

Abstract
The psychostimulant methamphetamine (MA) is a highly addictive drug that has surged in popularity over the last decade in North America. A burgeoning number of clandestine drug laboratories has led to dramatic increases in MA production, which have resulted in significant public health, legal and environmental problems. Current evidence indicates that exposure to MA is neurotoxic, and neuroimaging studies confirm that long-term use in humans may lead to extensive neural damage. These physiological changes are commonly associated with persistent forms of cognitive impairment, including deficits in attention, memory and executive function. In the present review, we provide a comprehensive description of the factors relating to MA use and the major health-related consequences, with an emphasis on MA-induced psychosis. It is hoped that increased knowledge of MA abuse will provide the basis for future treatment strategies.
Medical subject headings: substance use disorders, animal models, brain imaging (MRI), psychoses, methamphetamine
Abstract
La méthamphétamine (MA), un psychostimulant, est une drogue très toxicomanogène dont la popularité a grimpé en flèche au cours de la dernière décennie en Amérique du Nord. Des laboratoires clandestins de plus en plus nombreux ont entraîné des augmentations spectaculaires de la production de MA et, par conséquent, d'importants problèmes pour la santé publique, la loi et l'environnement. Les données actuelles indiquent que l'exposition à la MA est neurotoxique et des études de neuro-imagerie confirment que l'utilisation chronique chez l'être humain peut causer des dommages nerveux étendus. On établit couramment un lien entre ces changements physiologiques et des formes persistantes de déficience cognitive, y compris des déficits de l'attention, de la mémoire et de l'exécution. Nous présentons dans cette analyse critique une description détaillée des facteurs reliés à l'utilisation de la MA et ses principales répercussions sur la santé, en insistant sur la psychose causée par la MA. On espère qu'une meilleure connaissance de l'abus de MA servira de base à de futures stratégies de traitement.
Introduction
The illicit psychostimulant drugs, which include cocaine and the amphetamines as well as their derivatives, represent a highly addictive class of compounds. In recent years, there has been a dramatic increase in the use of certain drugs of this class. Among these, both methamphetamine (MA) and 3,4-methylenedioxymethamphetamine (MDMA, or “ecstasy”) have experienced a surge in popular use. MA (Fig. 1), can be synthesized by a straightforward 1-step process by reduction of ephedrine or pseudoephedrine,1 ingredients that are widely available in North America in nonprescription allergy medicine and through methods described in detail on the World Wide Web. The relative ease with which the primary ingredients of MA can be acquired and then converted into the final product has led to the widespread existence of numerous “mom-and-pop” laboratories,2 although larger criminal “super lab” organizations in Mexico, Canada and the United States continue to supply a large proportion of the high-purity drug.
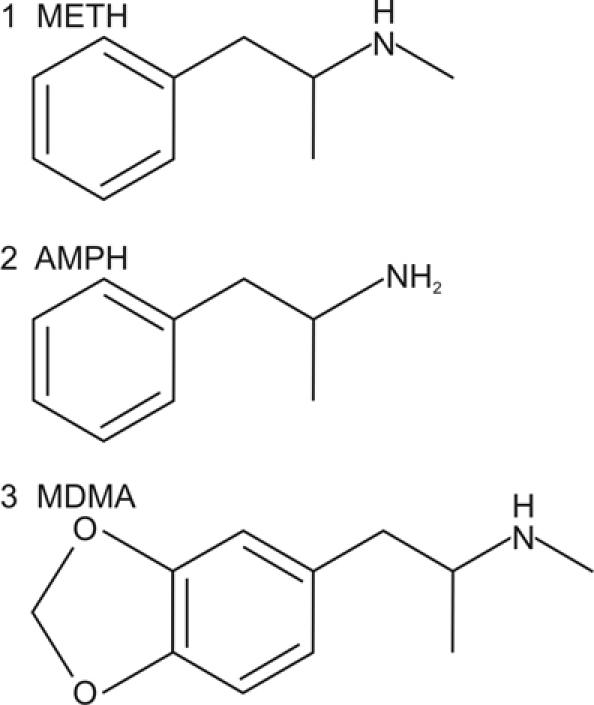
Fig. 1: Chemical structure of methamphetamine (1), as well as the closely related psychostimulants d-amphetamine (2) and 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) (3). AMPH = amphetamine; METH = methamphetamine
These factors have led to a so-called epidemic of MA addiction in certain regions of the US and Canada. The important long-term health-related consequences of this trend are underscored by evidence of the especially pernicious effects of MA exposure. Numerous preclinical and clinical studies demonstrate that MA exposure results in extensive neural damage, which is associated with cognitive impairment. At present, the treatment options for MA-induced psychosis and MA addiction are limited, and further clinical trials are required.
In this review, we aim to provide a broad overview of the current state of knowledge regarding MA and the effects of its use, presenting findings from the basic sciences and from clinical fields. We briefly describe the physiological effects of MA and summarize the major findings from the animal literature. Because most of the human studies on the effects of MA abuse have examined changes in vivo, we chose to examine the evidence for structural and molecular changes obtained from the neuroimaging techniques, magnetic resonance imaging (MRI) and positron emission tomography (PET). These changes are then compared with reported effects of MA abuse on cognition. After summarizing these data, we describe the social impact of MA abuse and the limited options for treating MA addiction.
Neurobiology of MA
MA is a psychostimulant drug that acts on the central nervous system (CNS) through a non-exocytotic mechanism, causing the release of monoamine neurotransmitters, including dopamine, norepinephrine and serotonin.1,3 Unlike cocaine, which works principally by blocking plasma membrane transporters that reuptake monoamines,4 MA exerts multiple pharmacological effects via different molecular processes (Fig. 2). The primary mechanisms by which the amphetamine class of drugs increase levels of monoamines (principally, dopamine) include the redistribution of catecholamines from synaptic vesicles to the cytosol5 and the reverse transport of neurotransmitter through plasma membrane transporters.6 In addition, amphetamines have been shown to block the activity of monoamine transporters,7 similar to cocaine, and to decrease the expression of dopamine transporters at the cell surface.8 There is also evidence that amphetamines can increase cytosolic levels of monoamines by inhibiting the activity of monoamine oxidase (MAO),9 as well as increase the activity and expression of the dopamine-synthesizing enzyme, tyrosine hydroxylase (TH).1,10 As a result of these combined mechanisms, amphetamines act as highly potent releasers of monoamines. Further, MA has a significantly greater elimination half-life than many other psychostimulants, such as cocaine, leading to behavioural and psychological effects that last substantially longer than these other drugs11 (8–13 h for MA v. 1–3 h for cocaine). MA also has a relatively high lipid solubility, allowing more rapid transfer of the drug across the blood–brain barrier.
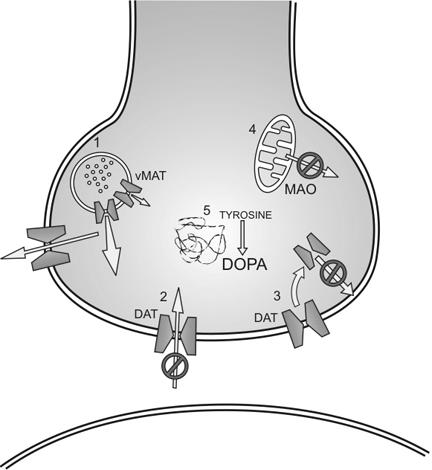
Fig. 2: Physiological mechanisms by which methamphetamine increases synaptic levels of monoamines, principally dopamine (DOPA). Mechanisms include the redistribution of catecholamines from synaptic vesicles to the cytosol (1) and the reverse transport of neurotransmitter through plasma membrane transporters. In addition, amphetamines have been shown to block the activity of monoamine transporters (2), similar to cocaine, and decrease expression of dopamine transporters at the cell surface (3). Amphetamines can increase cytosolic levels of monoamines by inhibiting the activity of monoamine oxidase (MAO) (4) and increase activity and expression of the tyrosine hydroxylase (5). DAT = dopamine transporter; vMAT = vesicular monoamine transporter.
The acute effects of MA on neurotransmitter release are feelings of euphoria, well-being and alertness,12 as well as increased libido and decreased appetite. Immediate somatic side effects of higher doses, which result partly from the effects of MA on epinephrine and norepinephrine release by the adrenal glands,13 may include increased blood pressure, hyperthermia, stroke, cardiac arrhythmia, stomach cramps and muscle tremor; acute negative psychological side effects include anxiety, insomnia, aggression, paranoia and hallucinations.2 We recently reviewed the intermediate-term negative effects of withdrawal from sustained higher doses of psychostimulant drugs. Terminating the administration of high doses of these drugs in humans and animals induces physiological and psychological effects that are opposite to the acute effects of the drug (Fig. 3); these include fatigue, anxiety, irritability, depression, inability to concentrate and even suicidality.15,16
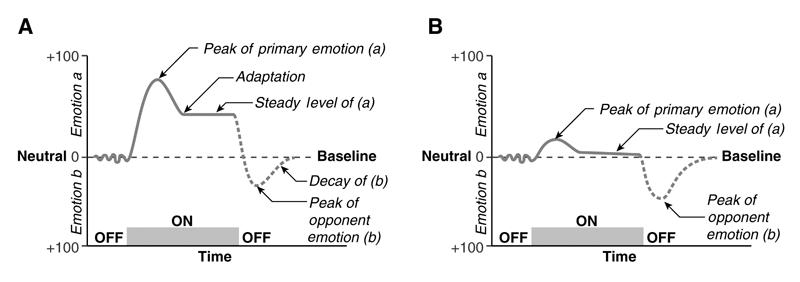
Fig. 3: (A) The central hypothesis of the opponent-process theory of motivation, as envisioned by Solomon and Corbit (14), is that emotions may be considered as pairs of opposites. Thus, when one emotion or affective state is experienced (Emotion a), an opposing emotion or affective state is triggered after a period of time (Emotion b). (B) With repeated stimulations, the opposing emotion or affective state increases in strength, decreasing the experience of the primary emotion or affective state and producing an enduring aftereffect.
Animal studies of MA exposure
Numerous preclinical reports used techniques such as in-vivo cerebral microdialysis to measure synaptic levels of neurotransmitters and demonstrated that exposure to amphetamines results in the rapid release of high levels of monoamines in the CNS,1,3 which are hypothesized to underlie the reinforcing properties of such drugs. Typically, both animals and humans will self-administer amphetamines until the drug supply is curtailed, or the individual will voluntarily cease further drug intake (often due to a lack of additional reinforcing effects of the drug arising from tolerance-related processes). During this early withdrawal period, animal studies have shown that synaptic levels of monoamines are decreased in limbic brain nuclei and that additional administration of non-contingent doses of amphetamines results in reduced levels of neurotransmitter release, compared with drug-naïve animals.16,17 This period is also associated with the onset of depressive-like symptoms, including anhedonia and decreased motivation.18–21 These temporary reductions in monoamines normally return to baseline levels over several days, as the psychological symptoms abate.16 However, the neurotoxic effects of amphetamines, including MA, are measured in terms of months or years rather than days.
Earlier studies identified a selective long-term loss of dopamine terminal markers in the brains of rodents treated with high doses of MA. These included decreased levels of dopamine, reduced TH activity and altered density of the dopamine transporter (reviewed in Sulzer et al1). More recent studies have demonstrated that there is a distinct anatomic degeneration of axon terminals in the rodent striatum following treatment with higher doses of MA.22 Despite the extensive loss of dopamine terminals, there is minimal evidence for actual loss of dopamine cell bodies, measured in the substantia nigra, pars compacta and ventral tegmental areas.23 It has been repeatedly observed that the nigrostriatal dopamine pathway is more vulnerable to the neurotoxic effects of MA than are the mesocorticolimbic dopaminergic projections from the ventral tegmental area to forebrain regions, such as the nucleus accumbens.24 It has been hypothesized that this phenomenon may be due to the greater concentration of dopamine transporters in the terminal regions of the nigrostriatal dopamine pathway (i.e., the striatum).25 The dopamine transporter plays a major role in MA-induced dopamine release, and genetically engineered mice without the dopamine transporter are significantly less vulnerable to the neurotoxic effects of MA26; dopamine uptake blockers, such as bupropion, decrease the magnitude of MA-induced neurotoxicity.27
The precise mechanisms by which MA exerts its neurotoxic effects on the dopamine system remain to be fully resolved. Nevertheless, there is strong evidence that endogenous dopamine is an important substrate. The depletion of dopamine from terminal regions by pretreatment with dopamine depleting agents, such as reserpine or α-methyl p-tyrosine (AMPT), decreases the neurotoxic effects of MA administration in rodents.28 A proposed mechanism includes the propensity for dopamine, in the presence of high doses of MA, to be rapidly and easily oxidized into reactive oxygen species (ROS), including quinones and semiquinones.29 Abundant evidence indicates that oxidative stress resulting from free radicals and ROS is necessary for the neurotoxic effects of MA on dopamine terminals, which may be mediated through the production of downstream neurotoxic compounds such as peroxynitrite (ONOO-). However, Itzhak and Achat-Mendes30 noted that the neuroprotective effects of dopamine depletion are body core temperature–sensitive; the palliative effects of pretreatment with reserpine and AMPT are mitigated when core temperature is raised.31 Alternative mechanisms of MA-induced neurotoxicity have been proposed to include activation of apoptotic biochemical cascades involving caspases, pro-apoptotic Bcl-2 family genes and the tumour suppressor gene p53. In addition, an increasing number of studies indicate the importance of both pro-and anti-inflammatory immune mediators, such as the cytokines, on the neurotoxic effects of MA on dopamine terminals.30
More recently, studies have focused on the neurotoxic effects of MA on serotonergic neurons. Similar to its effects on dopamine terminals, the administration of high doses of MA results in significant long-term reductions in markers of serotonergic terminals, which are most commonly measured by changes in the levels of serotonin and in the serotonin transporter.30,32 However, unlike the relatively discrete effects of MA neurotoxicity on dopamine neurons, which are largely restricted to the striatum, the effects of MA on serotonin neurons are much more diffuse. The list of regions exhibiting MA-induced serotonergic damage includes, but is not limited to, the perirhinal cortex, hippocampus, anterior cingulate cortex, caudate nucleus, nucleus accumbens and septum.33–35 The mechanism of MA-induced serotonergic toxicity is less well understood than it is for dopamine but is believed to be mediated, in large part, by the production of free radicals.32 Even less is known about the effects of high doses of MA on the norepinephrine pathways of the CNS, despite the greater capacity of MA to stimulate norepinephrine than either dopamine or serotonin release.36 Brunswick and colleagues reported decreases in levels of norepinephrine transporter binding sites in specific amygdaloid nuclei and in the dorsomedial hypothalamic nucleus after acute treatment with MA.33 Because the norepinephrine pathways of the brain play an important role in the regulation of arousal, motivation, attention and executive function,37,38 further study of this system is warranted.
It has also become clear from the animal literature that the neurotoxic effects of MA are strongly dependent on the type of dosing schedule. Most studies in rodents use acute exposures to multiple high doses of MA over 1 or 2 days (e.g., Itzhak et al,30 Belcher et al35) which typically result in a loss of about 30%–60% of dopamine terminal markers and are often accompanied by cognitive impairment.35,39 Human patterns of MA use are normally based on years of exposure to the drug.40–42 Thus, it may be argued that more homologous effects would be produced by exposing animals to longer regimens of MA or by requiring animals to self-administer the drug. These points are theoretically valid, although empirical evidence indicates that the neurotoxic effects of MA are significantly lessened in these latter types of study, perhaps due to the development of drug tolerance.43,44 A recent study showed that an escalating dose schedule of MA followed by a 1-day binge (0.1–4.0 mg/kg over 14 days plus 4 × 6 mg/kg at 2-h intervals) produced only an 11% decrease in dopamine levels, whereas the acute 1-day binge alone induced a 37% loss of dopamine;44 this is closer to results of postmortem human studies, which report decreases of 50%–61%.45 Because the aim of many preclinical studies is to attain the same postmortem degree of neurotoxicity in animals as in humans, there is strong rationale to continue using established and reliable regimens of MA administration that induce robust neurotoxic effects, although alternate MA regimens should be evaluated in the future. Further, it is unknown whether neurotoxic effects of MA use in humans result from longer-term exposure to low doses of the drug or from brief exposure to much higher doses.
The phenomenon of psychostimulant-induced “sensitization” refers to the enhanced physiological and behavioural response to a low dose of amphetamines after prior exposure to low, intermittent doses of this drug.46–48 Although psychostimulant sensitization may induce a range of molecular and behavioural changes, these are generally not considered neurotoxic. However, the findings from drug administration studies in animal paradigms indicate that even low, intermittent doses of MA may be able to induce subtle alterations in brain morphology and motivation, which may be relevant to earlier stages of MA drug abuse in humans. In addition, amphetamines have also been shown to “cross-sensitize” with stress, resulting in greater physiological responses to stress after prior exposure to amphetamines.48
Neurotoxic effects of MA use in humans
Postmortem human data regarding the effects of MA use on monoamine neurotoxicity are surprisingly sparse. Most postmortem human research has been performed by Kish and colleagues at the Centre for Addiction and Mental Health in Toronto, Ontario. In the first major evaluation of the effects of long-term MA use, Wilson and others noted that the mean striatal (nucleus accumbens, caudate, putamen) dopamine levels were reduced by 50%–61% in the brain of 12 long-term MA users, many of whom had died from drug overdose.49 In a follow-up study, this research group reported that dopamine levels were as severely depleted in MA users as in people with Parkinson's disease (PD) in the caudate but not in the putamen subdivision of the striatum.45 This finding may explain why PD–like symptoms are not more commonly observed in long-term MA users. Further evidence of dysregulation of the dopamine system was obtained in the same postmortem sample set; a 25%–30% decrease in the maximal extent of dopamine-induced stimulation of adenylyl cyclase activity was demonstrated, which was observed in the striatum of the MA users. These data showing reduced levels of dopamine and decreased dopamine receptor function linked to adenylyl cyclase in the striatum suggest that the physiological function of this brain region may be severely impaired in long-term MA users. Future studies, including different postmortem samples, are required to ascertain the effects of long-term MA use in humans on levels of other neurotransmitters and the molecular machinery responsible for neurotransmitter release.47,50–52
To date, most research on the neurotoxic effects of MA use in humans has been performed in vivo with neuroimaging techniques. Recent structural MRI studies in people with MA addiction have reported several morphological changes in the brain. The more prominent of these include a loss of grey matter in the cingulate, limbic and paralimbic cortices of MA abusers, as well as significantly smaller hippocampi and white-matter hypertrophy.41 Longer-term MA users also displayed an enlarged striatum42 and subtle alterations in cerebral vasculature.53 A recent study has identified shape changes of the corpus callosum in abstinent MA users, including increased curvature in the genu and decreased width in posterior midbody and isthmus areas, which connect frontal and parietal cortices.54
Several studies have used proton magnetic resonance spectroscopy (MRS) to examine changes in neuronal metabolites in specific brain regions in people with MA addiction (Table 1). This technique allows the measurement of markers of cellular integrity and function, which include N-acetyl aspartate (NAA), high-energy metabolic products (creatine [CR] and phosphocreatine [PCR]), cell membrane synthesis or degradation products (choline [CHO]) and glia markers (myo-inositol [MI] and CHO).57 Results from this body of research suggest that the effects of MA exposure are region-and metabolite-specific. In the first study of MA users, Ernst and others reported that there was an inverse correlation between levels of NAA in frontal white matter and the logarithm of lifetime MA used.55 Two more recent studies have noted statistically lower NAA/CR ratios in the anterior cingulate cortex,2,57 whereas CHO/CR and CHO/NAA ratios were significantly higher in MA abusers. Decreased levels of NAA, or reduced NAA/CR ratios, are associated with neuronal loss and clinical disease (e.g., Ende et al,59 Ernst et al60), whereas the CHO signal is associated with membrane synthesis and turnover,61 suggesting that increased CHO or CHO ratios may reflect compensatory responses to MA-induced damage.57 These combined findings indicate that exposure to MA may result in damage to frontal regions, with resulting adaptive processes.
Table 1
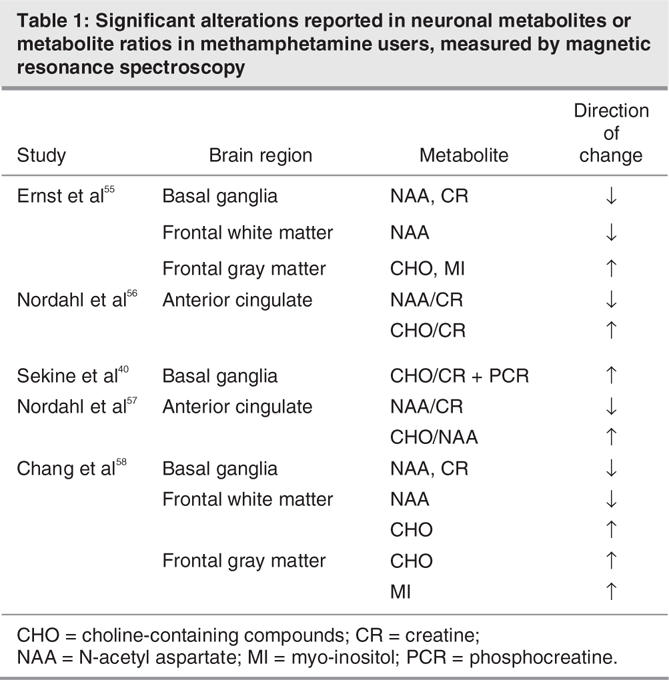
Data from MRS studies of the basal ganglia reveal a similar pattern of results, with significantly greater CHO/CR ratios in long-term MA users (non-drug using at the time of the study), compared with control subjects. This elevation was significantly correlated with duration of MA abuse and the severity of residual psychiatric symptoms.40 Levels of NAA and CR, the latter of which is commonly used as a standard reference in MRS studies, were both decreased in the basal ganglia of MA abusers.55 Similarly, levels of NAA and CR were also decreased in the basal ganglia of people with MA addiction with HIV, compared with non-drug abusing HIV patients.58 Thus, results from the basal ganglia broadly resemble MRS findings from the frontal brain regions and suggest that drug-induced neuronal damage does occur, with possible compensatory adaptive mechanisms involved in neuronal repair.
Positron emission tomography (PET) with radiolabelled ligands has been used to measure the levels of dopamine-relevant proteins in the brains of MA users in vivo. These studies have found consistent decreases in levels of the dopamine transporter in brain regions, including the orbitofrontal cortex, dorsolateral prefrontal cortex, striatum, nucleus accumbens and amygdala,62–65 which have been associated with cognitive impairment and severity of psychiatric symptoms. MA use for longer than 1 year was also associated with significantly lower levels of the dopamine D2 receptor in the striatum66 (however, see Iyo et al67). A recent study reported that the density of the serotonin transporter was significantly lower in global brain regions in longer-term MA abusers than in control subjects and that levels of the transporter were inversely related to duration of MA use. Further, people who abused MA were more aggressive, which was related to levels of the serotonin transporter in orbitofrontal, temporal and anterior cingulate areas.68 Measurement of glucose metabolism in MA users by PET has observed increased global metabolism but showed lower relative levels of striatal and thalamic metabolism.69,70 A global pattern of metabolic activity similar to that observed in major depressive disorder was also noted in MA users during early abstinence, which corresponds to the period of withdrawal that is associated with depressive-like features, such as dysphoria and anhedonia.15,71 It would be interesting to measure alternate PET indices of long-term MA use and withdrawal that are related to mood and cognitive dysregulation, such as serotonin synthesis,72 in this clinical population.
Although structural MRI and MRS have provided invaluable in-vivo information about the morphological and metabolite alterations in people who abuse MA, the results of the above studies are limited in their interpretation by several potential confounds. Perhaps the most important of these is lack of knowledge about the premorbid condition of MA abusers before the onset of addiction. Knowledge of brain morphology and/or metabolites before drug use, as well as functional indicators of these parameters, such as premorbid cognition, is sparse. Thus, it remains unknown whether the differences observed with MRI and MRS and functional deficits, such as cognitive impairment, between MA users and control subjects reflect a deleterious effect of drug exposure or a natural (but abnormal) condition of the brain that predisposes to addiction.40 Longitudinal studies that aim to observe changes at different times after admission to determine whether structural or metabolite alterations will reverse over time in the absence of the MA are complicated by extremely high rates of recidivism in people with addiction and uncertainty over self-reports of abstinence, drug dose or usage pattern.73,74 In addition, most MA addicts are poly-drug abusers, leaving open the question of whether observed changes are due to a specific drug or to its interaction with additional drugs.41 Many MA abusers with psychosis are treated with antipsychotic drugs, which have well-established effects on brain morphology. Members of our research group recently reported that schizophrenia patients treated with typical antipsychotic drugs exhibited increased basal ganglia volumes (measured by structural MRI) that are reversible following replacement with olanzapine (an atypical antipsychotic).75 More recently, we reported that typical antipsychotic drugs increase the volume of the thalamus.76 Thus, the use of animal paradigms will be invaluable in elucidating the influence of these potential confounds: cognition, brain structure and metabolites can be measured before specific treatment with MA and at predetermined times afterward, in the absence of further illicit or therapeutic drugs.
Cognitive effects of MA use
Numerous studies have confirmed that MA abuse is associated with cognitive impairment. Unlike the acute effects of a single low dose of MA, which can improve cognitive processing speed, attention, concentration and psychomotor performance,77,78 long-term exposure to MA may result in profound neuropsychological deficits (see Nordahl et al2). A recent study indicated that MA use was associated with a 40% prevalence of global neuropsychological impairment.79 Nevertheless, despite the broad nature of such deficits, some of the most consistent and severe changes include specific impairments in working memory, attention and executive function.80–86 It has been hypothesized that this specificity is due to the denser dopaminergic innervation of neural circuits that subserve these cognitive processes, including dopamine-rich fronto striatal thalamo cortical pathways,83 which are the primary substrate of neurotoxic doses of MA. People with a history of MA use displayed working memory deficits in such tasks as the immediate recall component of the auditory verbal learning test63 and took 18%–30% longer to complete the working memory components of the California computerized assessment package.87 Consistent with the greater distractibility of MA users, as widely observed in the clinic,82 attentional deficits have been noted in the Stroop Colour Word and Trail Making tests.88,89 Alternate procedures have demonstrated that the primary explicit attentional deficits in MA users may be related to reduced cognitive inhibition82 and an inability to suppress irrelevant task information.2 Impairments in executive function, which include the cognitive domains of abstract reasoning, planning and behavioural flexibility, are evident in MA users in the Stroop Interference Task.89 Episodic memory tasks typically have both a strategic-frontal component and an associative-hippocampal component; Woods and colleagues recently demonstrated that, although there is an episodic deficit in MA users in the Hopkins Verbal Learning Test, it is of a strategic (i.e., executive, planning and organizational) nature and is not purely mnemonic.83 Interestingly, the above deficits parallel, to some degree, the nature of cognitive impairment observed in attention-deficit hyperactivity disorder (ADHD) — a disorder that is frequently treated with amphetamines, including MA.90 Whether such similarity reflects an increased sensitivity to amphetamines in people with ADHD84 or a consequence of high doses of MA remains an ongoing matter for study.91
Extensive use of MA has also been repeatedly associated with deficits in episodic memory. These are most evident as impairment in word recall tasks, which measure recall at specific times after stimulus presentation.41,63,88 Unlike the cognitive domains of attention and executive function, which depend on the functional integrity of fronto-striatal circuits that are densely innervated by dopamine terminals, episodic memory relies predominantly on the hippocampus and related structures,92 where dopaminergic innervation is more sparse. However, animal studies indicate that MA is also neurotoxic to norepinephrine and serotonin terminals in the hippocampus, which play a critical role in regulating cognitive processing.93
One of the most prominent effects of MA abuse on cognitive function pertains to the development of drug-related psychosis. Aside from the sudden psychosis-inducing effects of high doses of MA, prior exposure to MA, following metabolism and excretion of the drug, can also lead to an enduring form of psychosis. Studies conducted in Japan, where high levels of MA use have been prevalent for decades, report that between 36% and 64% of MA users who have experienced psychotic symptoms continue to present with these symptoms for more than 10 days after the cessation of MA use, even though the MA is eliminated from the blood stream in less than 5 days.53 Another study investigating female inmates in Japan observed that 21% of those having experienced MA psychosis remained in a psychotic state for more than 6 months, whereas 49% returned to their premorbid state but experienced “flashbacks” (i.e., spontaneous recurrence of psychotic symptoms that would fit criteria for a paranoid-schizophrenia psychotic relapse) during their 15–20 months of incarceration.53 Studies in Japan show that MA users with MA psychosis are much more likely to experience psychotic symptoms again if they use MA and are also more likely to have a psychotic relapse when confronted with stressful situations, even years after cessation of MA use.53 MA users with persistent or recurrent psychotic symptoms become vulnerable to environmental stress and may benefit from antipsychotic medication in a manner similar to individuals with schizophrenia.53
Genetics and MA abuse
Evidence from twin studies reveals that most forms of substance abuse exhibit a strong degree of heritability, with liabilities of up to 71% for certain classes of drugs.94 This finding strongly suggests that there are inherited functional variants of genes that predispose individuals to acquiring and maintaining drug use. Regarding MA specifically, several studies have reported significant associations between different genes and MA use or the development of MA-induced psychosis. To our knowledge, all significant associations have been obtained in Chinese and Japanese cohorts (Table 2), although other reports have been measured in samples with Czechoslovakians (Table 3). The list of genes related to MA use and drug-induced psychosis includes a number pertinent to the dopaminergic system, which represents an important substrate for both the reinforcing properties of MA and, presumably, most forms of psychosis. The Val158Met polymorphism of catechol-O-methytransferase (COMT), which represents a high-activity allele, occurred with significantly greater frequency in MA users than in control subjects. In the same group of Han Chinese from Taiwan, a haplotype of the 120-bp variable number tandem repeat (VNTR) polymorphism and the exon 3 VNTR in the dopamine D4 receptor was significantly associated with MA use.96 Moreover, there were significant interactions between the COMT and D4 receptor polymorphisms and MA use. The dopamine transporter has been investigated in several studies as a genetic locus with a possible link to MA use. In 2 studies, there was no association between the DAT 3′-VNTR and MA use or psychosis in Han Chinese,106,107 although a study in a Japanese sample noted that fewer repeat alleles of this 40-bp region were significantly linked to increased likelihood of long-term psychosis following drug termination.99 In another study of a Japanese cohort, the A1 allele of the TaqI A polymorphism of the dopamine D2 receptor was significantly associated with both the symptoms and progression of MA psychosis or abuse (see Harano et al102). In apparent contrast, there was no significant association between this polymorphism and MA use in a Chinese male cohort, although this group was selected to exclude subjects with psychosis.108 The TaqI A polymorphism of the dopamine receptor D2 was also not associated with MA dependence in a Czechoslovakian sample, although data about psychosis in this group were not provided.109
Table 2

Table 3
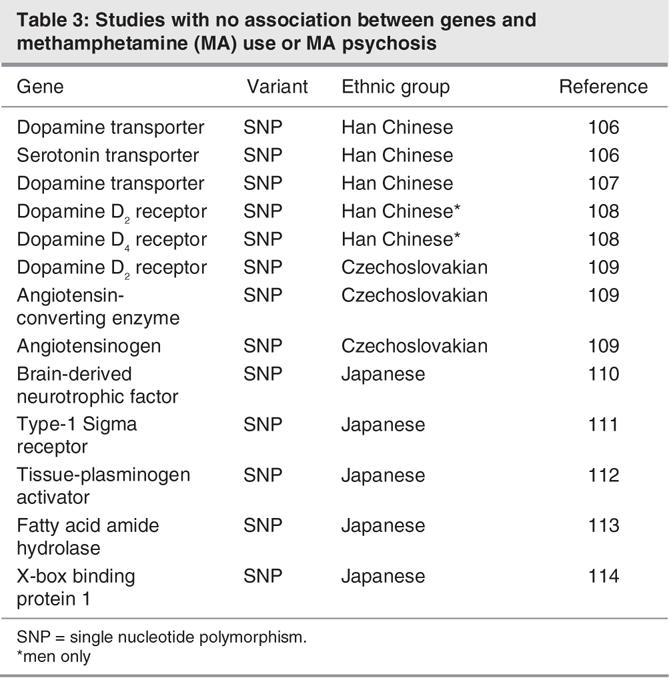
Additional genes associated with MA use include the gamma aminobutyric acid (GABA)A receptor gamma 2 subunit. A Japanese cohort of MA users, most of whom exhibited drug-induced psychosis, showed a significant association with a haplotype of the 2 single nucleotide polymorphisms (SNP), 315C > T and 1128 + 99C > A for the GABAA receptor gamma 2 subunit.100 In a study of Han Chinese, multiple haplotypes, all of which contained rs4480617, a novel SNP located at nucleotide-69 in the 5′-UTR of the (A) receptor gamma 2 subunit gene, were significantly associated with MA use but not MA-induced psychosis.101 Interestingly, significant genetic links in this study were only evident for female MA users. Other examples of significant sex-dependent, genetic associations with MA use include the genes glutathione S-transferase M198 and α-synuclein,105 both of which are present only in women. The reasons for these sex-dependent effects are unknown but they indicate that the nature of genetic factors on MA use and psychosis is likely to be more complicated than initially thought.
The MA problem
According to the World Health Organization, at least 35 million people regularly used MA worldwide in 1996, making it the second most commonly used illicit drug after cannabis.115 More recent surveys indicate that MA is the fastest-growing illicit drug in North America.116 MA abuse among youth is especially common, leading some to describe the MA problem as an “epidemic,” particularly in the Western regions of the US and Canada.117–119 The MA phenomenon started on the West Coast118,120,121 and has spread to other Canadian cities.119,122 In Vancouver, BC, 57% of “street kids” (adolescents under 19 years of age who are living on the street or who are involved with street life to a significant extent123) reported having used MA more than 10 times in their lifetime.124 Similar statistics have been reported in Victoria, BC, with 47% of street kids and 10% of all high school students surveyed reported having used the drug. Particular ethnographic groups may be at increased risked for the effects of MA. For instance, Aboriginal groups represent approximately 27% of transient population of Vancouver's Downtown Eastside,125 an area with among the highest rates of polysubstance abuse in North America. This contrasts with a demographic population frequency of 2% for Aboriginals in Vancouver as a whole.126 MA abuse within the male homosexual and bisexual community has become a major concern, particularly because of the increased rate of high-risk sexual behaviours associated with MA abuse;127,128 furthermore, long-term MA use can exacerbate the cognitive impairments associated with HIV.79 Increasing evidence indicates that prenatal exposure to MA results in severe morphological changes in the brain, with associated cognitive deficits.129 Recreational users (i.e., “ravers”) often take MA either voluntarily or inadvertently in combination with other drugs (i.e., MDMA).130
Forty-five percent of youth in Vancouver mentioned being able to obtain MA in less than 24 hours,131 and the drug can be absorbed in multiple ways: smoked (vapourized), snorted, intravenously injected or swallowed. Typically, users can maintain their dependence for less than Can $5 a day and experience intoxication effects for 6–16 hours.131 A recent qualitative survey conducted among street youth in Vancouver described how people intoxicated with MA believe the drug “makes them feel normal” and helps them cope with traumatic experiences and depression, but it also makes them feel more reactive and threatened by authority figures. Most of them perceived an important deterioration in their quality of life due to MA abuse.132 Other studies support an increase in violence that is linked to MA abuse.133 The survey reported that more than 80% of the street youth who regularly abused MA also suffered from MA psychosis and that psychotic symptoms increased with duration of MA use. Of great concern is the finding that repeated psychotic episodes may lead to increased treatment resistance, with symptoms becoming less responsive to medication following successive relapses and, in some cases, residual symptoms appearing that were not present before relapse.134
The recent rise in popularity of MA use is also reflected in US government crime statistics. The Drug Enforcement Agency reports that authorities siezed 1370 kg of MA along the Mexico–US border in 2001, compared with only 6.5 kg in 1992.135 In addition, the number of treatment admissions to publicly funded treatment facilities for MA has increased from approximately 20 000 in 1993 to over 110 000 in 2003.136 It was recently estimated that 600 000 people in the US use the drug on a weekly basis.137 Within Canada, the number of MA laboratories dismantled by Canadian law enforcement has increased, with 24 laboratories seized in 2000 and 39 in 2003. In 2000, the first “super laboratory” was uncovered in Vancouver, BC.138 In addition to the mental health problems involved with the longer-term consumption of MA, use of the drug is associated with numerous additional physical problems, including dental caries, infection, heart failure and malnutrition.139 A recent survey by the US National Association of Counties reported that “methamphetamine was responsible for more emergency department visits than any other drug and that the need for treatment programmes is growing dramatically.”140 Even the manufacture of the drug itself has dire consequences. In the US, the Hazardous Substances Emergency Events Surveillance (HSEES) system, which monitors data about the public health consequences (e.g., morbidity, mortality and evacuations) of acute hazardous substances in 16 American states, reported that 1791 of the 40 349 events reported to the HSEES system during January 2000 to June 2004 were associated with illicit MA production.116 This proportion was considerably higher in certain Western states, such as Washington and Oregon (399 and 246 events, respectively; figures were unavailable for California). These numbers are clinically significant; for example, a review of patients admitted to a burns unit in rural Iowa revealed that a substantial proportion were related to MA use or production, with a mean treatment cost of US$77 580 per patient.141
Treatment of MA-related disorders
The earliest trials of addiction to amphetamines focused on agonist-like replacement pharmacotherapies, similar to the opiate–methadone model. Dextroamphetamine has been the drug of choice in these studies, and although the literature is comprised mainly of uncontrolled, retrospective studies, there have been mixed results (summarized in Grabowski et al142), with one indication that dextroamphetamine substitution produced beneficial effects in a subgroup of patients.143 The only randomized controlled trial of dextroamphetamine substitution, which used 60 mg daily over 12 weeks in 41 participants, demonstrated a trend toward efficacy that did not reach significance.142,144 Over the past decade, several trials have hoped to capitalize on the successes in treating cocaine addiction by replicating these studies and substituting MA dependence. To this end, compounds including selective serotonin reuptake inhibitors (SSRI), tricyclic antidepressant drugs, MAO inhibitors, the GABAergic drugs gabapentin and baclofen, and the antinauseant drug ondansetron have been studied with inconclusive or negative results.145
The efficacy of the second-generation atypical antipsychotics have been described only anecdotally to date; however, a National Institute on Drug Abuse (NIDA)-sponsored phase 1 study is currently underway with aripiprazole,146 and risperidone and quetiapine are being tested in a phase IV head-to-head trial for comorbid MA use in schizophrenia.147 As mentioned above, MA causes a general loss of dopaminergic terminals and transmission in the CNS. Thus agents that increase dopamine levels may be effective in treating MA addiction. By this reasoning, the antidepressant selegiline, an MAO-B inhibitor that increases dopaminergic neurotransmission, has progressed to a Phase II, controlled, double-blind, NIDA-sponsored study.148 Similarly, a recent laboratory-controlled study of bupropion (a dopamine reuptake inhibitor) noted significantly reduced acute MA-induced subjective effects of doses up to 30 mg i.v. and decreased cue-induced craving in a small sample of MA abusers. However, as the authors of this study observed, the impact of these findings with respect to future outpatient trials may be limited by the relatively low dose of MA used and the capacity for higher doses of MA to overcome the modest effects found in this trial.149
Lobeline, a tobacco plant derivative that inhibits uptake of dopamine into synaptic vesicles, is currently entering Phase I clinical trials.148 A safety and efficacy trial involving 30 subjects was recently completed with vigabatrin, an antiepileptic drug. This drug is an irreversible inhibitor of GABA-transaminase and has shown preliminary effectiveness in cocaine dependence. Of the 18 patients who completed the study, 16 tested negative for MA for the duration of the 6-week trial.150 Additional compounds soon to enter or be involved in Phase I trials include the acetylcholinesterase inhibitor rivastigmine, the antiepileptic topirimate, the wake promoting GABA/glutaminergic agent modafinil and the experimental dopamine uptake inhibitor GBR 12909.148 Several compounds are in later stages of preclinical development, representing diverse approaches to treating MA abuse. Among the more promising of these platforms is immunopharmacotherapy, which is based on the generation or administration of antibodies that are capable of binding to MA before it can reach the brain.151,152
Finally, the effectiveness of psychotherapy and behavioural treatments has reliably demonstrated a clinical benefit with minimal potential for side effects. Much attention has recently been devoted to creating a standardized protocol. The “Matrix” model, a manualized 16-week intensive multi-component cognitive–behavioural/addictions model recently underwent a multisite trial involving 978 patients. Matrix participants initially showed significantly better attendance and longer periods of drug abstinence; however, at follow up, these differences became nonsignificant.153 Further research is underway to determine which of the many facets of this psychosocial intervention program were most effective.
Regarding treatment for MA-induced psychosis, there is a paucity of data and, currently, no controlled trials for the treatment of post-drug psychosis. The standard of care parallels the management of acute psychosis from other etiologies, such as schizophrenia. By extension, the current trend is for initial treatment with antipsychotics, with a bias toward the atypical antipsychotics as first-line treatment in Canada. Evidence for atypical antipsychotic use is limited to isolated case reports, suggesting some measure of efficacy.154,155 The length of appropriate pharmacological intervention is largely unstudied, and no consistent guidelines exist in the literature. A small case series indicated that antipsychotic treatment beyond the acute psychotic episode may protect against future psychotic episodes, even at very low doses.156 These data, however, remain to be replicated.
Current research indicates that people presenting with co-occurring disorders, such as MA-induced psychosis, warrant specific treatments that deal with both the psychosis and the addiction issue.157,158 Indeed, a comprehensive review of treatment programs for people with drug abuse and mental health problems revealed that the best outcomes stem from programs that are considered evidence-based and that integrate mental health and substance abuse treatment.157,159 Most MA treatments investigated so far have used only an “addiction” treatment model for stimulant dependence and have excluded people with comorbid mental health problems, such as persistent or recurrent psychotic symptoms.160–162 Further research is required to develop a more thorough understanding of the profiles of people who suffer from persistent or recurrent MA psychosis, in order to address their specific needs.
Conclusion
MA is a highly addictive psychostimulant drug, whose abuse has reached epidemic proportions in many parts of the US and Canada. Current patterns of use include higher rates of MA abuse in Western regions of North America, although availability and use of the drug appears to be spreading eastward. Longer-term use of MA can result in substantial cognitive deficits, especially to memory, attention and executive function, possibly from neurotoxicity. Studies of the neurochemistry and structural morphology of the brain in MA users reveal numerous alterations, some of which show a direct relation to functional changes in behaviour and cognition. At present, the best understood factors influencing MA use are environmental, although various candidate genes may predispose to MA addiction and drug-induced psychosis. Priorities for further research include better knowledge of the progressive neurobiological effects of MA use, as well as treatment and health care strategies for MA users.
We aimed to study MA psychosis from a clinical, biomedical and health services perspective. Specifically, we evaluated the treatment needs of people with MA psychosis in terms of addiction and clinical and psychosocial issues, as well as neurocognitive deficits, and to document their service use and pathways to care. Such a descriptive and exploratory study will enable us to gather the information that will undoubtedly lead to the development and validation of effective treatments for this population.
Acknowledgments
Drs. Barr and Lecomte are Michael Smith Foundation for Health Research (MSFHR) Scholars; Dr. Lecomte is a Canadian Institutes of Health Research (CIHR) Young Investigator. The Centre for Complex Disorders is partly funded by an Infrastructure Grant to Dr. Honer from MSFHR. This review was partly supported by CIHR Operating Grants. We received funding from the CIHR operating grants to AMB and TL and from BC Mental Health and Addictions Services.
Footnotes
Contributors: All authors contributed to the conception and design of the review. Drs. Barr, Panenka, Lang and Lecomte wrote the article; Drs. MacEwan, Thornton and Honer reviewed it. All authors gave final consent for the article to be published.
Competing interests: None declared for Drs. Barr, Panenka, MacEwan, Thornton, Lang and Lecomte. Dr. Honer has acted as a paid consultant for AstraZeneca and has received speaker fees from AstraZeneca, In Silico Biosciences, Janssen, Wyeth and Solvay. He has received travel assistance from Eli Lilly and AstraZeneca.
Correspondence to: Dr. Alasdair M. Barr, Centre for Complex Disorders, Department of Psychiatry, University of British Columbia, Vancouver General Hospital Research Pavilion, 828 W 10th Ave., Vancouver BC V5Z 1L8; fax 604 875-4376; albarr@interchange.ubc.ca
References
- 1.Sulzer D, Sonders MS, Poulsen NW, et al. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol 2005;75:406-33. [DOI] [PubMed]
- 2.Nordahl TE, Salo R, Leamon M. Neuropsychological effects of chronic methamphetamine use on neurotransmitters and cognition: a review. J Neuropsychiatry Clin Neurosci 2003;15:317-25. [DOI] [PubMed]
- 3.Kuczenski R, Segal DS, Cho AK, et al. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci 1995;15:1308-17. [DOI] [PMC free article] [PubMed]
- 4.Izenwasser S. The role of the dopamine transporter in cocaine abuse. Neurotox Res 2004;6:379-83. [DOI] [PubMed]
- 5.Brown JM, Hanson GR, Fleckenstein AE. Regulation of the vesicular monoamine transporter-2: a novel mechanism for cocaine and other psychostimulants. J Pharmacol Exp Ther 2001;296:762-7. [PubMed]
- 6.Khoshbouei H, Wang H, Lechleiter JD, et al. Amphetamine-induced dopamine efflux. A voltage-sensitive and intracellular Na+-dependent mechanism. J Biol Chem 2003;278:12070-7. [DOI] [PubMed]
- 7.Schmitz Y, Lee CJ, Schmauss C, et al. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci 2001;21:5916-24. [DOI] [PMC free article] [PubMed]
- 8.Saunders C, Ferrer JV, Shi L, et al. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci U S A 2000;97:6850-5. [DOI] [PMC free article] [PubMed]
- 9.Mantle TJ, Tipton KF, Garrett NJ. Inhibition of by amphetamine and related compounds. Biochem Pharmacol 1976;25:2073-7. [DOI] [PubMed]
- 10.Mandell AJ, Morgan M. Amphetamine induced increase in tyrosine hydroxylase activity. Nature 1970;227:75-6. [DOI] [PubMed]
- 11.Busto U, Bendayan R, Sellers EM. Clinical pharmacokinetics of non-opiate abused drugs. Clin Pharmacokinet 1989;16:1-26. [DOI] [PubMed]
- 12.Hart CL, Ward AS, Haney M, et al. Methamphetamine self-administration by humans. Psychopharmacology (Berl) 2001;157:75-81. [DOI] [PubMed]
- 13.Makisumi T, Yoshida K, Watanabe T, et al. Sympatho-adrenal involvement in methamphetamine-induced hyperthermia through skeletal muscle hypermetabolism. Eur J Pharmacol 1998;363:107-12. [DOI] [PubMed]
- 14.Solomon RL, Corbit JD. An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol Rev 1974;81:119-45. [DOI] [PubMed]
- 15.Barr AM, Markou A. Psychostimulant withdrawal as an inducing condition in animal models of depression. Neurosci Biobehav Rev 2005;29:675-706. [DOI] [PubMed]
- 16.Barr AM, Markou A, Phillips AG. A ‚crash' course on psychostimulant withdrawal as a model of depression. Trends Pharmacol Sci 2002;23:475-82. [DOI] [PubMed]
- 17.Di Ciano P, Blaha CD, Phillips AG. Inhibition of dopamine efflux in the rat nucleus accumbens during abstinence after free access to d-amphetamine. Behav Brain Res 2002;128:1-12. [DOI] [PubMed]
- 18.Barr AM, Zis AP, Phillips AG. Repeated electroconvulsive shock attenuates the depressive-like effects of d-amphetamine withdrawal on brain reward function in rats. Psychopharmacology (Berl) 2002;159:196-202. [DOI] [PubMed]
- 19.Barr AM, Phillips AG. Increased successive negative contrast in rats withdrawn from an escalating-dose schedule of D-amphetamine. Pharmacol Biochem Behav 2002;71:293-9. [DOI] [PubMed]
- 20.Barr AM, Fiorino DF, Phillips AG. Effects of withdrawal from an escalating dose schedule of d-amphetamine on sexual behavior in the male rat. Pharmacol Biochem Behav 1999;64:597-604. [DOI] [PubMed]
- 21.Barr AM, Phillips AG. Withdrawal following repeated exposure to d-amphetamine decreases responding for a sucrose solution as measured by a progressive ratio schedule of reinforcement. Psychopharmacology (Berl) 1999;141:99-106. [DOI] [PubMed]
- 22.Miller DB, O'Callaghan JP. Elevated environmental temperature and methamphetamine neurotoxicity. Environ Res 2003;92:48-53. [DOI] [PubMed]
- 23.Ricaurte GA, Seiden LS, Schuster CR. Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res 1984;303:359-64. [DOI] [PubMed]
- 24.Cass WA. Decreases in evoked overflow of dopamine in rat striatum after neurotoxic doses of methamphetamine. J Pharmacol Exp Ther 1997;280:105-13. [PubMed]
- 25.Eisch AJ, Gaffney M, Weihmuller FB, et al. Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res 1992;598:321-6. [DOI] [PubMed]
- 26.Fumagalli F, Gainetdinov RR, Valenzano KJ, et al. Role of dopamine transporter in methamphetamine-induced neurotoxicity: evidence from mice lacking the transporter. J Neurosci 1998;18:4861-9. [DOI] [PMC free article] [PubMed]
- 27.Marek GJ, Vosmer G, Seiden LS. Dopamine uptake inhibitors block long-term neurotoxic effects of methamphetamine upon dopaminergic neurons. Brain Res 1990;513:274-9. [DOI] [PubMed]
- 28.Lew R, Malberg JE, Ricaurte GA, et al. Evidence for and mechanism of action of neurotoxicity of amphetamine related compounds. In: Kostrzewa RM, editor. Highly selective neurotoxins: basic and clinical applications. Totowa: Humana Press Inc; 1997.
- 29.Sulzer D, Zecca L. Intraneuronal dopamine-quinone synthesis: a review. Neurotox Res 2000;1:181-95. [DOI] [PubMed]
- 30.Itzhak Y, Achat-Mendes C. Methamphetamine and MDMA (ecstasy) neurotoxicity: ‚of mice and men.' IUBMB Life 2004;56:249-55. [DOI] [PubMed]
- 31.Yuan J, Callahan BT, McCann UD, et al. Evidence against an essential role of endogenous brain dopamine in methamphetamine-induced dopaminergic neurotoxicity. J Neurochem 2001;77:1338-47. [DOI] [PubMed]
- 32.Hanson GR, Rau KS, Fleckenstein AE. The methamphetamine experience: a NIDA partnership. Neuropharmacology 2004;47(Suppl 1): 92-100. [DOI] [PubMed]
- 33.Brunswick DJ, Benmansour S, Tejani-Butt SM, et al. Effects of high-dose methamphetamine on monoamine uptake sites in rat brain measured by quantitative autoradiography. Synapse 1992;11:287-93. [DOI] [PubMed]
- 34.Armstrong BD, Noguchi KM. The neurotoxic effects of 3,4-methylenedioxymethamphetamine (MDMA) and methamphetamine on serotonin, dopamine, and GABA-ergic terminals: an in-vitro autoradiographic study in rats. Neurotoxicology 2004;25:905-14. [DOI] [PubMed]
- 35.Belcher AM, O'Dell SJ, Marshall JF. Impaired object recognition memory following methamphetamine, but not p-chloroamphetamine-or d-amphetamine-induced neurotoxicity. Neuropsychopharmacology. 2005;30:2026-34. [DOI] [PubMed]
- 36.Rothman RB, Baumann MH, Dersch CM, et al. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001;39: 32-41. [DOI] [PubMed]
- 37.Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 2003;42:33-84. [DOI] [PubMed]
- 38.Dalley JW, Cardinal RN, Robbins TW. Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev 2004;28:771-84. [DOI] [PubMed]
- 39.Schroder N, O'Dell SJ, Marshall JF. Neurotoxic methamphetamine regimen severely impairs recognition memory in rats. Synapse 2003;49:89-96. [DOI] [PubMed]
- 40.Sekine Y, Minabe Y, Kawai M, et al. Metabolite alterations in basal ganglia associated with methamphetamine-related psychiatric symptoms. A proton MRS study. Neuropsychopharmacology 2002;27: 453-61. [DOI] [PubMed]
- 41.Thompson PM, Hayashi KM, Simon SL, et al. Structural abnormalities in the brains of human subjects who use methamphetamine. J Neurosci 2004;24:6028-36. [DOI] [PMC free article] [PubMed]
- 42.Chang L, Cloak C, Patterson K, et al. Enlarged striatum in abstinent methamphetamine abusers: a possible compensatory response. Biol Psychiatry 2005;57:967-74. [DOI] [PMC free article] [PubMed]
- 43.Segal DS, Kuczenski R, O'Neil ML, et al. Prolonged exposure of rats to intravenous methamphetamine: behavioral and neurochemical characterization. Psychopharmacology (Berl) 2005;180:501-12. [DOI] [PubMed]
- 44.Segal DS, Kuczenski R, O'Neil ML, et al. Escalating dose methamphetamine pretreatment alters the behavioral and neurochemical profiles associated with exposure to a high-dose methamphetamine binge. Neuropsychopharmacology 2003;28:1730-40. [DOI] [PubMed]
- 45.Moszczynska A, Fitzmaurice P, Ang L, et al. Why is parkinsonism not a feature of human methamphetamine users? Brain 2004; 127: 363-70. [DOI] [PubMed]
- 46.Tenn CC, Kapur S, Fletcher PJ. Sensitization to amphetamine, but not phencyclidine, disrupts prepulse inhibition and latent inhibition. Psychopharmacology (Berl) 2005;180:366-76. [DOI] [PubMed]
- 47.Sawada K, Barr AM, Nakamura M, et al. Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry 2005;62:263-72. [DOI] [PubMed]
- 48.Barr AM, Hofmann CE, Weinberg J, et al. Exposure to repeated, intermittent d-amphetamine induces sensitization of HPA axis to a subsequent stressor. Neuropsychopharmacology 2002;26:286-94. [DOI] [PubMed]
- 49.Wilson JM, Kalasinsky KS, Levey AI, et al. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med 1996;2:699-703. [DOI] [PubMed]
- 50.Barr AM, Young CE, Sawada K, et al. Abnormalities of presynaptic protein CDCrel-1 in striatum of rats reared in social isolation: relevance to neural connectivity in schizophrenia. Eur J Neurosci 2004;20:303-7. [DOI] [PubMed]
- 51.Barr AM, Song C, Sawada K, et al. Tolerance to the anhedonic effects of lipopolysaccharide is associated with changes in syntaxin immunoreactivity in the nucleus accumbens. Int J Neuropsychopharmacol 2003;6:23-34. [DOI] [PubMed]
- 52.Sawada K, Young CE, Barr AM, et al. Altered immunoreactivity of complexin protein in prefrontal cortex in severe mental illness. Mol Psychiatry 2002;7:484-92. [DOI] [PubMed]
- 53.Iyo M, Namba H, Yanagisawa M, et al. Abnormal cerebral perfusion in chronic methamphetamine abusers: a study using 99MTc-HMPAO and SPECT. Prog Neuropsychopharmacol Biol Psychiatry 1997;21:789-96. [DOI] [PubMed]
- 54.Oh JS, Lyoo IK, Sung YH, et al. Shape changes of the corpus callosum in abstinent methamphetamine users. Neurosci Lett 2005;384:76-81. [DOI] [PubMed]
- 55.Ernst T, Chang L, Leonido-Yee M, et al. Evidence for long-term neurotoxicity associated with methamphetamine abuse: a 1H MRS study. Neurology 2000;54:1344-9. [DOI] [PubMed]
- 56.Nordahl TE, Salo R, Possin K, et al. Low N-acetyl-aspartate and high choline in the anterior cingulum of recently abstinent methamphetamine-dependent subjects: a preliminary proton MRS study. Psych Res 2002;116:43-52. [DOI] [PubMed]
- 57. Nordahl TE, Salo R, Natsuaki Y, et al. Methamphetamine users in sustained abstinence: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry 2005;62:444-52. [DOI] [PubMed]
- 58.Chang L, Ernst T, Speck O, et al. Additive effects of HIV and chronic methamphetamine use on brain metabolite abnormalities. Am J Psychiatry 2005;162:361-9. [DOI] [PMC free article] [PubMed]
- 59. Ende GR, Laxer KD, Knowlton RC, et al. Temporal lobe epilepsy: bilateral hippocampal metabolite changes revealed at proton MR spectroscopic imaging. Radiology 1997;202:809-17. [DOI] [PMC free article] [PubMed]
- 60.Ernst T, Chang L, Melchor R, et al. Frontotemporal dementia and early Alzheimer disease: differentiation with frontal lobe H-1 MR spectroscopy. Radiology 1997;203:829-36. [DOI] [PubMed]
- 61.Tedeschi G, Bertolino A, Lundbom N, et al. Cortical and subcortical chemical pathology in Alzheimer's disease as assessed by multislice proton magnetic resonance spectroscopic imaging. Neurology 1996;47:696-704. [DOI] [PubMed]
- 62.Volkow ND, Chang L, Wang GJ, et al. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci 2001;21:9414-8. [DOI] [PMC free article] [PubMed]
- 63.Volkow ND, Chang L, Wang GJ, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry 2001;158:377-82. [DOI] [PubMed]
- 64.McCann UD, Wong DF, Yokoi F, et al. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci 1998;18:8417-22. [DOI] [PMC free article] [PubMed]
- 65.Sekine Y, Iyo M, Ouchi Y, et al. Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am J Psychiatry 2001;158:1206-14. [DOI] [PubMed]
- 66.Volkow ND, Chang L, Wang GJ, et al. Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry 2001;158:2015-21. [DOI] [PubMed]
- 67.Iyo M, Sekine Y, Mori N. Neuromechanism of developing methamphetamine psychosis: a neuroimaging study. Ann N Y Acad Sci 2004;1025:288-95. [DOI] [PubMed]
- 68.Sekine Y, Ouchi Y, Takei N, et al. Brain serotonin transporter density and aggression in abstinent methamphetamine abusers. Arch Gen Psychiatry 2006;63:90-100. [DOI] [PubMed]
- 69.Volkow ND, Chang L, Wang GJ, et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am J Psychiatry 2001;158:383-9. [DOI] [PubMed]
- 70.Wang GJ, Volkow ND, Chang L, et al. Partial recovery of brain metabolism in methamphetamine abusers after protracted abstinence. Am J Psychiatry 2004;161:242-8. [DOI] [PubMed]
- 71.London ED, Simon SL, Berman SM, et al. Mood disturbances and regional cerebral metabolic abnormalities in recently abstinent methamphetamine abusers. Arch Gen Psychiatry 2004;61:73-84. [DOI] [PubMed]
- 72.Rosa-Neto P, Diksic M, Okazawa H, et al. Measurement of brain regional alpha-[11C]methyl-L-tryptophan trapping as a measure of serotonin synthesis in medication-free patients with major depression. Arch Gen Psychiatry 2004;61:556-63. [DOI] [PubMed]
- 73.Huber A, Ling W, Shoptaw S, et al. Integrating treatments for methamphetamine abuse: a psychosocial perspective. J Addict Dis 1997;16:41-50. [DOI] [PubMed]
- 74.Simon SL, Richardson K, Dacey J, et al. A comparison of patterns of methamphetamine and cocaine use. J Addict Dis 2002;21:35-44. [DOI] [PubMed]
- 75.Lang DJ, Kopala LC, Vandorpe RA, et al. Reduced basal ganglia volumes after switching to olanzapine in chronically treated patients with schizophrenia. Am J Psychiatry 2004;161:1829-36. [DOI] [PubMed]
- 76.Khorram B, Lang DJ, Kopala LC, et al. A longitudinal study on the affects of typical versus atypical antipsychotic drugs on thalamic volume in schizophrenia [abstract]. Schizophr Bull 2005;31:395. [DOI] [PubMed]
- 77.Johnson BA, Roache JD, Ait-Daoud N, et al. Effects of isradipine on methamphetamine-induced changes in attentional and perceptual-motor skills of cognition. Psychopharmacology (Berl) 2005;178: 296-302. [DOI] [PubMed]
- 78.Mohs RC, Tinklenberg JR, Roth WT, et al. Methamphetamine and diphenhydramine effects on the rate of cognitive processing. Psychopharmacology (Berl) 1978;59:13-9. [DOI] [PubMed]
- 79.Rippeth JD, Heaton RK, Carey CL, et al. Methamphetamine dependence increases risk of neuropsychological impairment in HIV infected persons. J Int Neuropsychol Soc 2004;10:1-14. [DOI] [PubMed]
- 80.Gonzalez R, Rippeth JD, Carey CL, et al. Neurocognitive performance of methamphetamine users discordant for history of marijuana exposure. Drug Alcohol Depend 2004;76:181-90. [DOI] [PubMed]
- 81.Simon SL, Dacey J, Glynn S, et al. The effect of relapse on cognition in abstinent methamphetamine abusers. J Subst Abuse Treat 2004;27:59-66. [DOI] [PubMed]
- 82.Salo R, Nordahl TE, Possin K, et al. Preliminary evidence of reduced cognitive inhibition in methamphetamine-dependent individuals. Psychiatry Res 2002;111:65-74. [DOI] [PubMed]
- 83.Woods SP, Rippeth JD, Conover E, et al. Deficient strategic control of verbal encoding and retrieval in individuals with methamphetamine dependence. Neuropsychology 2005;19:35-43. [DOI] [PubMed]
- 84.Sim T, Simon SL, Domier CP, et al. Cognitive deficits among methamphetamine users with attention deficit hyperactivity disorder symptomatology. J Addict Dis 2002;21:75-89. [DOI] [PubMed]
- 85.Simon SL, Domier CP, Sim T, et al. Cognitive performance of current methamphetamine and cocaine abusers. J Addict Dis 2002;21:61-74. [DOI] [PubMed]
- 86.Paulus MP, Hozack NE, Zauscher BE, et al. Behavioral and functional neuroimaging evidence for prefrontal dysfunction in methamphetamine-dependent subjects. Neuropsychopharmacology 2002;26:53-63.11751032
- 87.Chang L, Ernst T, Speck O, et al. Perfusion MRI and computerized cognitive test abnormalities in abstinent methamphetamine users. Psychiatry Res 2002;114:65-79. [DOI] [PubMed]
- 88.Simon SL, Domier C, Carnell J, et al. Cognitive impairment in individuals currently using methamphetamine. Am J Addict 2000;9:222-31. [DOI] [PubMed]
- 89.Kalechstein AD, Newton TF, Green M. Methamphetamine dependence is associated with neurocognitive impairment in the initial phases of abstinence. J Neuropsychiatry Clin Neurosci 2003;15:215-20. [DOI] [PubMed]
- 90.Halpern JH. Treatment of attention-deficit/hyperactivity disorder. JAMA 1999;281:1491. [DOI] [PubMed]
- 91.Matsumoto T, Yamaguchi A, Asami T, et al. Drug preferences in illicit drug abusers with a childhood tendency of attention deficit/hyperactivity disorder: a study using the Wender Utah Rating Scale in a Japanese prison. Psychiatry Clin Neurosci 2005;59:311-8. [DOI] [PubMed]
- 92.Eichenbaum H. The hippocampus and declarative memory: cognitive mechanisms and neural codes. Behav Brain Res 2001;127:199-207. [DOI] [PubMed]
- 93.Sweatt JD. Hippocampal function in cognition. Psychopharmacology (Berl) 2004;174:99-110. [DOI] [PubMed]
- 94.Kendler KS, Karkowski L, Prescott CA. Hallucinogen, opiate, sedative and stimulant use and abuse in a population-based sample of female twins. Acta Psychiatr Scand 1999;99:368-76. [DOI] [PubMed]
- 95.Ikeda M, Iwata N, Suzuki T, et al. Positive association of AKT1 haplotype to Japanese methamphetamine use disorder. Int J Neuropsychopharmacol 2005;9:77-81. [DOI] [PubMed]
- 96.Li T, Chen CK, Hu X, et al. Association analysis of the DRD4 and COMT genes in methamphetamine abuse. Am J Med Genet B Neuropsychiatr Genet 2004;129:120-4. [DOI] [PubMed]
- 97.Hashimoto T, Hashimoto K, Matsuzawa D, et al. A functional glutathione S-transferase P1 gene polymorphism is associated with methamphetamine-induced psychosis in Japanese population. Am J Med Genet B Neuropsychiatr Genet 2005;135:5-9. [DOI] [PubMed]
- 98.Koizumi H, Hashimoto K, Kumakiri C, et al. Association between the glutathione S-transferase M1 gene deletion and female methamphetamine abusers. Am J Med Genet B Neuropsychiatr Genet 2004;126:43-5. [DOI] [PubMed]
- 99.Ujike H, Harano M, Inada T, et al. Nine-or fewer repeat alleles in VNTR polymorphism of the dopamine transporter gene is a strong risk factor for prolonged methamphetamine psychosis. Pharmacogenomics J 2003;3:242-7. [DOI] [PubMed]
- 100.Nishiyama T, Ikeda M, Iwata N, et al. Haplotype association between GABAA receptor gamma2 subunit gene (GABRG2) and methamphetamine use disorder. Pharmacogenomics J 2005;5:89-95. [DOI] [PubMed]
- 101.Lin SK, Chen CK, Ball D, et al. Gender-specific contribution of the GABA(A) subunit genes on 5q33 in methamphetamine use disorder. Pharmacogenomics J 2003;3:349-55. [DOI] [PubMed]
- 102.Harano M, Uchimura N, Abe H, et al. A polymorphism of DRD2 gene and brain atrophy in methamphetamine psychosis. Ann N Y Acad Sci 2004;1025:307-15. [DOI] [PubMed]
- 103.Ide S, Kobayashi H, Tanaka K, et al. Gene polymorphisms of the mu opioid receptor in methamphetamine abusers. Ann N Y Acad Sci 2004;1025:316-24. [DOI] [PubMed]
- 104.Cheng CY, Hong CJ, Yu YW, et al. Brain-derived neurotrophic factor (Val66Met) genetic polymorphism is associated with substance abuse in males. Brain Res Mol Brain Res 2005;140:86-90. [DOI] [PubMed]
- 105.Kobayashi H, Ide S, Hasegawa J, et al. Study of association between alpha-synuclein gene polymorphism and methamphetamine psychosis/dependence. Ann N Y Acad Sci 2004;1025:325-34. [DOI] [PubMed]
- 106.Hong CJ, Cheng CY, Shu LR, et al. Association study of the dopamine and serotonin transporter genetic polymorphisms and methamphetamine abuse in Chinese males. J Neural Transm 2003;110:345-51. [DOI] [PubMed]
- 107.Liu HC, Lin SK, Liu SK, et al. DAT polymorphism and diverse clinical manifestations in methamphetamine abusers. Psychiatr Genet 2004;14:33-7. [DOI] [PubMed]
- 108.Tsai SJ, Cheng CY, Shu LR, et al. No association for D2 and D4 dopamine receptor polymorphisms and methamphetamine abuse in Chinese males. Psychiatr Genet 2002;12:29-33. [DOI] [PubMed]
- 109.Sery O, Vojtova V, Zvolsky P. The association study of DRD2, ACE and AGT gene polymorphisms and metamphetamine dependence. Physiol Res 2001;50:43-50. [PubMed]
- 110.Itoh K, Hashimoto K, Shimizu E, et al. Association study between brain-derived neurotrophic factor gene polymorphisms and methamphetamine abusers in Japan. Am J Med Genet B Neuropsychiatr Genet 2005;132:70-3. [DOI] [PubMed]
- 111.Inada T, Iijima Y, Uchida N, et al. No association found between the type 1 sigma receptor gene polymorphisms and methamphetamine abuse in the Japanese population: a collaborative study by the Japanese Genetics Initiative for Drug Abuse. Ann N Y Acad Sci 2004;1025:27-33. [DOI] [PubMed]
- 112.Iwata N, Inada T, Harano M, et al. No association is found between the candidate genes of t-PA/plasminogen system and Japanese methamphetamine-related disorder: a collaborative study by the Japanese Genetics Initiative for Drug Abuse. Ann N Y Acad Sci 2004;1025:34-8. [DOI] [PubMed]
- 113.MoritaY, Ujike H, Tanaka Y, et al. A nonsynonymous polymorphism in the human fatty acid amide hydrolase gene did not associate with either methamphetamine dependence or schizophrenia. Neurosci Lett 2005;376:182-7. [DOI] [PubMed]
- 114.Morita Y, Ujike H, Tanaka Y, et al. The X-box binding protein 1 (XBP1) gene is not associated with methamphetamine dependence. Neurosci Lett 2005;383:194-8. [DOI] [PubMed]
- 115.World Health Organization (WHO). Amphetamine-like stimulants: a report from the WHO meeting an amphetamines, MDMA and other psychostimulants. Geneva: Substance Abuse Department, WHO; 1997.
- 116.Centers for Disease Control and Prevention. Acute public health consequences of methamphetamine laboratories–16 states, January 2000-June 2004. MMWR Morb Mortal Wkly Rep 2005;54:356-9. [PubMed]
- 117.Freese TE, Obert J, Dickow A, et al. Methamphetamine abuse: issues for special populations. J Psychoactive Drugs 2000;32:177-82. [DOI] [PubMed]
- 118.Rawson RA, Anglin MD, Ling W. Will the methamphetamine problem go away? J Addict Dis 2002;21:5-19. [DOI] [PubMed]
- 119.Centre for Addiction and Mental Health (CAMH). Ontario student drug use survey. Toronto: CAMH; 1999.
- 120.Anglin MD, Burke C, Perrochet B, et al. History of the methamphetamine problem. J Psychoactive Drugs 2000;32:137-41. [DOI] [PubMed]
- 121.Derlet RW, Heischober B. Methamphetamine. Stimulant of the 1990s? West J Med 1990;153:625-8. [PMC free article] [PubMed]
- 122.Kalasinsky KS, Hugel J, Kish SJ. Use of MDA (the “love drug”) and methamphetamine in Toronto by unsuspecting users of ecstasy (MDMA). J Forensic Sci 2004;49:1106-12. [PubMed]
- 123.The McCreary Centre Society. Adolescent health survey: province of BC. Burnaby (BC): The McCreary Centre Society; 1998.
- 124.Pacific Community Resources. Lower mainland youth drug use survey. Surrey (BC): Pacific Community Resources; 2002. Available: www.pcrs.ca/Images/Agency%20Documents/Publications/Drug%20Survey.pdf (accessed 2005 Mar 16).
- 125.Evans L. Walking beside people in Vancouver downtown eastside [lecture]. Drug abuse and major mental illness: clinical neurosciences conference; 2006 Feb 24; Vancouver.
- 126.Statistical profile of aboriginal peoples 2001. Vancouver: BC Stats; 2005. Available: www.bcstats.gov.bc.ca/data/cen01/abor/CR15.pdf (accessed 2006 Mar 16).
- 127.Frosch D, Shoptaw S, Huber A, et al. Sexual HIV risk among gay and bisexual male methamphetamine abusers. J Subst Abuse Treat 1996;13:483-6. [DOI] [PubMed]
- 128.Lee SJ, Galanter M, Dermatis H, et al. Circuit parties and patterns of drug use in a subset of gay men. J Addict Dis 2003;22:47-60. [DOI] [PubMed]
- 129.Chang L, Smith LM, LoPresti C, et al. Smaller subcortical volumes and cognitive deficits in children with prenatal methamphetamine exposure. Psychiatry Res 2004;132:95-106. [DOI] [PubMed]
- 130.Zhao H, Brenneisen R, Scholer A, et al. Profiles of urine samples taken from ecstasy users at rave parties: analysis by immunoassays, HPLC, and GC-MS. J Anal Toxicol 2001;25:258-69. [DOI] [PubMed]
- 131.Canadian Community Epidemiology Network on Drug Use — Vancouver Site, Addictive Drug Information Council (ADIC). Methamphetamine environmental scan summit: final report. Richmond (BC): CCENDU and ADIC; 2003. Available: www.ccsa.ca/ccendu/pdf/methamph_summary_2003.pdf (accessed 2006 Mar 16).
- 132.Bungay V, Malchy L, Buxton J, et al. Life with jib: a snapshot study of street youth's use of crystal methamphetamine. Addiction Theory and Research. In press.
- 133.Cohen JB, Dickow A, Horner K, et al. Abuse and violence history of men and women in treatment for methamphetamine dependence. Am J Addict 2003;12:377-85. [PubMed]
- 134.Birchwood M, Spencer E. Early intervention in psychotic relapse. Clin Psychol Rev 2001;21:1211-26. [DOI] [PubMed]
- 135.US Drug Enforcement Agency. Drug trafficking in the United States. September 2006. Available: www.usdog.gov/dea/concern/drug_trafficking.html.
- 136.US Drug Enforcement Agency. Drug trafficking in the United States. September 2006. Available: www.usdoj.gov/dea/concern/18862/meth.html.
- 137.Roehr B. Half a million Americans use methamphetamine every week. BMJ 2005;331:476. [DOI] [PMC free article] [PubMed]
- 138.Canada–United States border drug threat assessment. Ottawa: Public Safety and Emergency Preparedness Canada; 2004. Available: www.psepc-sppcc.gc.ca/prg/le/bs/uscabdta-en.asp (accessed 2006 Mar 16).
- 139.Richards JR, Bretz SW, Johnson EB, et al. Methamphetamine abuse and emergency department utilization. West J Med 1999;170:198-202. [PMC free article] [PubMed]
- 140.Tanne JH. Methamphetamine epidemic hits middle America. BMJ 2006;332:382. [DOI] [PMC free article] [PubMed]
- 141.Danks RR, Wibbenmeyer LA, Faucher LD, et al. Methamphetamine-associated burn injuries: a retrospective analysis. J Burn Care Rehabil 2004;25:425-9. [DOI] [PubMed]
- 142.Grabowski J, Shearer J, Merrill J, et al. Agonist-like, replacement pharmacotherapy for stimulant abuse and dependence. Addict Behav 2004;29:1439-64. [DOI] [PubMed]
- 143.Sherman JP. Dexamphetamine for “speed” addiction. Med J Aust 1990;153:306. [DOI] [PubMed]
- 144.Shearer J, Wodak A, Mattick RP, et al. Pilot randomized controlled study of dexamphetamine substitution for amphetamine dependence. Addiction 2001;96:1289-96. [DOI] [PubMed]
- 145.Vocci F, Ling W. Medications development: successes and challenges. Pharmacol Ther 2005;108:94-108. [DOI] [PubMed]
- 146.National Institute on Drug Abuse. Assessment of interactions between methamphetamine and aripiprazole - 1. 2005. Available: www.clinicaltrials.gov/ct/gui/show/NCT00089440 (accessed 2006 Mar 16).
- 147.National Institute of Drug Abuse. Seroquel therapy for substance use disorders comorbid with schizophrenia. 2005. Available: www.clinicaltrials.gov/ct/gui/show/NCT00208143 (accessed 2006 Mar 16).
- 148.Office of Rare Diseases. Biennial and annual report on the rare diseases research activities at the National Institutes of Health FY 2004. Bethesda (MD): National Institutes of Health; 2004. Available: http://rarediseases.info.nih.gov/html/reports/Fy2004/nida.html (accessed 2006 Mar 16); 2005.
- 149.Newton TF, Roache JD, De La Garza R, et al. Bupropion reduces methamphetamine-induced subjective effects and cue-induced craving. Neuropsychopharmacology 2006;31:1537-44. [DOI] [PubMed]
- 150.Brodie JD, Figueroa E, Laska EM, et al. Safety and efficacy of gamma-vinyl GABA (GVG) for the treatment of methamphetamine and/or cocaine addiction. Synapse 2005;55:122-5. [DOI] [PubMed]
- 151.Meijler MM, Matsushita M, Wirsching P, et al. Development of immunopharmacotherapy against drugs of abuse. Curr Drug Discov Technol 2004;1:77-89. [DOI] [PubMed]
- 152.Daniels JR, Wessinger WD, Hardwick WC, et al. Effects of anti-phencyclidine and anti-(+)-methamphetamine monoclonal antibodies alone and in combination on the discrimination of phencyclidine and (+)-methamphetamine by pigeons. Psychopharmacology (Berl) 2006;185:36-44. [DOI] [PubMed]
- 153.Rawson RA, Marinelli-Casey P, Anglin MD, et al. A multi-site comparison of psychosocial approaches for the treatment of methamphetamine dependence. Addiction 2004;99:708-17. [DOI] [PubMed]
- 154.Misra LK, Kofoed L, Oesterheld JR, et al. Olanzapine treatment of methamphetamine psychosis. J Clin Psychopharmacol 2000;20:393-4. [DOI] [PubMed]
- 155.Misra L, Kofoed L. Risperidone treatment of methamphetamine psychosis. Am J Psychiatry 1997;154:1170. [DOI] [PubMed]
- 156.Sato M, Chen CC, Akiyama K, et al. Acute exacerbation of paranoid psychotic state after long-term abstinence in patients with previous methamphetamine psychosis. Biol Psychiatry 1983;18:429-40. [PubMed]
- 157.Drake RE, Mueser KT. Managing comorbid schizophrenia and substance abuse. Curr Psychiatry Rep 2001;3:418-22. [DOI] [PubMed]
- 158.Mueser KT, Noordsy DL, Drake RE, et al. Integrated treatment for dual disorders — a guide to effective practice. New York: The Guilford Press; 2003.
- 159.Torrey WC, Drake RE, Cohen M, et al. The challenge of implementing and sustaining integrated dual disorders treatment programs. Community Ment Health J 2002;38:507-21. [DOI] [PubMed]
- 160.Rawson R, Gonzales R, Brethen P. Treatment of methamphetamine use disorders: an update. J Subst Abuse Treat 2002;23:145-50. [DOI] [PubMed]
- 161.Worsley JN, Moszczynska A, Falardeau P, et al. Dopamine D1 receptor protein is elevated in nucleus accumbens of human, chronic methamphetamine users. Mol Psychiatry 2000;5:664-72. [DOI] [PubMed]
- 162.Cretzmeyer M, Vaughan Sarrazin M, Huber DL, et al. Treatment of methamphetamine abuse: research findings and clinical directions. J Subst Abuse Treat 2003;24:267-77. [DOI] [PubMed]