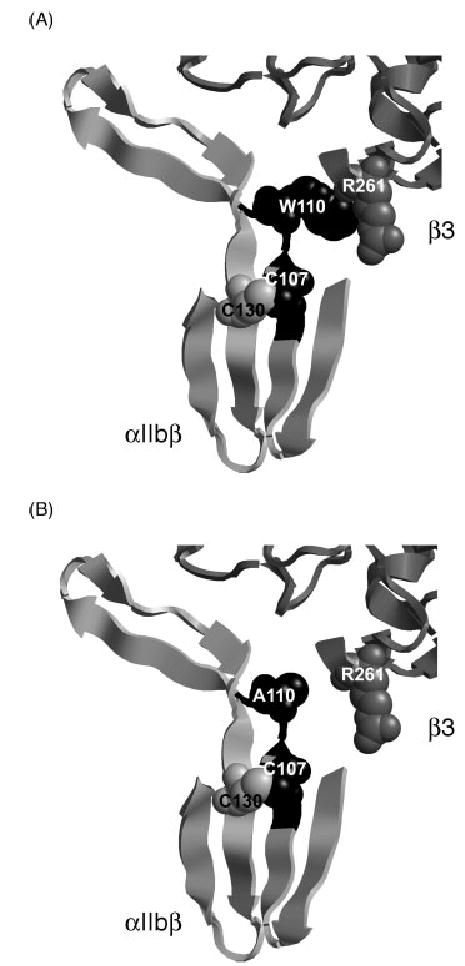
Model of the interface between blade 2 of the αIIbβ-propeller and βA domain of β3. Blade 2 of the αIIbβ-propeller (PDB 1TXV) shown in black are the six amino acids that are deleted in the Palestinian mutation. Trp110 of αIIb and Arg261 of β3 are shown as balls (A). Also depicted are Cys107 and Cys130 that are normally juxtaposed by disulfide bond and predictably are disrupted by replacing Cys107 by Ser. Note that the contact between Trp110 and Arg261 is disrupted when Trp110 is replaced by Ala (B). The figure was made using RasMol program and was based on the crystal structure of αIIbβ3 [10].
In three other Palestinian patients with GT, three novel mutations were identified, each in the homozygous state. Two of the three novel mutations were small deletions, i.e. G2374 in αIIb gene and TT1616–7 in β3 gene, respectively. Both mutations cause a shift in the reading frame and create a premature stop codon. The defects conferred by such stop codons can result in accelerated decay of mRNA, or in production of truncated proteins with removal of essential domains necessary for αIIbβ3 complex formation and its processing [22,25].
The third novel mutation is a point mutation (−3C → G) at the acceptor splice site of intron 14 and exon 15. The mutation creates a tandem repeat of AG at the termination of intron 14, which results in cryptic splicing and insertion of the original AG acceptor into exon 15. Disruption of highly conserved sequences like the intron 14 acceptor site, frequently gives rise to exon skipping unless, as in this case, the alteration produces a new consensus splice site very close to the original AG acceptor [26]. The insertion of AG into exon 15 creates a shift in the reading frame that leads to an aberrant sequence of amino acids from residue 742 and an extension of the β3 cytoplasmic tail by 40 amino acids. Surface expression of αIIbβ3 was relatively preserved on the surface of the platelets of the patient, but the patient’s variant αIIbβ3 failed to bind fibrinogen following activation by ADP or collagen (Fig. 1D). These data suggest that the aberrant β3 cytoplasmic tail disrupted inside out signaling. Of note, the conserved β3 NPxY sequence (amino acid 744–747) that was implicated in binding talin and β3-endonexin in the process of inside out signaling [27], was altered by the patient’s mutation. Elimination of NPxY motif as a result of truncation at residue 724 found in a patient with variant GT [28] or by the artificial substitution Tyr747Ala were previously shown to impair the normal inside-out signaling pathway [27–29]. Another mutation, a Ser752Pro substitution, was also shown to cause variant GT probably by causing impaired inside-out signaling [30]. It is likely that the aberrant amino acid sequence giving rise to Ser752Arg in the present study was an additional cause for abnormal inside-out signaling.
In conclusion, our data define a new common ancestral cluster of GT families confined to Palestinians and highlight the importance of a region in blade 2 of the β propeller of αIIb that is essential for normal αIIbβ3 maturation. Three novel mutations causing GT are presented of which one, in the β3 gene, is a variant related to an extended and aberrant cytoplasmic tail of β3 that appears to disrupt inside-out signaling.
Acknowledgments
We thank Dr Peter Newman for providing αIIb and β3 cDNAs, Victoria Kovalski and Sali Usher for their excellent technical assistance, Michael Begleiter, Children’s Mercy Hospital, Kansas City, for providing DNA of a GT patient, and Dr Mark Ginsberg, University of California, San Diego, for providing anti LIBS6 antibody. This study was supported by a Grant 19278 of Dr B. S. Coller from the Heart, Lung, and Blood Institute, USA.
Footnotes
To cite this article: Rosenberg N, Hauschner H, Peretz H, Mor-Cohen R, Landau M, Shenkman B, Kenet G, Coller BS, Awidi AA, Seligsohn U. A 13-bp deletion in αIIb gene is a founder mutation that predominates in Palestinian-Arab patients with Glanzmann thrombasthenia.
Author contributions
The roles of each one of the authors of this paper were as follows:
N. Rosenberg, design of the study, performance of most of the experiment, interpretation of data and writing; H. Hauschner, design and performance of mutagenesis, expression of mutations and fibrinogen binding to BHK cells; H. Peretz, design of the study, detection of one mutation and interpretation of data; R. Mor-Cohen, analysis of the founder effect and aging of the mutation; M. Landau, analysis of the crystal structure and modeling of the mutated proteins; B. Shenkman, design and performance the fibrinogen binding assay; G. Kenet, diagnosis of GT patients in four families, compiling clinical data and blood sampling; B.S. Coller, design of the study, interpretation and critical review of the paper; A.A. Awidi, diagnosis of GT patients in 15 families in Jordan, collection of samples and critical review of the paper; U. Seligsohn, design of the study, interpretation of data and writing of the manuscript.
References
- 1.Seligsohn U, Rososhansky S. A Glanzmann’s thrombasthenia cluster among Iraqi Jews in Israel. Thromb Haemost. 1984;52:230–1. [PubMed] [Google Scholar]
- 2.Khanduri U, Pulimood R, Sudarsanam A, Carman RH, Jadhav M, Pereira S. Glanzmann’s thrombasthenia. A review and report of 42 cases from South India. Thromb Haemost. 1981;46:717–21. [PubMed] [Google Scholar]
- 3.Schlegel N, Gayet O, Morel-Kopp MC, Wyler B, Hurtaud-Roux MF, Kaplan C, McGregor J. The molecular genetic basis of Glanzmann’s thrombasthenia in a Gypsy population in France: identification of a new mutation on the alpha IIb gene. Blood. 1995;86:977–82. [PubMed] [Google Scholar]
- 4.Awidi AS. Rare inheritaed bleeding disorders secondary to coagulation factors in Jordan: A nine-year study. Acta Haematol. 1992;88:11–3. doi: 10.1159/000204587. [DOI] [PubMed] [Google Scholar]
- 5.Newman PJ, Seligsohn U, Lyman S, Coller BS. The molecular basis of Glanzmann thrombasthenia in the Iraqi-Jewish and Arab populations in Israel. Proc Natl Acad Sci USA. 1991;88:3160–4. doi: 10.1073/pnas.88.8.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmed MA, Al-Sohaibani MO, Al-Mohaya SA, Sumer T, Al-Sheikh EH, Knox-Macaulay H. Inhibited bleeding disorders in eastern province of Saudi Arabia. Acta Haematol. 1988;79:202–6. doi: 10.1159/000205808. [DOI] [PubMed] [Google Scholar]
- 7.Rosenberg N, Yatuv R, Orion Y, Zivelin A, Dardik R, Peretz H, Seligsohn U. Glanzmann thrombashenia caused by an 11.2-kb deletion in the glycoprotein IIIa (β3) is a second mutation in Iraqi Jews that stemmed from a distinct founder. Blood. 1997;89:3654–62. [PubMed] [Google Scholar]
- 8.Nurden AT, Nurden P. Rebuttal: the French Gypsy mutation does not give rise to a particularly mild form of Glazmann’s thrombasthenia. J Thromb Haemost. 2003;1:2459. doi: 10.1046/j.1538-7836.2003.0468g.x. (Letter) [DOI] [PubMed] [Google Scholar]
- 9.Xiong J-P, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin αVβ3. Science. 2001;294:339–45. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao T, Takagi J, Coller BS, Wang JH, Springer TA. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature. 2004;432:59–67. doi: 10.1038/nature02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dardik R, Kaufman Y, Savion N, Rosenberg N, Shenkman B, Varon D. Platelets mediate tumor cell adhesion to subendothelium under flow conditions: Involvement of platelets GPIIb-IIIa and tumor cell αv integrins. Int J Cancer. 1997;70:201–7. doi: 10.1002/(sici)1097-0215(19970117)70:2<201::aid-ijc11>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg N, Dardik R, Rosenthal E, Zivelin A, Seligsohn U. Mutations in the αIIb and β3 genes that cause Glanzmann thrombasthenia can be distinguished by a simple procedure using transformed B-lymphocytes. Thromb Haemost. 1998;79:244–8. [PubMed] [Google Scholar]
- 13.Peretz H, Seligsohn U, Zwang E, Coller BS, Newman PJ. Detection of the Glanzmann’s thrombasthenia mutations in Arab and Iraqi-Jewish patients by polymerase chain reaction and restriction analysis of blood or urine samples. Thromb Haemost. 1991;66:500–4. [PubMed] [Google Scholar]
- 14.Yatuv R, Rosenberg N, Dardik R, Brenner B, Seligsohn U. Glanzmann thrombasthenia in two Iraqi-Jewish siblings is caused by a novel splice junction mutation in the glycoprotein IIb. Blood Coag Fibrin. 1998;9:285–98. doi: 10.1097/00001721-199804000-00011. [DOI] [PubMed] [Google Scholar]
- 15.Jin Y, Dietz HC, Nurden A, Bray PF. Single-strand conformation polymorphism analysis is a rapid and effective method for the identification of mutations and polymorphisms in the gene for glycoprotein IIIa. Blood. 1993;82:2281–8. [PubMed] [Google Scholar]
- 16.French DL, Coller BS, Usher S, Berkowitz R, Eng C, Seligsohn U, Peretz H. Prenatal diagnosis of Glanzmann thrombasthenia using the polymorphic markers BRCA1 and THRA1 on chromosome 17. Br J Haematol. 1998;102:582–7. doi: 10.1046/j.1365-2141.1998.00798.x. [DOI] [PubMed] [Google Scholar]
- 17.Coller BS, Cheresh DA, Asch E, Seligsohn U. Platelet vitronectin receptor expression differentiates Iraqi-Jews from Arab patients with Glanzmann thrombasthenia in Israel. Blood. 1991;77:75–83. [PubMed] [Google Scholar]
- 18.Risch N, de Leon D, Ozelius L, Kramer P, Almasy L, Singer B, Fahn S, Breakefield X, Bressman S. Genetic analysis of idiopathic torsion dystonia in Ashkenazi Jews and their recent descent from a small founder population. Nat Genet. 1995;9:152–9. doi: 10.1038/ng0295-152. [DOI] [PubMed] [Google Scholar]
- 19.Reeve JP, Rannala B. DMLE+: Bayesian linkage disequilibrium gene mapping. Bioinformatics. 2002;18:894–5. doi: 10.1093/bioinformatics/18.6.894. [DOI] [PubMed] [Google Scholar]
- 20.Rannala B, Bertorelle G. Using linkage markers to infer the age of a mutation. Hum Mut. 2001;18:87–100. doi: 10.1002/humu.1158. [DOI] [PubMed] [Google Scholar]
- 21.Rosenberg N, Landau M, Luboshitz J, Rechavi G, Seligsohn U. A novel Phe171Cys mutation in integrin αIIb causes Glanzmann thrombasthenia by abrogating αIIbβ3 complex formation. J Thromb Heamost. 2004;2:1167–75. doi: 10.1111/j.1538-7836.2004.00758.x. [DOI] [PubMed] [Google Scholar]
- 22.Vinciguerra C, Bordet JC, Beaune G, Grenier C, Dechavanne M, Negrier C. Description of 10 new mutations in platelet glycoprotein IIb (αIIb) and glycoprotein IIIa (β3) genes. Platelets. 2001;12:486–95. doi: 10.1080/095371001317126383. [DOI] [PubMed] [Google Scholar]
- 23.Kamata T, Tieu KK, Irie A, Springer TA, Takada Y. Amino acid residues in the alpha IIb subunit that are critical for ligand binding to integrin alpha IIb beta 3 are clustered in the beta-propeller model. J Biol Chem. 2001;276:44275–83. doi: 10.1074/jbc.M107021200. [DOI] [PubMed] [Google Scholar]
- 24.Tamura T, Hato T, Yamanouchi J, Fujita S. Critical residues for ligand binding in blade 2 of the propeller domain of the integrin alpha IIb subunit. Thromb Haemost. 2004;91:111–8. doi: 10.1160/TH03-06-0392. [DOI] [PubMed] [Google Scholar]
- 25.Yatuv R, Rosenberg N, Zivelin A, Peretz H, Trakhtenbrot L, Seligsohn U. Identification of a region in glycoprotein IIIa involved in subunit association with glycoprotein IIb: farther lessons from Iraqi-Jewish Glanzmann thrombasthenia. Blood. 2001;98:1063–9. doi: 10.1182/blood.v98.4.1063. [DOI] [PubMed] [Google Scholar]
- 26.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 27.Claderwood DA. Talin controls integrin activation. Biochem Soc Trans. 2004;32:434–7. doi: 10.1042/BST0320434. [DOI] [PubMed] [Google Scholar]
- 28.Wang R, Shattil SJ, Ambruso DR, Newman PJ. Truncation of the cytoplasmic domain of β3 in a variant form of Glanzmann thrombasthenia abrogates signaling through the integrin αIIbβ3 complex. J Clin Invest. 1997;100:2393–403. doi: 10.1172/JCI119780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Toole TE, Ylanne J, Culley BM. Regulation of integrin affinity states through an NPXY motif in the beta subunit cytoplasmic domain. J Biol Chem. 1995;270:8553–8. doi: 10.1074/jbc.270.15.8553. [DOI] [PubMed] [Google Scholar]
- 30.Chen YP, Djaffar I, Pidard D, Steiner B, Cieutat AM, Caen JP, Rosa JP. Ser-752>Pro mutation in the cytoplasmic domain of integrin β3 subunit and defective activation of platelet integrin αIIbβ3 (glycoprotein IIb-IIIa) in a variant Glanzmann thrombasthenia. Proc Natl Acad Sci USA. 1992;89:10169–73. doi: 10.1073/pnas.89.21.10169. [DOI] [PMC free article] [PubMed] [Google Scholar]