

Abstract
Misfolding and aggregation of proteins is a common thread linking a number of important human health problems. The misfolded and aggregated proteins are inducers of cellular stress and activators of immunity in neurodegenerative diseases. They might posses clear cytotoxic properties, being responsible for the dysfunction and loss of cells in the affected organs. Despite the crucial importance of protein misfolding and abnormal interactions, very little is currently known about the molecular mechanism underlying these processes. Factors that lead to protein misfolding and aggregation in vitro are poorly understood, not to mention the complexities involved in the formation of protein nanoparticles with different morphologies (e.g. the nanopores) in vivo. A better understanding of the molecular mechanisms of misfolding and aggregation might facilitate development of the rational approaches to prevent pathologies mediated by protein misfolding. The conventional tools currently available to researchers can only provide an averaged picture of a living system, whereas much of the subtle or short-lived information is lost. We believe that the existing and emerging nanotools might help solving these problems by opening the entirely novel pathways for the development of early diagnostic and therapeutic approaches. This article summarizes recent advances of the nanoscience in detection and characterization of misfolded protein conformations. Based on these findings we outline our view on the nanoscience development towards identification intracellular nanomachines and/or multicomponent complexes critically involved in protein misfolding.
Keywords: protein misfolding, protein aggregation, conformational disease, nanomedicine, atomic force microscopy, force spectroscopy, single molecule analyses
PROTEIN MISFOLDING AND DISEASES
Misfolding and aggregation of proteins is a common thread linking a number of important human health problems associated with protein deposition diseases, including neurodegenerative disorders such as Parkinson’s disease, Down’s syndrome, Alzheimer’s and Huntington’s diseases, systemic and localized amyloidoses and transmissible encephalopathies [Dobson, 2004b]. The first and perhaps most important elements in most neurodegenerative processes are misfolded and aggregated proteins. These are inducers of cellular stress and activators of immunity in neurodegenerative diseases, which affect neuronal dysfunction and loss. All together, the accumulation of abnormal protein aggregates exert toxicity by disrupting intracellular transport, overwhelming protein degradation pathways, and/or disturbing vital cell functions. In addition, the formation of inclusion bodies is known to represent a major problem in the recombinant production of therapeutic proteins [Fink, 1998]. Formulation of these therapeutic proteins into delivery systems and their in vivo delivery are often complicated by protein association [Demidov, 2004]. Finally, since protein refolding is frequently accompanied by transient association of partially folded intermediates, the propensity to aggregate is considered a general characteristic of the majority of partially folded proteins [Dobson, 2004b; Ptitsyn, 1995a; Ptitsyn, 1995b; Ptitsyn et al., 1995; Segel et al., 1999]. Thus, protein folding abnormalities and subsequent events underlie a multitude of pathologies and difficulties with protein therapeutic applications. Current demographic trends indicate that need for age-related and other degenerative disorders and macromolecule therapeutics will be at the forefront of future medical developments. The field of medicine therefore can be dramatically advanced by establishing a fundamental understanding of key factors leading to the misfolding and self-aggregation of proteins involved in the various protein folding pathologies.
Schematically the transformations of the protein from normal folded state to misfolded and aggregated forms are shown in Figure 1. Protein misfolding leads to the formation of the aggregates of different morphologies – protofilaments (A); annular aggregates (B) and fibrils (C). These morphologies are typically analyzed by electron microscopy (EM) or atomic force microscopy (AFM). The AFM images obtained in our lab are shown in the figure. Despite the crucial importance of protein misfolding and abnormal interactions, very little is currently known about the molecular mechanism underlying these processes. Factors that lead to protein misfolding and aggregation in vitro are poorly understood, not to mention the complexities involved in the formation of protein nanoparticles with different morphologies, e.g. spherical oligomers and nanopores. For example, images A–C in Figure 1 show that different misfolded states leads to different aggregates, but currently this is purely hypothetical view with very little support. Although it is well known that the same protein under pathological conditions can lead to the formation of fibrillar, pore-like, spherical, or amorphous aggregates with diverse biological consequences [Uversky, 2003], the conditions leading to misfolding and the formation of such abnormal complexes are unclear, let alone their prevention. The mechanisms underlying aggregation in vivo in biological systems are even less clearly understood due to the experimental difficulties in monitoring aggregates in their natural environment. Without deep understanding of the protein misfolding and aggregation phenomena there is no hope for the elaboration of appropriate early diagnostic tools and the development of efficient therapeutics capable of preventing the development of protein deposition diseases.
Figure 1.

Scheme of protein misfolding and aggregation. In the scheme normally folded protein can undergo conformational transitions into various misfolded states that may aggregate in different morphologies. AFM images of Aβ peptide aggregated into protofilaments, toroids (rings) and fibrils are shown in plates (A), (B) and (C) respectively.
Sstructure of self-assembled nano-aggregates
The fibrillogenesis can be studied using a wide arsenal of modern biophysical approaches developed for analysis of protein structure, conformational transitions and folding (reviewed in [Makin and Serpell, 2005; Stromer and Serpell, 2005]). Such relatively low resolution techniques as transmission EM and AFM are excellent tools for distinguishing various morphologies of self-assembled aggregates; these studies allowed to arrange aggregates in several families, protofilaments (subfibrils), oligomers and fibrils [Inouye and Kirschner, 2005; Jeyashekar et al., 2005; Makin and Serpell, 2005; Stromer and Serpell, 2005] (see also Figure 1A–C). These structural studies were instrumental in understanding the toxic effects of different morphologies. It has been also noted that the formation of amyloid-like fibrils does not represent the only pathological hallmark of “conformational” or protein deposition diseases [Bellotti et al., 1999; Dobson, 2004b; Kelly, 1998[[Dobson, 1999 #5; Rochet and Lansbury, 2000; Uversky and Fink, 2004]. In several neurodegenerative disorders (as well as in numerous in vitro experiments) the protein depositions are composed of the amorphous aggregates, cloud-like inclusions without defined structure (e.g., [Jeyashekar et al., 2005]). Similarly, soluble oligomers represent another alternative final product of the aggregation process. The choice between three aggregation pathways, fibrillation, amorphous aggregate formation or oligomerization, is not well defined. However, the aggregation pathways depend on the amino acid sequence (could be modified by mutations) and by the protein environment. Irrespective the chosen pathway, a first stage of the aggregation process is assumed to be the transition into misfolding states (the structural transformation of soluble proteins into the “sticky” aggregation-prone precursor or intermediate(s)). Furthermore, intermediate might contain different amount of ordered structure even for the same protein undergoing different aggregation processes. Schematically this model is shown in Figure 2. It has been also pointed out that the variations in the amount of the ordered structure in the amyloidogenic precursor might be responsible for the formation of fibrils with distinct morphologies [Smith et al., 2003]. Note that there are several investigations favoring the idea that the deposited proteinacous inclusions (such as senile plaques in AD brains or Lewy bodies or Lewy neurites in PD brains) are not toxic, but the formation of some protofibrillar structures is responsible for the toxicity [Lashuel et al., 2002a; Lashuel et al., 2002b; Mucke et al., 2000; Selkoe, 1997; Urbanc et al., 2002].
Figure 2.

Folded protein (A) adopts an unfolded state (B) that is the transient state for the misfolded conformation (C).
High resolution direct structural techniques such as x-ray crystallography and NMR are capable of providing structural details of stable conformations of proteins at the angstrom level resolution. However, this level resolution for amyloids has been achieved only very recently due to the enormous experimental problems associated with studies of protein aggregates. Initial the cross beta sheet organization within fibrils was resolved by the x-ray diffraction studies from β-amyloid fibrils [Serpell, 2000]. The fibril appears to be composed of several protofilaments. Each of these protofilaments is composed of β-sheet structure in which hydrogen bonding occurs along the length of the fibril and the β-strands run perpendicular to the fibril axis. M. F. Perutz proposed the water filled nanotubes model of amyloid fibril in which polypeptide chains fold to β-strands which form a cylindrical sheet of 3 nm diameter that is folded to form a hollow cylinder filled with water [Perutz et al., 2002a]. Proposed first for the fibrils formed by poly Glu polymer (a model system for Huntington’s protein) [Perutz et al., 2002b], the model can be used also to explain the peculiarities of the fibril structure of amyloid β (Aβ) peptide, α-synuclein and prion proteins.
A structural model for amyloid fibrils formed by the 40-residue Aβ peptide was proposed based on the results of the solid state NMR spectroscopy [Balbach et al., 2002; Petkova et al., 2004; Petkova et al., 2002]. These studies showed that the first 10 residues of Aβ (1–40) within the fibril are not structured; however the rest of the protein except for the 25–29 region adopts antiparallel β-sheet conformation. Residues 25–29 contain a bend of the peptide backbone that brings the two β-sheets in contact through side chain-side chain interactions. Similar approach has recently been applied for the analysis of structure of amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p (Ure2p(10)(-)(39)) [Chan et al., 2005]. It has been shown that Ure2p(10)(-)(39) fibrils contain in-register parallel β-sheets. Furthermore, the hydrogen bonding between side chain amide groups of Gln18 residues was in favor of “polar zippers” proposed by M. F. Perutz for stabilization of amyloid fibrils formed by peptides with glutamine- and asparagine-rich sequences, such as Ure2p(10)(-)(39) [Perutz et al., 2002a; Perutz et al., 2002b].
Recently, a series of studies were published where x-ray crystallography was successfully applied to decipher the structural details of amyloid fibrils at the angstrom level of resolution. The electron and x-ray diffraction data from filaments of Ure2p demonstrated the 4.7 Å reflection that is typical for the cross-beta structure and highly indicative of amyloid [Baxa et al., 2005]. A structural model of the murine PrP small β-sheet was obtained by the analysis of the 19-mer comprising the two β-strands 127–133 and 159–164 linked by a four-residue sequence with high turn propensity [Croixmarie et al., 2005]. According to EM, this 19-residue peptide spontaneously forms very long single fibrils. The X-ray diffraction revealed an average arrangement of the hairpin peptides into a structure that can be approximated by an empty-core cylinder. The hairpins are oriented perpendicular to the cylinder axis with a 130 Å helix pitch. Furthermore, the structure consists of two β-sheet ribbons wound around a cylinder and assembled into a single fibril with a hairpin orientation perpendicular to the fibril axis. Molecular dynamics simulations revealed the zipper-like network of polar interactions between the edges of the two ribbons, including the partially buried water molecules [Croixmarie et al., 2005]. High resolution x ray-crystallographic data were used to determine the atomic structure of the cross-beta spine from microcrystals formed by a seven-residue peptide segment [Nelson et al., 2005]. The spine is a double β-sheet, with each sheet formed from parallel segments stacked in register. Side chains protruding from the two sheets form a dry, tightly self-complementing steric zipper, holding the sheets. Within each sheet, every segment is bound to its two neighboring segments through stacks of both backbone and side-chain hydrogen bonds [Nelson et al., 2005].
The combination of the available structural information on Aβ (1–42) fibrils with detailed amide hydrogen-exchange measurements, pairwise mutagenesis, thioflavin T (ThioT) binding, and high-resolution cryoelectron microscopy was recently used to determine a 3D structure of Aβ(1–42) fibrils [Luhrs et al., 2005]. According to the model, residues 1–17 are disordered, residues 18–42 form a β-strand-turn-β-strand motif that contains two intermolecular, parallel, in-register β -sheets that are formed by residues 18–26 (β1) and 31–42 (β2). In addition, at least two molecules of Aβ (1–42) are involved in the repeating structure of a protofilament. Moreover, intermolecular side-chain contacts are formed between the odd-numbered residues of strand β1 of the two adjacent peptides. This interaction pattern leads to partially unpaired beta-strands at the fibrillar ends, which explains the sequence selectivity, the cooperativity, and the apparent unidirectionality of A β fibril growth providing a structural basis for constructing the fibrils inhibitors. Amyloid fibrils have been formed in vitro from several disease-associated and disease-unrelated proteins and peptides (reviewed in [Uversky and Fink, 2004]).
PROTEIN MISFOLDING AND AGGERAGTION. UNANSWERED QUESTIONS
There is growing number of evidence supporting the conclusion that the ability to form fibrils is a generic property of the polypeptide chain; i.e., many proteins, perhaps all, are potentially able to form amyloid fibrils under appropriate conditions [Dobson, 1999; Dobson, 2004b]. This means that amyloidogenic polypeptides are unrelated in terms of sequence or structure. The experiments with probing of aggregated forms of proteins with antibodies showed that there is a class of structure specific antibodies that recognize protein aggregates (e.g., fibrils), but do not bind to a monomeric form of the same protein (monomers) [O’Nuallain and Wetzel, 2002]. Importantly, among those structure-specific antibodies are those that distinguish between different aggregated morophologies of the same protein [Glabe, 2004; Kayed et al., 2003]. These findings suggest that aggregated forms of protein have different epitopes; in other words, their structural characteristics are different. Importantly, it was also established that some antibodies were able to recognize aggregates formed by different proteins. The striking conclusion based on these experiments was that the same structural morphologies but formed but different proteins are recognized by the same structure-specific antibody. These studies lead to the intriguing conclusion that aggregated protein might have common structural motifs even if they are not structurally close prior to fibrillation, being rich in β-sheet, α-helix, β-helix, or contain both α-helices and β-sheets, be globular proteins with rigid 3D-structure or belong to the realm of natively unfolded (or intrinsically unstructured) proteins [Uversky and Fink, 2004]. Despite these differences, amyloid fibrils have similar structural features. In fact, based on the results of extensive structural studies on several amyloidogenic proteins a general hypothesis of fibrillogenesis has been formulated: structural transformation of a polypeptide chain into a partially folded conformation represents an important prerequisite for protein fibrillation [Uversky and Fink, 2004]. Schematically this pathway is shown in Figure 2.
In protein misfolding diseases, potentially pathogenic misfolded and aggregated forms of a protein can form in different ways:
The protein may have an intrinsic propensity to assume a pathologic conformation, which becomes evident with aging or at persistently high concentrations;
The replacement of a single amino acid in the protein, as occurs in hereditary amyloidosis, can increase propensity of protein to misfold and, thus, represents another obvious mechanism of amyloidogeneity;
Often, proteolytic digestion of the protein precursor might produce an amyloidogenic fragment;
Interactions (or impaired interactions) with some endogenous factors (e.g., chaperones, intracellular or extracellular matrixes, other proteins) can change conformation of a pathogenic protein and increase its propensity to misfold and aggregate;
Exposure to internal or external toxins can induce conformational changes in a given protein and facilitate its misfolding and aggregation;
Malfunction of the antioxidant defense system can lead to the increased production of free radicals, which might induce oxidative modification of a pathogenic protein, or its binding partners;
Impaired posttranslational modifications (phosphorylation, advanced glycation, deamidation, racemization, etc.) might change protein conformation, facilitate its misfolding and promote aggregation;
Impaired functioning of proteasome or other proteolytic systems might result in the dramatic increase in the local concentration of the pathogenic protein.
These mechanisms can act independently or in association with one another. In addition to the intrinsic amyloidogenic potential of the pathogenic protein, other factors may act synergistically in amyloid deposition.
What information is lacking for a quantitative description of the system?
Despite the crucial importance of protein misfolding and abnormal interactions, very little is currently known about the molecular and intra-cellular mechanisms underlying these processes. Furthermore, factors that lead to protein misfolding and aggregation in vitro are poorly understood, including the complexities involved in the formation of protein nanoparticles with different morphologies. Although it is well known that incubation of a protein under non-physiological conditions can lead to the formation of fibrillar, pore-like, spherical, or amorphous nano-ensembles with diverse biological consequences, the physiological insults leading to misfolding and the formation of such abnormal complexes are unclear. The mechanisms underlying aggregation in biological systems are even less well understood due to the difficulties in monitoring aggregates in vivo. Clearly, a full understanding of the molecular mechanisms of misfolding and aggregation is essential for the development of rational approaches to prevent protein misfolding that lead to self-assembly in nano-ensembles. Thus, one needs to elaborate novel approaches to determine the molecular basis of protein misfolding and aggregation, and from this fundamental knowledge, to formulate a predictive molecular model that incorporates the cellular nanomachinery and physiological processes involved in protein deposition disorders. In this way, we can establish crucial enabling insights to catalyze the development of new therapeutic advances and novel nanotechnologies for diagnosis.
Misfolded conformations of proteins differ from folded and other aberrant protein conformations by their increased propensity to interact with each other leading to the formation of nano-aggregates. The structure of individual protein molecules within well ordered aggregates can be partially elucidated by traditional structural techniques, including X-ray crystallography, NMR, circular dichroism, fluorescence, and IR spectroscopies (reviewed in [Dobson, 2004a; Dobson, 2005]). However, none of these techniques is capable of sensing the misfolded conformation of the protein prior to aggregation. Apparently, the conformation of misfolded protein preceding the aggregation is different from what one can see in aggregates, but to what extent this difference might cause the disease is not clear. The vast majority of current experimental approaches to analyze protein misfolding and aggregation are based on traditional, ensemble techniques that describe the conformational behavior of the entire system and thus do not allow investigators to distinguish between conformational changes in individual protein molecules prior aggregation and changes induced by protein-protein interaction. This leaves open the question on the effect of different factors on folding/misfolding of an individual protein molecule. Conventional structural tools do not allow for the measurement of protein interaction forces or the kinetics of interconversion among different protein conformations in a single protein molecule. The ability to measure these parameters is critical to achieve a quantitative understanding of protein misfolding and aggregation at the nanoscale level. Thus, new experimental tools and approaches are crucial for understanding the protein misfolding phenomenon.
What tools are in place and what additional ones will be required to develop?
Structural methods such as x-ray crystallography, NMR, electron microscopy, and atomic force microscopy (AFM) have provided useful data regarding the secondary structure of proteins in nano-assemblies and the morphologies of self-assembled aggregates. However, we still lack a mechanistic understanding of the processes leading to the misfolding and of the interactions between misfolded conformations leading to protein self-assembly in nano-aggregates. This includes the capability to identify the conformation(s) of a misfolded protein prior to the onset of formation of nano-ensembles. Indeed, there may be several non-canonical protein conformations that exist transiently and that transform from one into the other. To address these questions, we need to utilize new “thinking” and new techniques capable of probing transient conformations of single protein molecules.
The tools currently available to researchers and clinicians essentially provide a population-based picture of a living-dynamical system. Because most studies have involved these ensemble techniques, which average over an enormous number of molecules or freeze the complex system in time, we lack insight into the manifold opportunities for conformational change available to an individual molecule in the complex environment of a cell or tissue. It is evident that the mechanisms of life and disease involve dynamic molecular-scale interactions—most of which occurs under some level of physical stress. An apparently simple process, as examined by existing techniques, may actually have many steps and a variety of important intermediate states—each with their own unique dynamics and response to stress. These intermediate states are likely to be only marginally stable making their transient existence difficult to measure. However, they can be very important to control several pathways. Thus, it is vitally important to establish ultrasensitive methods capable of probing protein conformations and weak intermolecular interactions over a wide range of time scales.
It has been already mentioned that there is rapidly growing evidence that the pathogenesis of protein misfolding diseases is not a result of protein deposition in a form of visible aggregates, which, in fact, represent a final stage of the multi-step molecular association cascade. Rather, the earlier steps in the series of conformational changes and protein-protein interactions are directly tied to pathogenesis. There is now increased understanding of the pathways involved in protein association, and some recent clues have emerged as to the molecular mechanisms of cellular toxicity [Ross and Poirier, 2004]. Targeting these early events of development of protein misfolding diseases will lead to the development of novel approaches toward rational diagnostics and therapeutics of these disorders.
NANOSCIENCE APPROACHES FOR THE PROTEIN MISFOLDING
As we noted above, the major problem with probing misfolded protein conformations is that they are transient states, and the vast majority of traditional structural techniques are not amenable to these systems. Important advances over the last few years have demonstrated that single molecule biophysics techniques can visualize or probe intermediates, follow molecular scale events in real time, and measure a wide range of intermolecular interactions. We think that exploiting new and established single molecule methods, in conjunction with complementary ensemble techniques and a set of theoretical approaches enabling quantitative analysis of experimental data with testable predictions for conformational properties and the potential for aggregation of misfolded states of the protein represents a novel approach (nanotool) for analysis of transient conformations of proteins. This nanotool will provide the scientific resources needed to develop innovative nanomedicine approaches for understanding, diagnosis, prevention, and cure of protein misfolding diseases. The section below outlines very recent advances in the nanoscience applications to probe the protein misfolding phenomenon [Karsai et al., 2005; Kellermayer et al., 2005; Kransnoslobodtsev, 2005; McAllister et al., 2005]. We believe that further studies utilizing this approach and other nanoimaging tools will lead to understanding the misfolding phenomenon at the nanoscale level and thus open prospects for the development of entirely new prospects for curing the disease at the very early stages.
Molecular mechanisms of protein misfolding
The approach proposed in our recent paper [McAllister et al., 2005] is based on the hypothesis that misfolded conformations of proteins differ from folded and other aberrant protein conformations by their increased propensity to interact with each other leading to the formation of nano-aggregates. To test this hypothesis and to measure the interprotein interactions, we used the AFM force spectroscopy approach, in which proteins were anchored to the substrate surface and the AFM tip (Figure 3A). When the tip approaches to the surface, the tethered proteins can interact forming a complex (Figure 3B). The forces holding the complex are measured by pulling the complex apart (Figure 3C). The force is low if the complex is weak (normal state of the protein; Figure 3D; the force curve is shown as insert d on this scheme), but the forces increase if the protein adopt a misfolded conformation as illustrated by scheme E in Figure 3 and insert e in this scheme. Various protein conformations can be induced by changing the environmental conditions (e.g., solvent), and the conditions facilitating the protein aggregation were primary conditions for detecting misfolded conformations of the protein. In the AFM force spectroscopy experiments, the interprotein interaction is measured by performing a series of such approach-retraction cycles at various locations at the substrate. This approach is capable of probing of the interprotein interactions at the single molecule level and enables one intimate detection and quantitative characterization of conformational states without interference from neighboring molecules. Using the force spectroscopy approach we were able to monitor directly the strength of the interprotein interaction depending on the protein conformation. We tested this idea using three different proteins, α-synuclein, amyloid β peptide and lysozyme [McAllister et al., 2005] using pH as a factor stimulating conformational changes in proteins. The proteins were covalently linked to amino-functionalized mica and Si3N4 AFM probes via glutaraldehyde crosslinking. The results of the force spectroscopy assay for monitoring interprotein interactions for lysozyme are shown in Figure 4. This figure summarizing the force spectroscopy results obtained at different pH values shows that intermolecular interactions for lysozyme increase at pH below 4, but the interaction drops at more acidic pH. To test the assumption that the maximum in the intermolecular forces corresponds to the formation of misfolded protein states, we performed experiments on growing the fibrils. The protein solution (10mg/ml) was incubated at 57°C in glycine buffer (0.15M) at pH2.0, pH2.7 and pH3.7. The image of the sample prepared at pH2.0 is shown in Figure 5A Fibrils are seen easily, although the sample prepared at these conditions is characterized by the appearance of short fibrils and small globular aggregates. The sample prepared at pH2.7 has predominately fibrillar morphology (Figure 5B) with fibrils as long as several microns. The incubation at pH3.7 did not lead to the formation of fibrils or large aggregates (Figure 5C). This observation is consistent with the force spectroscopy data (Fig. 4) showing that at pH3.7 the interprotein interaction is quite low. Similar results were retrieved for other proteins and the data obtained show that the interaction between all the proteins increases sharply with a decrease in pH. AFM imaging showed that at pH values corresponding to maximum interprotein interaction, the rate of protein aggregation increases dramatically. These studies illustrate the power of nanomechanical tools for unraveling molecular mechanisms of protein misfolding and fibrils formation.
Figure 3.
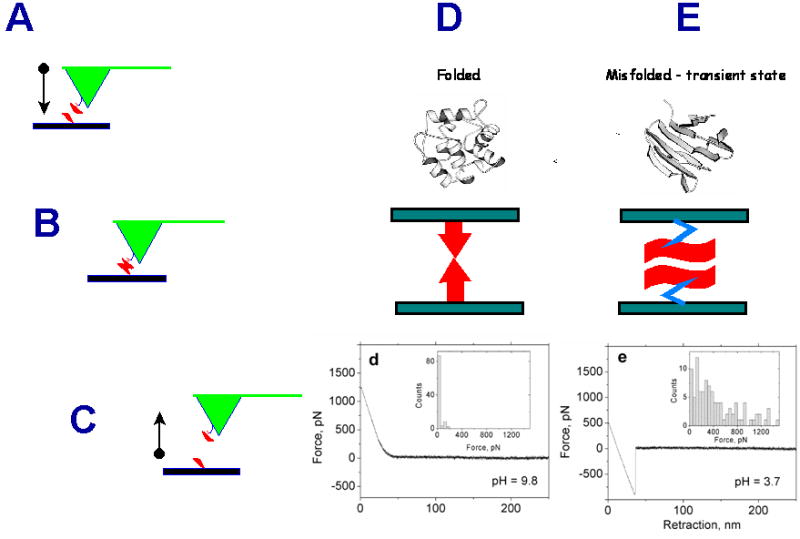
Scheme explaining the nanoprobing approach for the detection and analysis of misfolded states of the protein. Proteins anchored to the substrate surface and the AFM tip (A) are brought into the contact (B) and then pulled apart (C). The forces holding the complex depend on the protein conformation. They are low for normally folded proteins (the force curve “d”). Large rupture force (e) corresponds to a misfolded sate of the protein (D).
Figure 4.
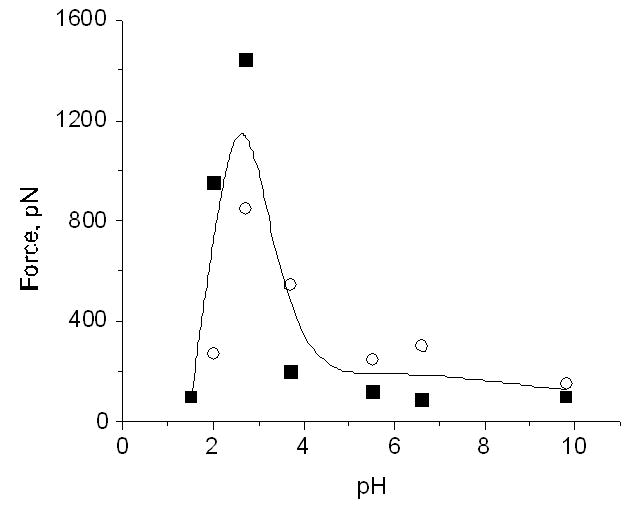
The dependence of the rupture forces on pH for lysozyme. See paper [McAllister et al., 2005] for details.
Figure 5.

AFM images of fibrils formed by lysozyme grown at pH2.0 (A), pH2.7 (B) and pH3.7 (C).
Although the proposed approach allowed us to study various proteins using the same immobilization strategy, glutaraldehyde reaction takes place at all amine-containing moieties (e.g., lysine and arginine) in addition to the protein N-termini. To avoid this ambiguity in the protein tethering that can complicate the single molecule analysis of rupture events, we used an alternative approach, in which Aβ peptide was bound to the surface at N-terminal only [Kransnoslobodtsev, 2005]. We took advantage of the fact that Aβ peptide does not contain cysteins and incorporated this amino acid to the N-terminus of the peptide allowing for the use of thiol-specific chemistry for the protein immobilization. We selected N-terminus because N-terminal part of the peptide is not critically involved in various conformational transitions of this peptide including the formation of β-sheet conformations within amyloid fibrils (e.g., [Tycko, 2003]). We synthesized maleimide-PEG-silatrane to functionalize mica (MAS mica) and the AFM tip surfaces with maleimide capable of binding SH-terminated peptide
The data obtained for pH3.7 are shown in Figure 6. An intensive peak at the beginning of the force curve corresponding to the short-range adhesion forces between the tip and mica surfaces typically observed at acidic pH values is accompanied by a rupture event (indicated with an arrowhead) with the step size ~20 pN that is substantially larger the noise level. The histogram for the data obtained from the analysis of series of such events is shown as insert (i) in Figure 6. The mean value for this distribution is 17±3 pN. Note that control experiments with Aβ-peptide at the mica surface or the tip only did not show these types of interactions. Given a finite size of the MAS-PEG linkers and similar size of the peptide (10–15 nm total), it is reasonable to attribute the second, small peak to the rupture of a single Aβ-Aβ pair. Additional evidence supporting this interpretation comes from the analysis of this particular part of the force-distance curve. First, the profile of this curve is typical for stretching of various polymers and this section can be fitted by the worm like chain (WLC) or exponential stretching models [Gutsmann et al., 2004]. The fit to the exponential model of this section of the force distance curve (a red straight line in the semi-logarithmic scale) is shown as insert (ii) above the curve. Second, the entire stretching range is ca. 15 nm. The extension measured from the series of force curves was 13.8±1.7 nm was close to the expected value for a full extension of 12 amino acids of the N-terminus of Aβ peptide that are not involved in the peptide folding [Tycko, 2003] and 5 PEG moieties of the MAS linker. Additional proofs for these estimates were obtained from the experiments with stretching of the surface-immobilized Aβ peptide via MAS chemistry with the tip terminated with glutaraldehyde. During the tip-surface contact covalent bonds between the residues with free amino groups of the peptide (the closest to the N-terminal is Lys-16) and the tip immobilized glutaraldehyde can be formed allowing the stretching the Aβ peptide molecule between the anchors. The stretching experiments showed the elastic-type profile of the force curve with the stretch value 7.8–8.5 nm; this value is very close to the half of the stretching effect observed for stretching Aβ-Aβ pairs. Similar data were obtained for pulling experiments for pH2.0 and the mean value for the rupture forces was 22±4 pN (data not shown) that is very close to the value obtained at pH 3.7. Overall, the data on the pH-dependent interaction obtained are consistent with our earlier data [McAllister et al., 2005] suggesting the Aβ-peptide misfolding at acidic pH, but here we were able to measure the interactions between individual Aβ-Aβ pairs.
Figure 6.

The data for pulling Aβ 1-40 peptides N-terminated with cysteine and immobilized on the maleimide-silatrane functionalized mica surfaces and the AFM tips. The force curve is approximated by the WLC (thin black curve). The insert shows the histogram generated from a series of the force curves. See paper [Kransnoslobodtsev, 2005] for specifics.
We anticipate that further exploration of this approach combining experimental AFM force spectroscopy studies with a thorough theoretical analysis of protein-protein interactions will be instrumental in identifying common patterns for the protein misfolding pathways. The use of ultrasensitive biomembrane and optical trap force probes will provide important insights into structural metastability and transition kinetics under small forces and on long time scales, thereby significantly extending the dynamic range of AFM force spectroscopy approach.
However, the conformations of the proteins before they were brought into the contact and after the complex formation may be different; therefore alternative experimental approaches have to be employed for structural analysis of the proteins conformations before they are brought into the contact. The dynamic force spectroscopy (DFS) approach in which the protein mechanical properties are probed by applying pulling forces at selected sites within the protein is one of the promising single molecule techniques. Importantly, recent studies [Dietz and Rief, 2004; Dietz and Rief, 2006] showed that the DFS approach enable the characterization of the protein structure at the sub-nanometer scale level.
The force spectroscopy data, translated into prominent barriers in the energy landscape governing the stability of protein conformation and the strength of pair-wise interactions, will provide the essential experimental foundation for molecular modeling and theoretical analysis of misfolded conformations and for elucidating the underlying mechanisms in protein misfolding. The computer modeling approaches will allow one to predict structural instabilities at atomic level, important interaction sites, and the refolding patterns in protein dynamics giving rise to misfolding pathways. Altogether, experimental and theoretical studies will provide the nano-tools capable of predicting the formation of misfolded states of the protein. Ultimately, this model should be extended to establish a holistic algorithm for predicting protein self-assembly into mesoscopic aggregates.
What is the time scale for the protein misfolding? How homogeneous are misfolded states for individual proteins? What are the dynamics of misfolded protein molecules at each misfolded state? These fundamental questions can be explored through the use of single molecule fluorescence microscopy utilizing the Förster (fluorescence) resonance energy transfer (FRET) phenomenon. It has become increasingly clear that state-of-the-art single molecule FRET offers a powerful new approach to understanding protein dynamic structure (e.g., [Allen et al., 2004; Haas, 2005; Harms et al., 2003; Jager et al., 2005; Kapanidis et al., 2004; Kuzmenkina et al., 2005; Schuler et al., 2002; Xie et al., 2004]). It enables evaluation of molecules as they explore time and space. This technique allows observation of the dynamic behavior of individual protein molecules at various folding states, to explore heterogeneity between molecules, and determine mechanisms of their interactions. These studies will be instrumental in extending the measurements to the intracellular environment where individual molecules will be viewed as they move inside the cell, carry out specific functions, or behave as components of larger systems.
In summary, the recent nanoimaging works illustrate that single molecule biophysics techniques are capable of probing transiently existing intermolecular interactions. We believe that this approach based on the combination of new and established single molecule methods with complementary ensemble techniques will provide the scientific resources needed to develop innovative nanoimaging tools for understanding the protein misfolding phenomenon and thus pave the way for building the nanoscale model for the early stage of the development of protein misfolding diseases.
Protein self-assembly in nanostructures
It is widely accepted that assembly of misfolded proteins into aggregates is a key step in further development of the diseases (e.g., [Golde, 2005; Jeyashekar et al., 2005]). Although among all aggregate morphologies fibrils attracted the major interests in various physico-chemical and structural studies, the growing data indicate that pre-amyloid oligomers, rather than fibrils, are the pathogenic species (e.g., [Glabe, 2005; Veerhuis et al., 2005] and references therein). One of the hypotheses is that such oligomers within membranes can form hollow structures, pores, functioning as aberrant channels, allowing small ions to stream through, thus killing the cells [Hartley et al., 1999]. Such channels were formed within phospholipids bilayers by such amyloidogenic proteins as Aβ40 and Aβ42 [Rhee et al., 1998], α-synuclein, IAPP, polyglutamine as well as the prion protein (PrP 106–126). In all cases only the spherical amyloid oligomers induced membrane currents that increased proportionally to the amyloid peptide concentration; neither monomeric nor fibrillar forms increased the current [Kayed et al., 2004]. In addition, membrane currents were shut down by addition of anti-oligomer antibodies. These pore-like structures within membranes were imaged directly. α-Synuclein protofibrils have been shown to form pore-like assemblies on the surface of brain-derived vesicles [Rochet et al., 2004]. Donut-shaped protrusions were observed by AFM on lipid bilayers after reconstitution of Aβ42 in DOPC [Lin et al., 2001]. Recent studies with the use of direct imaging with AFM, optical spectroscopy, and electrophysiological techniques showed that variety of amyloid-related molecules (Aβ40, α-synuclein, ABri, ADan, serum amyloid A, and amylin) in reconstituted membranes form morphologically compatible ion-channel-like structures and behave as single channels. Protofibrils with toroidal shape were shown to be formed spontaneously during the incubation of several monomeric proteins [Lashuel et al., 2002a; Lashuel et al., 2003; Lashuel et al., 2002b]. The sizes of these toroidal aggregates vary in a broad range as it is seen in the AFM images shown in Figure 1B.
Interesting molecular modeling computer simulations in attempt to test the hypothesis on the formation of the pore structure formed by β-amyloid peptides were performed by S. Sherman and L. Kinarsky (in preparation). Modeling was based on the assumption that the ion channels can be formed by multimeric Aβ protein containing four to eight Aβ monomers [Durell et al., 1994]. The best score was obtained for a model that consisted of 10 antiparallel β-strands formed by five consecutive Aβ 7–40 peptides with an added GG linker between the Aβ subunits (Figure 7). In this model, Aβ40 forms β-hairpin-like structures bent near positions 24–26, which is consistent with the NMR [D’Ursi et al., 2004; Tycko, 2003; Tycko, 2004] and molecular dynamics simulation data [Ma and Nussinov, 2002]. From this model, the inner diameter of the 10-stranded β-barrel was estimated to be 11–13 Å, whereas its outer diameter was 22–25 Å. Multimeric channel-like structures of Aβ consisting of four and six monomers incorporated into planar lipid bilayers were detected with AFM [Bhatia et al., 2000; Lin et al., 2001], although the resolution was not sufficient to retrieve structural parameters.
Figure 7.
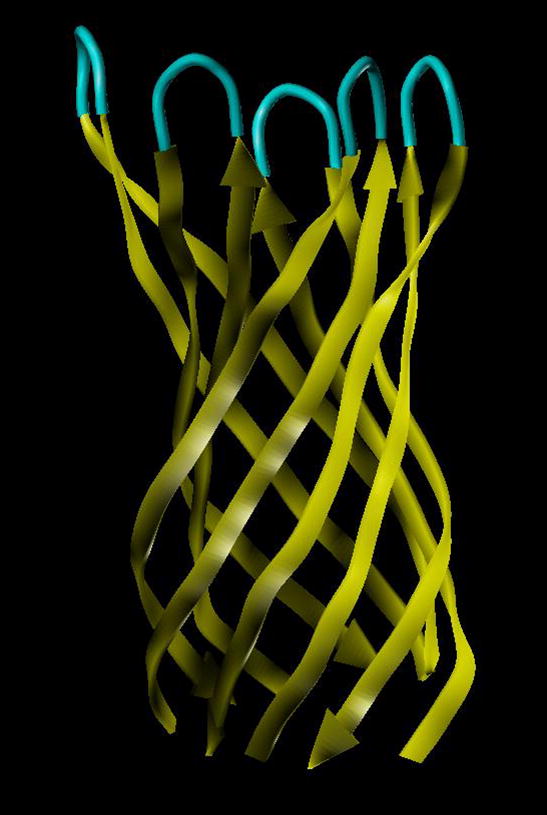
Ribbon-tube representation of the three-dimensional structural model of the β-barrel transmembrane protein consisting of 10 antiparallel β-strands formed by the five adjacent Aβ peptides.
Although the channel hypothesis is very attractive, some experimental data do not support this interesting model. For example, paper of Kayed et al [Kayed et al., 2004] reported the results that soluble oligomers from several types of amyloid-related proteins specifically increased lipid bilayer conductance regardless of the sequence, but this effect was observed without any evidence of discrete channel or pore formation or ion selectivity. The neutron scattering data indicated that thinning of the membranes rather than the formation of the pores is likely responsible for observed conductivity effect of the oligomers. Different laboratories use different criteria for the characterization of initial protein samples and the procedures for their aggregations, preparation of the complexes with membranes, so partially the controversy in the data can be explained by this factor. Further studies with the use of imaging methods such as high-resolution nanoimaging approaches applied for studies of membrane proteins [Engel and Müller, 2000; Fotiadis et al., 2003; Fotiadis et al., 2004a; Fotiadis et al., 2004b; Fotiadis et al., 2002] will help understanding the mechanisms of the interaction of amyloidogenic proteins with membranes and thus resolve the controversies. In addition to imaging, AFM was very instrumental as a nanoprobing tool for unraveling structure-function relationship for membrane channels. In fact, single-molecule force-spectroscopy was employed to unfold and refold single sodium-proton antiporters (NhaA) of Escherichia coli from membrane patches [Kedrov et al., 2004]. Dynamic force spectroscopy analyses revealed different unfolding patterns for the C-terminal or the N-terminal ends of the protein. Interestingly, after unfolding of ten of the 12 helices, the extracted polypeptide was allowed to refold back into the membrane. Recent AFM studies of porous structures formed by misfolded proteins [Quist et al., 2005] are very encouraging and provide a ground for optimism for unraveling the role of this nanoporous structures in development of the diseases.
In addition to oligomeric forms of the amyloid-related proteins, the interest to larger aggregates, nanofibrils in particular, remains high. First, the plaque deposits consist of the fibrils primarily; therefore the structural analysis of these aggregates is needed for understanding their formation. Second, amyloid fibrils are natural nanomaterials with a number interesting physico-chemical properties attractive for using them as biomaterials for a number of practical purposes. The fibrils are quite resistant to protease treatment, an attractive property for biomaterials. The recent findings that fibrils are not toxic for cells ease a potential biohazard concern with such applications. Such nanoimaging techniques as electron microscopy and AFM are the most useful instrumental techniques for understanding the structural organization of the Aβ fibrils (reviewed in [Makin and Serpell, 2005; Stromer and Serpell, 2005]), although our knowledge about the structure of fibrils formed by other proteins is less clear. What are the mechanical properties of amyloid fibrils? How strong is the interaction between protofibrils within the fibrils? These are very important questions that need to be addressed before using the nanofibrils as potential biomaterials. Electron microscopy and AFM images of the nanofibrils formed by the majority of different proteins showed that they are quite straight filaments of the several nanometers wide as shown in Figure 1C (see also [Makin and Serpell, 2005; Stromer and Serpell, 2005]). According to the polymer statistics, the flexibility of linear polymer is characterized by the persistence length P: the larger the persistence length, the stiffer the polymer [Flory, 1953]. For example persistence length of the DNA double helix (diameter is 2 nm) is ca. 45 nm [Lu et al., 2001]. The RecA-DNA fibrils is 15 times stiffer – persistence length is ~700 nm [Stasiak, 1992]. The amyloid fibrils have a comparable width, but they are so straight that their persistence length is approaching to infinity suggesting that these polymeric fibrils are very stiff. Surprisingly, the AFM images of the carbon nanofibrils revealed a similar curvature tempting us to hypothesize that stiffness of these two nanofibrils is comparable. The stiffness of carbon nanotubes was probed in the single molecule bending experiments in which the fibril was bent by the AFM tip [Salvetat et al., 1999]. Similar experiments can be applied to the amyloid fibrils and the hypothesis of their rigidity can be tested directly.
Recently AFM was applied to measure the interaction between the protofibrils within the fibril [Karsai et al., 2005; Kellermayer et al., 2005]. In these experiments, the AFM tip was pressed against the fibril selected at the previously imaged area to create a strong interaction between the tip and the protofibril. The tip was pulled away from the fibril and in so doing to measure the forces stabilizing the structure of the fibril or/and the unzipping the protofibril within the fibril. Schematically this idea is shown as inset in Figure 8. Experiments performed with fibrils formed by Aβ peptides 1–40 and 25–35 showed that this nanopulling approach is capable of probing the mechanics of amyloid fibrils. Different sets of force spectroscopy data obtained for these peptides can be explained by different unzipping mechanisms of β-sheets for both peptides. Acetylation of Lys 28 in Aβ 25-35 led to a dramatic decrease of the rupture forces, although both peptides form morphologically undistinguishable fibrils [Karsai et al., 2005]. Computer simulations based on a simple two-state model suggest that the decreased unzipping forces can be explained by a destabilized zippered state of the fibril. We have performed similar pulling experiments with fibrils formed by α-synuclein protein. The results are shown in Figure 8 in which pulling experiments at 6 selected points were performed; they are numbered in Figure 8A and probing points are indicated with red crosses and arrows. If the tip is attached to filament of the fibril, we will be able to pull a filament from the fibril [Karsai et al., 2005; Kellermayer et al., 2005]. Image B (in color) shows the results of such pulling experiments. Unambiguously identified gaps on fibrils are indicated in image B with white arrows and numbered according to the previous image (A). The filaments were removed from the fibril at 4 from 6 total points. The force measurements (Figure 8C) revealed a characteristic extension-rupture pattern that is the indication of unzipping of fibrils (extension part of the force curve) followed by the rupture event [Karsai et al., 2005; Kellermayer et al., 2005]. If the tip peels of the filament from the fibrils, the damages to the fibrils are observed (points 2, 4, 5 and 6; Figure 8B). However, the rupture of the tip-fibril contact may not lead to the damage; therefore we did not observe damages to the fibril at points 1 and 3. Force curves do not allow distinguishing between these two options, but the conclusion can be made based on the analysis of the images taken before and after pulling. The experiments were performed with the fibrils cross-linked to the functionalized APS-mica surface via glutaraldehyde [McAllister et al., 2005]. In contrast to the results of [Kellermayer et al., 2005] we did not observe extended plateaus on the force curves. Such force curves were detected in the case when the fibrils were immobilized on the functionalized APS-surface via Van der Waals and electrostatic interactions without cross-linking (Figure 8D). Based on these findings we suggest that the extended plateaus are due to the sliding of the fibrils along the surface rather than peeling off the protofibril or their unzipping. The nature of the tip-sample contacts formed during pressing the tip against the surface is still unclear and can depend on the sample type and the tip material. Similar pulling experiments with DNA molecules typically required applying forces in the range or several nN to provide a strong contact between the tip and the DNA molecule immobilized on the surface [Clausen-Schaumann et al., 2000]. We were able to apply much smaller force (ca. 100 pN) to attach the filament to the tip. The approaches utilizing the formation of very specific contacts between the tip and the fibril and thus providing the data for further quantitative analysis need to be developed. The use of tips functionalized with chemically reactive is one of promising ways. Note in this regard positive results on the use of maleimide terminated AFM probes for covalent bonding of proteins at their cysteine moieties [Kransnoslobodtsev, 2005].
Figure 8.
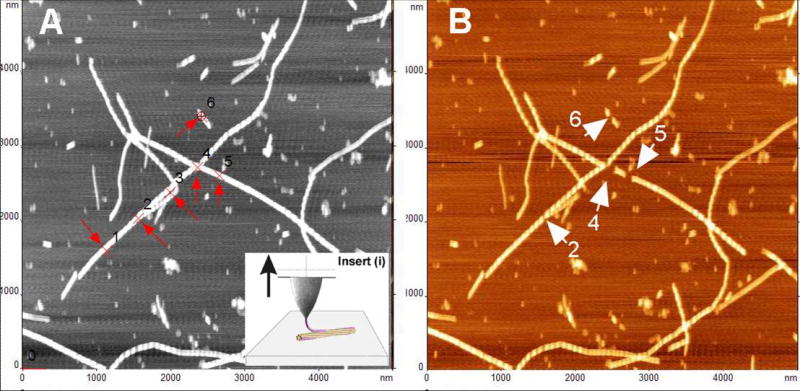
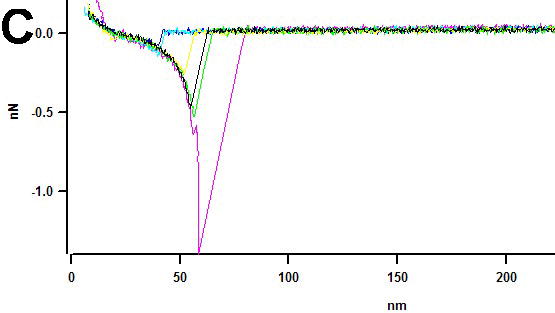
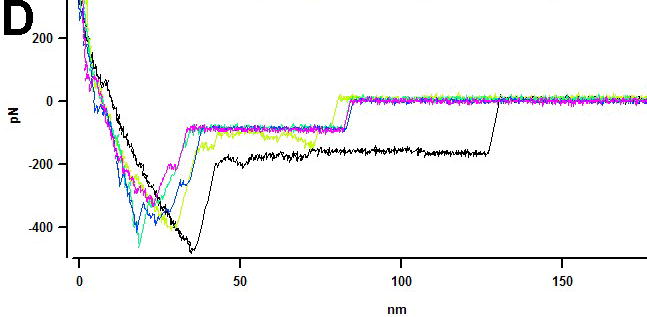
AFM pulling experiments of a-synuclein fibrils (see insert (i)). Pulling points (1–6) are indicated with red arrows in image (A) obtained before pulling. Image (B) was taken after pulling. Damaged sections of fibrils in image (B) are indicated with white arrows and numbered according to image (A). (C) Force curves for pulling the fibrils cross-linked to the APS-mica surface (trigger – 100pN, dwell time – 2s). The tip spring constant k=67.31 pN/nm. (D) Force curves for pulling of the non-covalently bound fibrils (trigger 500 pN, dwell time 2 s). The tip spring constant k=51.69 pN/nm. Pulling was done in PBS buffer.
Prospects for applications of the nanoimaging for understanding molecular interactions involved in protein misfolding
The structure-function paradigm states that the specific functionality of a given protein is predetermined by the unique spatial positioning of amino acid side chains and prosthetic groups in a defined 3-D structure. However, many proteins under physiological conditions adopt dynamic ensembles of interconverting conformations rather than a defined 3-D structure [Dunker et al., 2001; Dyson and Wright, 2005; Fink, 2005; Tompa, 2002; Uversky, 2002a; Uversky, 2002b; Uversky et al., 2000; Wright and Dyson, 1999]. These flexible or intrinsically disordered proteins/regions are involved in a multitude of vital functions such as recognition and interaction with different binding partners, protein modification and cellular transport [Dunker et al., 2002a; Dunker et al., 2002b; Dunker et al., 2005; Dunker et al., 2001; Dyson and Wright, 2005; Iakoucheva et al., 2002; Oldfield et al., 2005b; Tompa, 2002; Uversky, 2002a; Uversky, 2002b; Uversky et al., 2005]. They are more common in eukarytotes than in prokaryotes [Dunker et al., 2000; Oldfield et al., 2005a; Ward et al., 2004]. Eukaryotes seem to have opted to use disordered proteins/regions to create flexibility in function. The price for the plasticity is occasional toxic misfolding events which appear to be handled quite well until past reproductive age when there is no longer any effective selective pressure. In fact, a large number of proteins associated with misfolding diseases (e.g., α-synuclein, τ protein, prion protein, etc.) are intrinsically disordered or have significant disordered segments [Uversky and Fink, 2004]. Regions of intrinsic disorder become structured upon binding with a partner.
Thus two groups of proteins involved in conformational diseases should be distinguished, those that approach misfolding from a natively folded structure, which is destabilized by mutation as in the classical amyloidoses (TTR, calcitonin, IgG light chain, serum amyloid A, lysozyme, cystatin C, etc.), myocilin, CFTR or by proteolysis (Aβ) and those approaching misfolding from an intrinsically disordered structure (α-synuclein, τ protein, prion protein, polyQ proteins). Obviously, the misfolding pathways of the two groups would be different. Furthermore, the cell’s ability to recognize and deal with the unfolded/initial misfolded state should differ between the two groups. Those proteins that are built with a classical native hydrophobic stabilization are recognized early by the eukaryotic protein fidelity machinery while the highly polar disordered proteins/regions lack the critical clustering of hydrophobic residues and may only be picked up at a later, less reversible stage in aggregation.
We hypothesize that a disordered state of the proteins is a precursor for a misfolded conformation [Uversky and Fink, 2004]. A rationale for this hypothesis stems from the fact that misfolding of the protein requires a dramatic re-arrangement of the protein structure that can readily occur in a disordered state. Intramolecular interaction of specific protein regions will facilitate the next step in the formation of a misfolded conformation. Understanding the basis for the misfolding and inappropriate recognition will facilitate the rational design of approaches aimed at inhibiting and controlling the misrecognition events as well as modeling and manipulating the recognition process within cells. We anticipate that the experimental approaches for analyzing of disordered protein conformations will be adapted from the set of nanotools described above.
The interaction of monomeric or oligomeric forms of misfolded proteins with chaperones and co-chaperones has the potential to perturb the balance between refolding, degradation, and cellular signaling controlled by these complexes. Aggregated protein species are notoriously resistant to soluble proteases and may inhibit or sequester chaperone systems. It has been shown that Alzheimer’s β-peptide interacts with human hsp70 and the co-chaperone BAG-1 and postulated that such interactions may be exacerbated by oligomeric and/or fibrillar forms of Aβ. Alteration of the relative populations of fibrillar and spherical oligomeric huntingtin exon I containing the expanded polyglutamine stretch by hsp70-hsp40 chaperones has been also demonstrated [Muchowski and Wacker, 2005]. The use of single molecule imaging and probing techniques such as force microscopy, single molecule FRET, and two-color correlation spectroscopy, and tools such as conformation-specific antibodies as well as techniques for introducing protein complexes into cells will help better understanding the role of chaperone interaction with the misfolded proteins. By continuously following fibrils or oligomers and their interactions with chaperones at the single molecule level, one can learn if the pathway to (potentially toxic) oligomers goes through monomers or whether soluble oligomers can form directly from fibrils.
Various studies have shown that protein misfolding is promoted by oxidative damage (e.g. [Dawson and Dawson, 2003; Recchia et al., 2004]). The questions which are not answered as yet are: How different antioxidant proteins affect the conformation and structural dynamics of pathogenic proteins? Whether antioxidant proteins prevent the misfolding of previously oxidized and non-oxidized amyloidogenic proteins incubated in the presence of reactive oxygen species? We believe that these questions can be answered by the application of the nanotools described above.
Footnotes
Grant sponsors: NIH (GM62235 and 1-PN1 EY016593-01), NATO (LST.CLG.980194), the M. J. Fox Foundation for Parkinson’s Research (CFT03) (to YLL), the Programs of the Russian Academy of Sciences for the “Molecular and cellular biology” and “Fundamental science for medicine” (to VNU).
References
- Allen MW, Urbauer RJ, Zaidi A, Williams TD, Urbauer JL, Johnson CK. Fluorescence labeling, purification, and immobilization of a double cysteine mutant calmodulin fusion protein for single-molecule experiments. Anal Biochem. 2004;325:273–84. doi: 10.1016/j.ab.2003.10.045. [DOI] [PubMed] [Google Scholar]
- Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer’s beta-amyloid fibrils: evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance. Biophys J. 2002;83:1205–16. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxa U, Cheng N, Winkler DC, Chiu TK, Davies DR, Sharma D, Inouye H, Kirschner DA, Wickner RB, Steven AC. Filaments of the Ure2p prion protein have a cross-beta core structure. J Struct Biol. 2005;150:170–9. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Bellotti V, Mangione P, Stoppini M. Biological activity and pathological implications of misfolded proteins. Cell Mol Life Sci. 1999;55:977–91. doi: 10.1007/s000180050348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. Faseb J. 2000;14:1233–43. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- Chan JC, Oyler NA, Yau WM, Tycko R. Parallel beta-sheets and polar zippers in amyloid fibrils formed by residues 10–39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669–80. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen-Schaumann H, Seitz M, Krautbauer R, Gaub HE. Force spectroscopy with single bio-molecules. Curr Opin Chem Biol. 2000;4:524–30. doi: 10.1016/s1367-5931(00)00126-5. [DOI] [PubMed] [Google Scholar]
- Croixmarie V, Briki F, David G, Coic YM, Ovtracht L, Doucet J, Jamin N, Sanson A. A cylinder-shaped double ribbon structure formed by an amyloid hairpin peptide derived from the beta-sheet of murine PrP: an X-ray and molecular dynamics simulation study. J Struct Biol. 2005;150:284–99. doi: 10.1016/j.jsb.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–22. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Demidov VV. Nanobiosensors and molecular diagnostics: a promising partnership. Expert Rev Mol Diagn. 2004;4:267–8. doi: 10.1586/14737159.4.3.267. [DOI] [PubMed] [Google Scholar]
- Dietz H, Rief M. Exploring the energy landscape of GFP by single-molecule mechanical experiments. Proc Natl Acad Sci U S A. 2004;101:16192–7. doi: 10.1073/pnas.0404549101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz H, Rief M. Protein structure by mechanical triangulation. Proc Natl Acad Sci U S A. 2006;103:1244–7. doi: 10.1073/pnas.0509217103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson CM. Protein misfolding, evolution and disease. Trends Biochem Sci. 1999;24:329–32. doi: 10.1016/s0968-0004(99)01445-0. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Experimental investigation of protein folding and misfolding. Methods. 2004a;34:4–14. doi: 10.1016/j.ymeth.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Principles of protein folding, misfolding and aggregation. Semin Cell Dev Biol. 2004b;15:3–16. doi: 10.1016/j.semcdb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Dobson CM. Structural biology: prying into prions. Nature. 2005;435:747–9. doi: 10.1038/435747a. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Intrinsic disorder and protein function. Biochemistry. 2002a;41:6573–82. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Brown CJ, Obradovic Z. Identification and functions of usefully disordered proteins. Adv Protein Chem. 2002b;62:25–49. doi: 10.1016/s0065-3233(02)62004-2. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–48. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Lawson JD, Brown CJ, Williams RM, Romero P, Oh JS, Oldfield CJ, Campen AM, Ratliff CM, Hipps KW, Ausio J, Nissen MS, Reeves R, Kang C, Kissinger CR, Bailey RW, Griswold MD, Chiu W, Garner EC, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–71. [PubMed] [Google Scholar]
- Durell SR, Guy HR, Arispe N, Rojas E, Pollard HB. Theoretical models of the ion channel structure of amyloid beta-protein. Biophys J. 1994;67:2137–45. doi: 10.1016/S0006-3495(94)80717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ursi AM, Armenante MR, Guerrini R, Salvadori S, Sorrentino G, Picone D. Solution structure of amyloid beta-peptide (25–35) in different media. J Med Chem. 2004;47:4231–8. doi: 10.1021/jm040773o. [DOI] [PubMed] [Google Scholar]
- Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- Engel A, Müller DJ. Observing single biomolecules at work with the atomic force microscope. Nat Struct Biol. 2000;7:715–8. doi: 10.1038/78929. [DOI] [PubMed] [Google Scholar]
- Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- Fink AL. Natively unfolded proteins. Curr Opin Struct Biol. 2005;15:35–41. doi: 10.1016/j.sbi.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Flory P (1953): “Principles of Polymer Chemistry.” New York: Cornell University Press.
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. Atomic-force microscopy: Rhodopsin dimers in native disc membranes. Nature. 2003;421:127–8. doi: 10.1038/421127a. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Liang Y, Filipek S, Saperstein DA, Engel A, Palczewski K. The G protein-coupled receptor rhodopsin in the native membrane. FEBS Lett. 2004a;564:281–8. doi: 10.1016/S0014-5793(04)00194-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotiadis D, Qian P, Philippsen A, Bullough PA, Engel A, Hunter CN. Structural Analysis of the Reaction Center Light-harvesting Complex I Photosynthetic Core Complex of Rhodospirillum rubrum Using Atomic Force Microscopy. J Biol Chem. 2004b;279:2063–8. doi: 10.1074/jbc.M310382200. [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Scheuring S, Müller SA, Engel A, Müller DJ. Imaging and manipulation of biological structures with the AFM. Micron. 2002;33:385–97. doi: 10.1016/s0968-4328(01)00026-9. [DOI] [PubMed] [Google Scholar]
- Glabe CC. Amyloid accumulation and pathogensis of Alzheimer’s disease: significance of monomeric, oligomeric and fibrillar Abeta. Subcell Biochem. 2005;38:167–77. doi: 10.1007/0-387-23226-5_8. [DOI] [PubMed] [Google Scholar]
- Glabe CG. Conformation-dependent antibodies target diseases of protein misfolding. Trends Biochem Sci. 2004;29:542–7. doi: 10.1016/j.tibs.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Golde TE. The Abeta hypothesis: leading us to rationally-designed therapeutic strategies for the treatment or prevention of Alzheimer disease. Brain Pathol. 2005;15:84–7. doi: 10.1111/j.1750-3639.2005.tb00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutsmann T, Fantner GE, Kindt JH, Venturoni M, Danielsen S, Hansma PK. Force spectroscopy of collagen fibers to investigate their mechanical properties and structural organization. Biophys J. 2004;86:3186–93. doi: 10.1016/S0006-3495(04)74366-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas E. The study of protein folding and dynamics by determination of intramolecular distance distributions and their fluctuations using ensemble and single-molecule FRET measurements. Chemphyschem. 2005;6:858–70. doi: 10.1002/cphc.200400617. [DOI] [PubMed] [Google Scholar]
- Harms GS, Orr G, Montal M, Thrall BD, Colson SD, Lu HP. Probing conformational changes of gramicidin ion channels by single-molecule patch-clamp fluorescence microscopy. Biophys J. 2003;85:1826–38. doi: 10.1016/S0006-3495(03)74611-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–84. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iakoucheva LM, Brown CJ, Lawson JD, Obradovic Z, Dunker AK. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323:573–84. doi: 10.1016/s0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- Inouye H, Kirschner DA. Alzheimer’s beta-amyloid: insights into fibril formation and structure from Congo red binding. Subcell Biochem. 2005;38:203–24. doi: 10.1007/0-387-23226-5_10. [DOI] [PubMed] [Google Scholar]
- Jager M, Michalet X, Weiss S. Protein-protein interactions as a tool for site-specific labeling of proteins. Protein Sci. 2005;14:2059–68. doi: 10.1110/ps.051384705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyashekar NS, Sadana A, Vo-Dinh T. Protein amyloidose misfolding: mechanisms, detection, and pathological implications. Methods Mol Biol. 2005;300:417–35. doi: 10.1385/1-59259-858-7:417. [DOI] [PubMed] [Google Scholar]
- Kapanidis AN, Lee NK, Laurence TA, Doose S, Margeat E, Weiss S. Fluorescence-aided molecule sorting: analysis of structure and interactions by alternating-laser excitation of single molecules. Proc Natl Acad Sci U S A. 2004;101:8936–41. doi: 10.1073/pnas.0401690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsai A, Nagy A, Kengyel A, Martonfalvi Z, Grama L, Penke B, Kellermayer MS. Effect of lysine-28 side-chain acetylation on the nanomechanical behavior of alzheimer amyloid beta25-35 fibrils. J Chem Inf Model. 2005;45:1641–6. doi: 10.1021/ci0501701. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–9. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–6. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Kedrov A, Ziegler C, Janovjak H, Kuhlbrandt W, Muller DJ. Controlled unfolding and refolding of a single sodium-proton antiporter using atomic force microscopy. J Mol Biol. 2004;340:1143–52. doi: 10.1016/j.jmb.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Kellermayer MS, Grama L, Karsai A, Nagy A, Kahn A, Datki ZL, Penke B. Reversible mechanical unzipping of amyloid beta-fibrils. J Biol Chem. 2005;280:8464–70. doi: 10.1074/jbc.M411556200. [DOI] [PubMed] [Google Scholar]
- Kelly JW. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr Opin Struct Biol. 1998;8:101–6. doi: 10.1016/s0959-440x(98)80016-x. [DOI] [PubMed] [Google Scholar]
- Kransnoslobodtsev AV, Luda S. Shlyakhtenko, Egor Ukraintsev, Tatiana O. Zaikova John F. W. Keana and Yuri L. Lyubchenko (2005): Nanomedicine and Protein Misfolding Diseases. Nanomedicine: Nanotechnology, Biology, and Medicine in press. [DOI] [PMC free article] [PubMed]
- Kuzmenkina EV, Heyes CD, Nienhaus GU. Single-molecule Forster resonance energy transfer study of protein dynamics under denaturing conditions. Proc Natl Acad Sci U S A. 2005;102:15471–6. doi: 10.1073/pnas.0507728102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Neurodegenerative disease: amyloid pores from pathogenic mutations. Nature. 2002a;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Hartley DM, Petre BM, Wall JS, Simon MN, Walz T, Lansbury PT., Jr Mixtures of wild-type and a pathogenic (E22G) form of Abeta40 in vitro accumulate protofibrils, including amyloid pores. J Mol Biol. 2003;332:795–808. doi: 10.1016/s0022-2836(03)00927-6. [DOI] [PubMed] [Google Scholar]
- Lashuel HA, Petre BM, Wall J, Simon M, Nowak RJ, Walz T, Lansbury PT., Jr Alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J Mol Biol. 2002b;322:1089–102. doi: 10.1016/s0022-2836(02)00735-0. [DOI] [PubMed] [Google Scholar]
- Lin H, Bhatia R, Lal R. Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. Faseb J. 2001;15:2433–44. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- Lu Y, Weers B, Stellwagen NC. DNA persistence length revisited. Biopolymers. 2001;61:261–75. doi: 10.1002/bip.10151. [DOI] [PubMed] [Google Scholar]
- Luhrs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Dobeli H, Schubert D, Riek R. 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc Natl Acad Sci U S A. 2005;102:17342–7. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B, Nussinov R. Stabilities and conformations of Alzheimer’s beta -amyloid peptide oligomers (Abeta 16–22, Abeta 16–35, and Abeta 10–35): Sequence effects. Proc Natl Acad Sci U S A. 2002;99:14126–31. doi: 10.1073/pnas.212206899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makin OS, Serpell LC. Structures for amyloid fibrils. Febs J. 2005;272:5950–61. doi: 10.1111/j.1742-4658.2005.05025.x. [DOI] [PubMed] [Google Scholar]
- McAllister C, Karymov MA, Kawano Y, Lushnikov AY, Mikheikin A, Uversky VN, Lyubchenko YL. Protein Interactions and Misfolding Analyzed by AFM Force Spectroscopy. J Mol Biol. 2005;354:1028–42. doi: 10.1016/j.jmb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–8. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–8. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005a;44:1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- Oldfield CJ, Cheng Y, Cortese MS, Romero P, Uversky VN, Dunker AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry. 2005b;44:12454–70. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- O’Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci U S A. 2002;99:1485–90. doi: 10.1073/pnas.022662599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002a;99:5591–5. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perutz MF, Pope BJ, Owen D, Wanker EE, Scherzinger E. Aggregation of proteins with expanded glutamine and alanine repeats of the glutamine-rich and asparagine-rich domains of Sup35 and of the amyloid beta-peptide of amyloid plaques. Proc Natl Acad Sci U S A. 2002b;99:5596–600. doi: 10.1073/pnas.042681599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. Solid state NMR reveals a pH-dependent antiparallel beta-sheet registry in fibrils formed by a beta-amyloid peptide. J Mol Biol. 2004;335:247–60. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta -amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci U S A. 2002;99:16742–7. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptitsyn OB. How the molten globule became. Trends Biochem Sci. 1995a;20:376–9. doi: 10.1016/s0968-0004(00)89081-7. [DOI] [PubMed] [Google Scholar]
- Ptitsyn OB. Molten globule and protein folding. Adv Protein Chem. 1995b;47:83–229. doi: 10.1016/s0065-3233(08)60546-x. [DOI] [PubMed] [Google Scholar]
- Ptitsyn OB, Bychkova VE, Uversky VN. Kinetic and equilibrium folding intermediates. Philos Trans R Soc Lond B Biol Sci. 1995;348:35–41. doi: 10.1098/rstb.1995.0043. [DOI] [PubMed] [Google Scholar]
- Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Amyloid ion channels: a common structural link for protein-misfolding disease. Proc Natl Acad Sci U S A. 2005;102:10427–32. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia A, Debetto P, Negro A, Guidolin D, Skaper SD, Giusti P. Alpha-synuclein and Parkinson’s disease. Faseb J. 2004;18:617–26. doi: 10.1096/fj.03-0338rev. [DOI] [PubMed] [Google Scholar]
- Rhee SK, Quist AP, Lal R. Amyloid beta protein-(1–42) forms calcium-permeable, Zn2+-sensitive channel. J Biol Chem. 1998;273:13379–82. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Lansbury PT., Jr Amyloid fibrillogenesis: themes and variations. Curr Opin Struct Biol. 2000;10:60–8. doi: 10.1016/s0959-440x(99)00049-4. [DOI] [PubMed] [Google Scholar]
- Rochet JC, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. Interactions among alpha-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 2004;23:23–34. doi: 10.1385/jmn:23:1-2:023. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Salvetat J-P, Briggs GAD, Bonard J-M, Bacsa RR, Kulik AJ, Stoekli T, Burnham NA, Forró L. Elastic and Shear Moduli of Single-Walled Carbon Nanotube Ropes. Phys Rev Let. 1999;82:944–947. [Google Scholar]
- Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–7. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- Segel DJ, Eliezer D, Uversky V, Fink AL, Hodgson KO, Doniach S. Transient dimer in the refolding kinetics of cytochrome c characterized by small-angle X-ray scattering. Biochemistry. 1999;38:15352–9. doi: 10.1021/bi991337k. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genotypes, phenotypes, and treatments. Science. 1997;275:630–1. doi: 10.1126/science.275.5300.630. [DOI] [PubMed] [Google Scholar]
- Serpell LC. Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta. 2000;1502:16–30. doi: 10.1016/s0925-4439(00)00029-6. [DOI] [PubMed] [Google Scholar]
- Smith DP, Jones S, Serpell LC, Sunde M, Radford SE. A systematic investigation into the effect of protein destabilisation on beta 2-microglobulin amyloid formation. J Mol Biol. 2003;330:943–54. doi: 10.1016/s0022-2836(03)00687-9. [DOI] [PubMed] [Google Scholar]
- Stasiak A. Three-stranded DNA structure; is this the secret of DNA homologous recognition? Mol Microbiol. 1992;6:3267–76. doi: 10.1111/j.1365-2958.1992.tb02194.x. [DOI] [PubMed] [Google Scholar]
- Stromer T, Serpell LC. Structure and morphology of the Alzheimer’s amyloid fibril. Microsc Res Tech. 2005;67:210–7. doi: 10.1002/jemt.20190. [DOI] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27:527–33. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- Tycko R. Insights into the amyloid folding problem from solid-state NMR. Biochemistry. 2003;42:3151–9. doi: 10.1021/bi027378p. [DOI] [PubMed] [Google Scholar]
- Tycko R. Progress towards a molecular-level structural understanding of amyloid fibrils. Curr Opin Struct Biol. 2004;14:96–103. doi: 10.1016/j.sbi.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT. Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer’s disease. Proc Natl Acad Sci U S A. 2002;99:13990–5. doi: 10.1073/pnas.222433299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002a;11:739–56. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN. What does it mean to be natively unfolded? Eur J Biochem. 2002b;269:2–12. doi: 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- Uversky VN. A protein-chameleon: conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J Biomol Struct Dyn. 2003;21:211–34. doi: 10.1080/07391102.2003.10506918. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Fink AL. Conformational constraints for amyloid fibrillation: the importance of being unfolded. Biochim Biophys Acta. 2004;1698:131–53. doi: 10.1016/j.bbapap.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–27. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Uversky VN, Oldfield CJ, Dunker AK. Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recognit. 2005;18:343–84. doi: 10.1002/jmr.747. [DOI] [PubMed] [Google Scholar]
- Veerhuis R, Boshuizen RS, Familian A. Amyloid associated proteins in Alzheimer’s and prion disease. Curr Drug Targets CNS Neurol Disord. 2005;4:235–48. doi: 10.2174/1568007054038184. [DOI] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337:635–45. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293:321–31. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- Xie Z, Srividya N, Sosnick TR, Pan T, Scherer NF. Single-molecule studies highlight conformational heterogeneity in the early folding steps of a large ribozyme. Proc Natl Acad Sci U S A. 2004;101:534–9. doi: 10.1073/pnas.2636333100. [DOI] [PMC free article] [PubMed] [Google Scholar]