

Abstract
Objective
Long-term antidepressant drug exposure may regulate its target molecule — the serotonin transporter (SERT). This effect could be related to an individual's genotype for an SERT promoter polymorphism (human serotonin transporter coding [5-HTTLPR]). We aimed to determine the effects of fluoxetine exposure on human platelet SERT levels.
Method
We harvested platelet samples from 21 healthy control subjects. The platelets were maintained alive ex vivo for 24 hours while being treated with 0.1 μM fluoxetine or vehicle. The effects on SERT immunoreactivity (IR) were then compared. Each individual's SERT promoter genotype was also determined to evaluate whether fluoxetine effects on SERT were related to genotype.
Results
Fluoxetine exposure replicably altered SERT IR within individuals. Both the magnitude and the direction of effect were related to a person's SERT genotype. People who were homozygous for the short gene (SS) displayed decreased SERT IR, whereas those who were homozygous for the long gene (LL) demonstrated increased SERT IR. A mechanistic experiment suggested that some individuals with the LL genotype might experience increased conversion of complexed SERT to primary SERT during treatment.
Conclusions
These preliminary results suggest that antidepressant effects after longer-term use may include changes in SERT expression levels and that the type and degree of effect may be related to the 5-HTTLPR polymorphism.
Medical subject headings: antidepressants, gene expression, serotonergic mechanisms
Abstract
Objectif
L'exposition prolongée à des antidépresseurs peut en régulariser la molécule cible — le transporteur de la sérotonine (SERT). Cet effet pourrait être relié au génotype d'un sujet pour le polymorphisme du promoteur du SERT (transporteur de la sérotonine humaine codant [5-HTTLPR]). Nous voulions déterminer les effets de l'exposition à la fluoxétine sur les concentrations de SERT dans les plaquettes humaines.
Méthode
Nous avons prélevé des échantillons de plaquettes de 21 sujets témoins en bonne santé. On a maintenu les plaquettes vivantes ex vivo pendant 24 heures pendant qu'on les traitait à la fluoxétine 0,01 mM ou au moyen d'un véhicule. On a alors comparé les effets sur l'immunoréactivité (IR) du SERT. On a aussi déterminé le génotype promoteur du SERT de chaque individu pour déterminer s'il y avait un lien entre les effets de la fluoxétine sur le SERT et le génotype.
Résultats
L'exposition à la fluoxétine a modifié de façon répétable l'IR du SERT chez certains individus. On a établi un lien entre à la fois l'ordre de grandeur et l'orientation de l'effet et le génotype du SERT d'une personne. Chez les sujets homozygotes pour le gène court (SS), l'IR du SERT a diminué, tandis qu'elle augmentait chez les sujets homozygotes pour le gène long (LL). Une expérience mécanistique a indiqué que chez des sujets qui ont le génotype LL, la conversion du SERT complexé en SERT primaire pourrait augmenter au cours du traitement.
Conclusions
Ces résultats préliminaires indiquent que les effets antidépresseurs après une utilisation prolongée peuvent inclure des changements des taux d'expression du SERT et qu'il peut y avoir un lien entre le type et le degré de l'effet et le polymorphisme 5-HTTLPR.
Introduction
The serotonin transporter (SERT) is the target molecule for most of the antidepressant drugs currently in use — both the older tricyclic drugs and the newer selective serotonin reuptake inhibitors (SSRI). When these drugs bind to the SERT, they inhibit its normal function — the uptake of serotonin (5-hydroxytryptamine [5-HT]) from the synapse, leading to an accumulation of 5-HT in the synapse. This short-term effect is believed to be critical in causing eventual clinical improvement, although the entire mechanism responsible has not been elucidated. In addition to acute blockade, it is possible that changes may be induced in SERT function after prolonged exposure to antidepressant drugs. Recent studies employing sensitive measures of SERT expression over extended time periods have found that antidepressant exposure decreases SERT levels in the rat midbrain.1 This delayed long-term regulatory effect eventually has a much greater impact on actual synaptic levels of serotonin than the initial pharmacological blockade.2 It remains unclear whether such marked adaptive effects occur in humans.
Progressively diminishing SERT concentrations could decrease the SERT's ability to remove 5-HT from the synapse, thus continually increasing synaptic 5-HT concentrations and perhaps also damping temporal fluctuations. Alterations in SERT function could also have an effect on neuronal membrane potential, because ionic currents pass through the SERT during translocation of 5-HT, as well as during apparent resting intervals.3 From a clinical perspective, if further regulation of SERT function occurs in addition to simple uptake blockade, such a process could explain the extended time necessary for treatment response, as well as side effects that develop during the course of treatment. Because of these considerations, regulation of SERT function could be an important aspect of the clinical response to longer-term antidepressant exposure.
The present experiments evaluated SERT expression levels in healthy control platelets, harvested and maintained alive for 24 hours and treated with fluoxetine, to test the hypothesis that fluoxetine exposure would alter total SERT immunoreactivity (IR). We chose to study platelets because they express large numbers of SERT molecules and they can be obtained in blood samples. Further, the number and/or function of SERT molecules expressed in the human brain4–6 or peripherally7–9 may be influenced by a common polymorphism in the SERT promoter region (human serotonin transporter coding [5-HTTLPR]), although there are also reports showing no relation.10–12 Several studies have suggested that the genotype for the SERT polymorphism is related to eventual antidepressant response.13–21 For these reasons, we determined the SERT promoter genotype in the study subjects to discover whether it affected fluoxetine-related regulation of platelet SERT expression.
Methods
Twenty-one subjects were studied after providing informed consent, as approved by the Human Studies Committee of the Ann Arbor Veteran's Affairs Medical Center. Subjects were under 50 years of age (range 22–48 yr) and included 3 women and 18 men. No subject had a history of taking antidepressant medication.
Platelet preparation22
Platelets were harvested in a vacutainer tube containing sodium citrate (0.1 μM). An initial platelet-rich supernatant was obtained through mild centrifugation (160 × g for 10 min), followed by platelet separation with more intense centrifugation (12 000 × g for 10 min). The resulting pellet was washed twice for 10 minutes in phosphate buffer solution (PBS). The platelets were resuspended in PBS. One-half of each subject's platelets were treated with fluoxetine, 0.1 μM in PBS, and the other one-half were treated with PBS only, for 24 hours on a vortexer at 4°C, to decrease nonspecific degradation. The concentration of fluoxetine used was similar to serum levels found in human patients. After incubation, platelets were centrifuged 12 000 × g for 10 minutes, then lysed with radioimmunoprecipitation buffer with protease inhibitors and agitated for 30 minutes at 4°C. Total protein concentrations were determined spectroscopically with the Bio-Rad DC protein assay kit; individual gel loadings (further described below) were based on these determinations. Total protein concentrations were virtually identical between treated and untreated samples, indicating that treatment did not cause nonspecific degradation of total protein. An initial time course study found that preserving platelets for periods longer than 24 hours resulted in declining [3H]5-HT uptake rates (Fig. 1). One tube of frozen blood was sent for genotyping by the 5-HTTLPR (see below). At least 5 subjects were identified for each of the 3 genotypes.
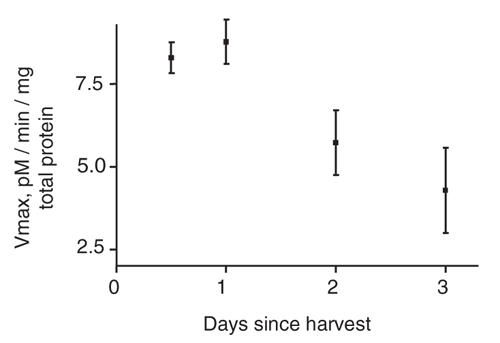
Fig. 1: Effect of platelet storage time on [3H]5-hydroxytryptamine uptake. There was a noticeable decline by the second day; thus additional experiments were performed within 24 hours. Each data point represents the results from 3 replicate tubes. The error bars display standard deviations. vMAX = maximal update velocity.
[3H]5-HT uptake assays
We assessed platelet uptake to determine whether the SERT remained functional during the 24-hour treatment period. Krebs Phosphate (KP) buffer (pH 7.4) was used and contained NaCl 120 mmoL/L, KCl 4.8 mmoL/L, MgSO4 1.4 mmoL/L, Na2HPO4 16 mM, glucose 11 mmoL/L, ascorbic acid 1.0 mmoL/L, pargyline 0.03 mmoL/L and CaCl2 1.2 mmoL/L. Cells were incubated at room temperature for 30 minutes to stablize in KP buffer before assay. [3H]5-HT (27.5 Ci/mmol, NEN, Boston, Mass.) was added in a final concentration of 10 nM to initiate uptake. The healthy platelets were kept at 37°C and shaken for 15 minutes. ([3H]5-HT uptake was linear for 15 minutes in time course experiments.) Uptake was terminated by removal of the reaction medium, followed by 3 washes with 1 mL of ice-cold buffer. Cells were dissolved in 500 μl 1% sodium dodecyl sulfate (SDS), and the solution was counted, as described above. Nonspecific uptake was determined in the presence of 10 μM (-)imipramine.
SERT IR quantification
SERT levels were measured by Western blot assays of SERT IR, with a method similar to that used with the dopamine transporter.23 This was done in preference to measuring radiolabelled serotonin uptake, or radioligand binding, because each of these would require the complete washing out of inhibitory levels of fluoxetine to below the nanomolar range, which could be difficult to accomplish without losing substantial amounts of platelets. After treatment incubation and total protein concentrations were determined, platelet protein samples (0.25 mg/gel lane, based on film response standard curve; see Fig. 2) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS–PAGE) with a 7.5% polyacrylamide solution (8 × 5 cm, 2 mm thick). Samples were then electrophoretically transferred to Immobilon-P membranes at 36 mA for 1 hour. Blots were incubated with 5% (w/v) dry milk in Tris-buffered saline with Tween ([TTBS] 0.1% Tween 20, 0.15 M NaCl, and 10 mmoL/L Tris-HCl, pH 7.4) for 1 hour. Each step was performed at room temperature. The primary antibody used was a rabbit anti-human serotonin transporter antibody at 1:4000 dilution (Chemicon, Inc, Temecula, Calif.), followed by a horseradish peroxidase-conjugated donkey anti-rabbit IgG second antibody (Amersham Life Sciences, Piscataway, NJ). The antigen-antibody complex was detected with the ECL plus Western blotting detection system at 1:2000 dilution (Amersham Life Sciences, Piscataway, NJ). An intense array of bands were consistently identified clustered near the 78 kDa standard, which was also present in samples from the human brain stem containing raphe nuclei (positive control), but not in the cerebellum (negative control). Multiple fluoxetine-treated and control samples from the same individual were assayed next to each other on the same gels. Film exposure times were varied to ensure that the dynamic range of the film was used. The relative optical densities of protein bands were digitized and quantitated with a microcomputer imaging analysis system (Microcomputer Imaging Devices, Ottawa, Ont.), and the results were expressed as a percentage change, compared with the vehicle-treated platelets, and analyzed with a 1-way analysis of variance (ANOVA) to test for genotype effect. No absolute measure of basal SERT levels was performed, because we have previously described the relation between SERT expression in the human brain5 and in platelets.8
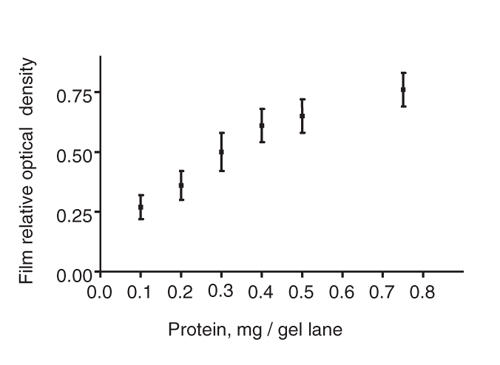
Fig. 2: Film response to varying levels of protein. Platelet protein samples from 3 people were pooled, and varying protein amounts were loaded into sample gels. Each data point represents the results from 3 to 4 sets of paired lanes. Further experiments used a loading of 0.25 mg total protein per gel lane, based on the linearity of relative optical densities detected around this loading.
SERT genotyping5,24
We used the Pure Gene extraction kit (Gentra, Redmond, Calif.) to extract DNA from whole blood. PCR for 5-HTTLPR was carried out in a 10 μl volume containing 50 ng of genomic template; 0.5 μM of each primer, one of which was 5′ fluorescently labelled; 200 μM of each dNTP; 1 PCRbuffer; 1.5 mmoL/L MgCl2; and 0.3 units of DyNAzymeTM EXT DNA polymerase (Finnzymes Oy, Espoo, Finland), with 0.5 M GC-melt (Clontech, Palo Alto, Calif.). Primer sequences were 5′-FAM-CTGAATGCCAGCACCTAACCCCTAATGT-3′ and 5′- GTTTCTTGGGGAATACTGGTAGGGTGCAAGGAGAA-3′. Samples were amplified on a 9700 thermal cycler (Applied Biosystems, Foster City, Calif.), with an initial heat-activation step at 96ºC for 12 minutes, followed by 40 to 45 cycles, with a denaturation step of 96ºC for 30 seconds; an annealing step of 68ºC for 45 seconds; and an extension step of 72ºC for 3 minutes for 5-HTTLPR, with a final extension step at 72ºC for 10 minutes. Post-PCR products were injected and detected by laser-induced fluorescence on an ABI PRISM 3730 Genetic Analyzer (Dallas, TX) at the University of Chicago DNA sequencing and genotyping core. Electropherograms were processed, and alleles were called with Genemapper software blind to platelet data.
Results
The changes in SERT IR induced by fluoxetine exposure were consistent within an experiment (see Fig. 3) and replicable over time in individuals (as shown in Fig. 4). Genotyping of each subject for the SERT promoter polymorphism found that, of the 21 individuals, 5 were homozygous for the long gene (LL), 10 were heterozygous (SL) and 6 were homozygous for the short gene (SS).
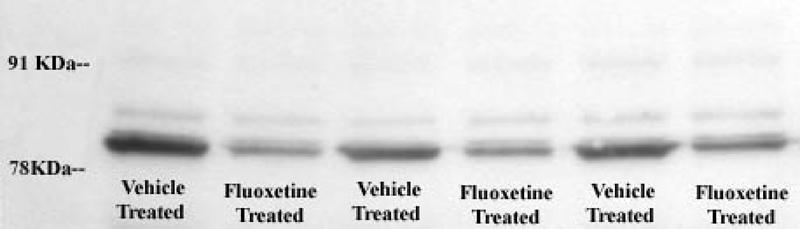
Fig. 3: Digitized image of a gel displaying platelet serotonin transporter (SERT) immunoreactivity (IR) and the effect of fluoxetine exposure (0.1 μM dose for 24 hours) in a typical short-gene subject. On Day 1 before treatment, 0.5 mL of platelet solution were pipetted into each test tube. Twenty-four hours later, total protein concentrations were determined in each membrane solution, and the gel loading concentrations were adjusted as indicated. There was no overall effect of fluoxetine treatment on total protein (t 0.128= 24, p = 0.90), nor was there any correlation between total protein and SERT IR (r2 = 0.07, df = 1,18, p = 0.40).
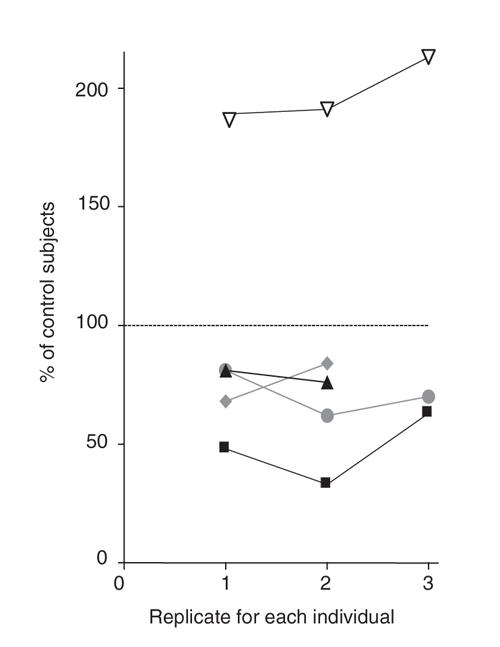
Fig. 4: Effects of repeated fluoxetine treatment (0.1 μM for 24 hours) on platelet serotonin transporter (SERT) immunoreactivity (IR) in the same people and replicability of fluoxetine effect on platelet SERT IR. Samples from 5 subjects were assayed at 2 or 3 time points, separated by 2 to 12 months. Each shape represents a unique individual, and the colours represent different genotypes. Black = subjects with an SS genotype, grey = subjects with an SL genotype, and white = subjects with the LL genotype. As shown in the graph, the changes induced were consistent within and between individuals. Each data point represents the results from 3 to 4 sets of paired lanes.
LL = homozygous with the long gene; SL = heterozygous; SS = homozygous with the short gene.
An initial inspection of the data was performed to determine whether age, sex or genotype affected fluoxetine-induced change in SERT IR. It was hypothesized that only genotype would have an effect. As the analyses below indicate, age and sex and their interactions with genotype were not statistically significant, whereas genotype alone was a significant factor.
Subjects' mean age was 32 years (standard deviation [SD] 9.6 yr), which did not correlate with fluoxetine-induced change in SERT IR (r2 = 0.01, df = 1,19, p = 0.97). Age did not vary between the SERT genotype groups: people with the SS gene averaged 33 (SD 4) years of age, the SL gene averaged 32 (SD 4) years of age and the LL gene averaged 29 (SD 5) years of age; these were not significantly different (1-way ANOVA, F2.18 = 0.331, p = 0.72).
Platelets from 3 females (one from each of the LL, SL and SS genotype groups) displayed, on average, a 9% (SD 11%) increase in SERT IR, whereas those from males showed a 0.5% (SD 36%) decrease, which was not significantly different (t20= 0.344, p = 0.43). The rank order in females was the same as for the larger group, with people with the SS genotype showing a decrease in SERT and with the LL genotype showing an increase.
There was an effect of SERT genotype on the response to fluoxetine treatment (Fig. 5), which was significant by 1-way ANOVA (F18 = 3.582, p < 0.05). Individuals with the LL genotype were significantly different from those with the SS genotype (Tukey's q = 3.705, p < 0.05), but people with the LS genotype were not significantly different from either homozygous group. On average, those with the SS genotype displayed approximately a 21% (SD 10%) decrease in the 78 kDa SERT IR band, whereas those with the LL genotype displayed a 35 (SD 21%) increase.
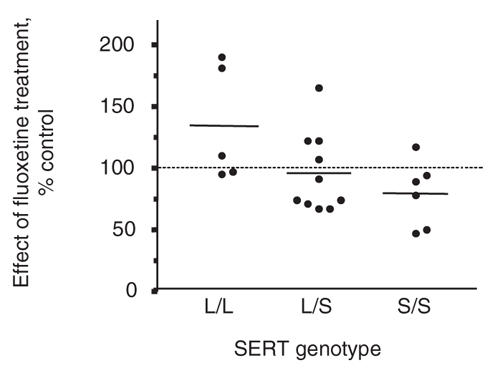
Fig. 5: Effect of fluoxetine treatment (0.1 μM for 24 hours) on platelet serotonin transporter (SERT) immunoreactivity in subjects grouped by SERT genotype. SERT genotype had a significant effect on the response to fluoxetine exposure (1-way analysis of variance; F2,18 = 3.582, p < 0.05). People who were homozygous with the long gene (LL) were significantly different from those homozygous with the short gene (SS) (Tukey's q = 3.705, p < 0.05).
LS = people with heterozygous genotypes.
Overall, there was no effect of fluoxetine treatment on SERT protein levels, at 38.3 μg/mL (SD 2.1) versus 39.4 mg/mL (SD 2.2) (t40 = 0.1367, 2-tailed, p = 0.83) and no difference on total protein, at 393 mg/mL (SD 41) versus 406 (SD 43) (t40 = 0.1324, 2-tailed, p = 0.87).
Discussion
Understanding the changes induced in SERT function by sustained antidepressant inhibition is important, because an individual's response to long-term uptake inhibition could be more important than immediate reuptake blockade in explaining eventual clinical response. In the present experiment, exposure to a high dose of the antidepressant drug fluoxetine altered platelet SERT IR in some people. Although there was considerable variation between individuals, the SERT IR response was replicable. Most significantly, the changes induced in SERT IR were related to an individual's SERT promoter genotype. The SERT promoter genotype is a genetic polymorphism that has been implicated in controlling antidepressant treatment response in several earlier studies and possibly in contributing to depressive symptoms.
In the current experiment, individuals with the SS genotype displayed reduced SERT IR in their platelets after exposure to fluoxetine, compared with vehicle-treated platelets. This suggests that, if parallel effects occur in the human brain, people with the SS genotype might display greater antidepressant effects–both therapeutic and adverse. The existing clinical literature consistently suggests that greater side effects occur in people with the SS genotype, since they are more likely to experience antidepressant-induced mania25 and antidepressant-induced insomnia and agitation.26 This is easy to conceptualize, because emergence of these side effects would seem more likely in people who display basally lower SERT levels4–9 and in those who further undergo an antidepressant-induced decline in SERT function, as suggested in the current study. In addition, other studies indicate that people with the SS genotype are more prone to rapid cycling,27 violence28 or violent suicide–adverse29 outcomes that are highly pertinent to antidepressant administration. Thus, further work is needed to confirm whether people with the SS genotype are more prone to these undesirable side effects and whether these are related to a downregulation of overall SERT concentrations.
In contrast, the preponderance of existing clinical treatment trials indicates that individuals witth the SS genotype have poorer clinical response to longer-term antidepressant treatment.13–19 (However, there are exceptions: opposite results were noted in 2 of 3 Asian samples evaluated,20,21 whereas Yu and others16 reported results in Han Chinese similar to the European samples.) These results and our own data are less easily explained. Perhaps increased side effects contribute to greater dropouts or mask improvement. Most likely, other unidentified compensatory effects are initiated, nullifying or inducing resistance to the therapeutic neurochemical effects. The decline in SERT IR seen in the platelets from individuals with the SS genotype could reflect degradative or nondestructive, conformational, oligomerization or complexation effects. Further, because the direction of change in SERT IR concentrations appeared to be opposite in people with the LL genotype, our data indicate that multiple distinct processes are likely contributing factors in individuals with each genotype.
The present results, although consistent and suggestive, are preliminary and were discovered in healthy individuals. Similar studies need to be performed in patients with depressive illness. This task is complicated by the fact that it is uncertain whether SERT function is altered10,30–32 or not20,33,34 by depressive illness before beginning antidepressant treatment. If SERT function is previously disturbed in patients with depression, this could obviously confound interpretation of the type of effect presently described in healthy control subjects.
Therapeutic effects are commonly considered slow to evolve, although 24 hours is a short time interval for modelling neuronal response to fluoxetine. However, total platelet half-life is only 10 days, and platelets have limited capacities for more modulated biochemical responses. It is possible that the effects seen in the present experiment after 24 hours might be magnified over time in platelets in vivo or in another model. Conversely, recent data, likely reflecting short-term increases in serotonin levels rather than more complex adaptations, indicate that clinical effects of antidepressant drugs can be detected earlier than previously thought.35–37
It remains to be determined whether the change in platelet SERT IR that we detected was related to a change in SERT function. Although likely, we did not confirm this association in the present study, owing to difficulties in consistently removing inhibitory amounts of fluoxetine. The relation between IR and function is not necessarily a direct one; most transporter molecules are held in reserve and are not functionally active in the outer membrane.38 Thus, individual differences in the regulation of SERT function may result from genetic variance affecting its trafficking, including the conformational structure of the SERT protein and interactions with phosphorylative and coupling enzymes that are responsible for recognizing and responding to SERT recycling signals.3
Despite the fact that platelets and brain neurons are highly distinct physiologically, binding site occupancy-induced molecular changes are likely similar in all membrane-embedded SERT molecules. Because the likely mechanisms do not appear to necessarily invoke complex regulatory loops, further experiments in platelets, and possibly dispersed cells, may illuminate the relevant cell physiology. Also, unlike studies of platelet 5-HT concentrations, the present technique avoids the possible confounding effect of variable 5-HT degradation, which is perturbed by fluctuations in intrinsic monoamine oxidase activity and by potential artifactual oxidation during harvesting or assay.39 Another strength of our study is that we avoided possible differences in platelet survival or the confounding effects of other internal influences by removing the platelets from the serum environment and controlling the drug exposure in vitro. We believe that further work is necessary to test the hypotheses that fluoxetine exposure alters SERT levels, function and clinical response to antidepressant drugs in platelets and in the brain in a manner related to SS or LL genotype.
Acknowledgments
This work was supported by a VA Merit Award and National Institutes of Health award DA15509.
Footnotes
Contributors: Dr. Little designed the study. All authors acquired the data, which Drs. Little and Cook analyzed. Dr. Little wrote the article. All authors critically reviewed the article and gave final approval for its publication.
Competing interests: None declared.
Correspondence to: Dr. Karley Y. Little, Laboratory of Affective Neuropharmacology/116-A, Ann Arbor V.A.M.C., 2215 Fuller Rd., Ann Arbor, MI 48105; fax 734 769-7410; kylittle@umich.edu
References
- 1.Benmansour S, Cecchi M, Morilak DA, et al. Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci 1999;19:10494-501. [DOI] [PMC free article] [PubMed]
- 2.Benmansour S, Owens WA, Cecchi M, et al. Serotonin clearance in vivo is altered to a greater extent by antidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter. J Neurosci 2002;22:6766-72. [DOI] [PMC free article] [PubMed]
- 3.Blakely RD, Ramamoorthy S, Schroeter S, et al. Regulated phosphorylation and trafficking of antidepressant-sensitive serotonin transporter proteins. Biol Psychiatry 1998;44:169-78. [DOI] [PubMed]
- 4.Heinz A, Higley JD, Gorey JG, et al. In vivo association between alcohol intoxication, aggression, and serotonin transporter availability in nonhuman primates. Am J Psychiatry 1998;155:1023-8. [DOI] [PubMed]
- 5.Little KY, McLaughlin DP, Zhang L, et al. Cocaine, ethanol, and genotype effects on human midbrain serotonin transporter binding sites and mRNA levels. Am J Psychiatry 1998;155:207-13. [DOI] [PubMed]
- 6.van Dyck CH, Malison RT, Staley JK, et al. Central serotonin transporter availability measured with [123I]beta-CIT SPECT in relation to serotonin transporter genotype. Am J Psychiatry 2004;161:525-31. [DOI] [PubMed]
- 7.Lesch KP, Bengel D, Heils A, et al. Association of anxiety-related traits with a polymorphism in the serotonin transporter gene regulatory region. Science 1996;274:1527-31. [DOI] [PubMed]
- 8.Stoltenberg SF, Twitchell G, Cook EH, et al. The serotonin transporter promoter polymorphism, peripheral indices of serotonin function and personality measures in families with alcoholism. Am J Med Genet 2002;114:230-4. [DOI] [PubMed]
- 9.Greenberg BD, Tolliver TJ, Huang SJ, et al. Genetic variation in the serotonin transporter promoter region affects serotonin uptake in human blood platelets. Am J Med Genet 1999;88:83-7. [PubMed]
- 10.Mann JJ, Huang YY, Underwood MD, et al. A serotonin transporter gene promoter polymorphism (5-HTTLPR) and prefrontal cortical binding in major depression and suicide. Arch Gen Psychiatry 2000;57:729-38. [DOI] [PubMed]
- 11.Willeit M, Stastny J, Pirker W, et al. No evidence for in vivo regulation of midbrain serotonin transporter availability by serotonin transporter promoter gene polymorphism. Biol Psychiatry 2001;50:8-12. [DOI] [PubMed]
- 12.Patkar AA, Berrettini WH, Mannelli P, et al. Relationship between serotonin transporter gene polymorphisms and platelet serotonin transporter sites among African-American cocaine-dependent individuals and healthy volunteers. Psychiatr Genet 2004;14:25-32. [DOI] [PubMed]
- 13.Smeraldi E, Zanardi R, Benedetti F, et al. Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Mol Psychiatry 1998;3:508-11. [DOI] [PubMed]
- 14.Pollock BG, Ferrell RE, Mulsant BH, et al. Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology 2000;23:587-90. [DOI] [PubMed]
- 15.Zanardi R, Benedetti F, DiBella D, et al. Efficacy of paroxetine in depression is influenced by a functional polymorphism within the promoter of serotonin transporter gene. J Clin Psychopharmacol 2000;20:105-7. [DOI] [PubMed]
- 16.Yu YW, Tsai SJ, Chen TJ, et al. Association study of the serotonin transporter promoter polymorphism and symptomatology and antidepressant response in major depressive disorders. Mol Psychiatry 2002;7:1115-9. [DOI] [PubMed]
- 17.Joyce PR, Mulder RT, Luty SE, et al. Age-dependent antidepressant pharmacogenomics: polymorphisms of the serotonin transporter and G protein beta3 subunit as predictors of response to fluoxetine and nortriptyline. Int J Neuropsychopharmacol 2003;6:339-46. [DOI] [PubMed]
- 18.Arias B, Catalan R, Gasto C, et al. 5-HTTLPR polymorphism of the serotonin transporter gene predicts non-remission in major depression patients treated with citalopram in a 12-weeks follow up study. J Clin Psychopharmacol 2003;23:563-7. [DOI] [PubMed]
- 19.Rausch JL, Johnson ME, Fei YJ, et al. Initial conditions of serotonin transporter kinetics and genotype: influence on SSRI treatment trial outcome. Biol Psychiatry 2002;51:723-32. [DOI] [PubMed]
- 20.Kim DK, Lim SW, Lee S, et al. Serotonin transporter gene polymorphism and antidepressant response. Neuroreport 2000;11:215-9. [DOI] [PubMed]
- 21.Yoshida K, Ito K, Sato K, et al. Influence of the serotonin transporter gene-linked polymorphic region on the antidepressant response to fluvoxamine in Japanese depressed patients. Prog Neuropsychopharmacol Biol Psychiatry 2002;26:383-6. [DOI] [PubMed]
- 22.Arora RC, Meltzer HY. A modified assay method for determining serotonin uptake in human platelets. Clin Chim Acta 1981;112:225-33. [DOI] [PubMed]
- 23.Little KY, Elmer LW, Zhong H, et al. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol 2002;61:436-45. [DOI] [PubMed]
- 24.Heils A, Teufel A, Petri S, et al. Allelic variation of human serotonin transporter gene expression. J Neurochem 1996;66:2621-4. [DOI] [PubMed]
- 25.Mundo E, Walker M, Cate T, et al. The role of serotonin transporter protein gene in antidepressant-induced mania in bipolar disorder: preliminary findings. Arch Gen Psychiatry 2001;58:539-44. [DOI] [PubMed]
- 26.Perlis RH, Mischoulon D, Smoller JW, et al. Serotonin transporter polymorphisms and adverse effects with fluoxetine treatment. Biol Psychiatry 2003;54:879-83. [DOI] [PubMed]
- 27.Rousseva A, Henry C, van den Bulke D, et al. Antidepressant-induced mania, rapid cycling and the serotonin transporter gene polymorphism. Pharmacogenomics J 2003;3:101-4. [DOI] [PubMed]
- 28.Retz W, Retz-Junginger P, Supprian T, et al. Association of serotonin transporter promoter gene polymorphism with violence: relation with personality disorders, impulsivity, and childhood ADHD psychopathology. Behav Sci Law 2004;22:415-25. [DOI] [PubMed]
- 29.Lin PY, Tsai G. Association between serotonin transporter gene promoter polymorphism and suicide: results of a meta-analysis. Biol Psychiatry 2004;55:1023-30. [DOI] [PubMed]
- 30.Neuger J, El Khoury A, Kjellman BF, et al. Platelet serotonin function in untreated major depression. Psychiatry Res 1999;85:189-98. [DOI] [PubMed]
- 31.Rosel P, Arranz B, Vallejo J, et al. Altered [3H]imipramine and 5-HT2 but not [3H]paroxetine binding sites in platelets from depressed patients. J Affect Dis 1999;52:225-33. [DOI] [PubMed]
- 32.Meyer JH, Houle S, Sagrati S, et al. Brain serotonin transporter binding potential measured with carbon 11–labeled DASB positron emission tomography: effects of major depressive episodes and severity of dysfunctional attitudes. Arch Gen Psychiatry 2004;61:1271-9. [DOI] [PubMed]
- 33.Little KY, McLaughlin DP, Ranc J, et al. Serotonin transporter binding sites and mRNA levels in depressed persons committting suicide. Biol Psychiatry 1997;41:1156-64. [DOI] [PubMed]
- 34.Hoehe MR, Wendel B, Grunewald I, et al. Serotonin transporter (5-HTT) gene polymorphisms are not associated with susceptibility to mood disorders. Am J Med Genet 1998;81:1-3. [DOI] [PubMed]
- 35.Posternak MA, Zimmerman M. Is there a delay in the antidepressant effect? A meta-analysis. J Clin Psychiatry 2005;66:148-58. [DOI] [PubMed]
- 36.Kemp AH, Gray MA, Silberstein RB, et al. Augmentation of serotonin enhances pleasant and suppresses unpleasant cortical electrophysiological responses to visual emotional stimuli in humans. Neuroimage 2004;22:1084-96. [DOI] [PubMed]
- 37.Harmer CJ, Bhagwagar Z, Perrett DI, et al. Acute SSRI administration affects the processing of social cues in healthy volunteers. Neuropsychopharmacology 2003;28:148-52. [DOI] [PubMed]
- 38.Carneiro AM, Blakely RD. Regulated alterations in platelet serotonin transporter localization and protein interactions, program no. 156.15, 2005. Washington, D.C.: Society for Neuroscience; 2005. Online.
- 39.Mulder EJ, Anderson GM, Kema IP, et al. Reactivity of serotonin in whole blood. Biol Psychiatry 2002;51:266-8. [DOI] [PubMed]