

Abstract
Purpose
Persistent hyperplastic primary vitreous (PHPV) is an idiopathic developmental eye disease associated with failed involution of the hyaloid vasculature. The present work addressed the pathogenesis of PHPV in a mouse model that replicates many aspects of the human disease.
Methods
Ophthalmoscopic and histologic analyses documented pathologic processes in eyes of mice lacking the Arf gene compared with Ink4a-deficient and wild-type control animals. Immunohistochemical staining, in situ hybridization, and RT-PCR demonstrated the expression of relevant gene products. Arf gene expression was determined by in situ hybridization using wholemounts of wild-type mouse eyes and by immunofluorescence staining for green fluores-cent protein (GFP) in Arf+/GFP heterozygous knock-in mouse eyes.
Results
Abnormalities in Arf−/− mice mimicked those found in patients with severe PHPV. The mice had microphthalmia; fibrovascular, retrolental tissue containing retinal pigment epithelial cells and remnants of the hyaloid vascular system; posterior lens capsule destruction with lens degeneration and opacity; and severe retinal dysplasia and detachment. Eyes of mice lacking the overlapping Ink4a gene were normal. Arf was selectively expressed in perivascular cells within the vitreous of the postnatal eye. Cells composing the retrolental mass in Arf−/− mice expressed the Arf promoter. The remnant hyaloid vessels expressed Flk-1. Its ligand, vascular endothelial growth factor (Vegf), was expressed in the retrolental tissue and the adjacent dysplastic neuroretina.
Conclusions
Arf−/− mice have features that accurately mimic severe PHPV. In the HVS, Arf expression in perivascular cells may block their accumulation or repress Vegf expression to promote HVS involution and prevent PHPV.
The hyaloid vascular system (HVS) in the primary vitreous of the mammalian eye provides an elegant example of developmentally regulated vascular regression. In the HVS, the hyaloid artery arises from the ophthalmic/central retinal artery posteriorly and branches in the vitreous to form the vasa hyaloidea propria (VHP) and the tunica vasculosa lentis (TVL), which envelops the lens.1,2 The pupillary membrane (PM) makes up the anterior component of this system.1,2 These vascular beds normally regress late in human fetal development and in the first several weeks of life in the mouse,1,2 to create the avascular cornea, lens, and secondary vitreous composed of collagens, fibronectin, and other extracellular matrix macromolecules.3
Persistent hyperplastic primary vitreous (PHPV) has long been used to describe a disease process associated with persistence of the hyaloid vascular structures in the vitreous.4,5 Recently, the nomenclature was revisited, and persistent fetal vasculature (PFV) was proposed to reflect more accurately the broader manifestations of failed regression of other vascular beds in the eye.1 (Because our mouse model has only developmental defects in the vitreous, we will use the term PHPV.) Depending on whether the major abnormalities are in the region of the TVL/VHP or the hyaloid artery, PHPV is subdivided into anterior or posterior forms, respectively.6,7 Most reported cases show overlap, though, with abnormalities in both anatomic compartments.6 Fibroblast-like cells, occasionally intermixed with pigmented cells, form a fibrovascular mass surrounding the remnants of the HVS.6,8 In anterior or combined PHPV, the retrolental tissue lies adjacent to a rent in the posterior lens capsule, typically resulting in cataract formation.4,6,8 In some cases, it can lead to intralenticular hemorrhage; lens swelling with secondary glaucoma; and total lens absorption, calcification, or even replacement by adipose tissue.6,8 In posterior or combined PHPV, the retina may be detached by congenital nonattachment or by traction from the retrolental tissue adjacent to the inner neuroretina.6,8 This may be associated with retinal folding, dysplasia, and reactive retinal pigment epithelial (RPE) cell accumulation.8
The broad clinical manifestations of PHPV depend on the anatomic region involved (i.e., anterior, posterior, or both) and the extent of residual hyaloid vessels. Typically, PHPV presents in children as unilateral microphthalmia, leukokoria, or cataract and is associated with retinal folding and detachment.6 Visual acuity can be nearly normal, but is 20/200 or less at diagnosis in most cases of posterior PHPV.7 Depending on its severity, surgical treatment focuses on vision preservation by lensectomy, vitrectomy, or membranectomy to prevent the sequelae of glaucoma and phthisis. Occasionally, enucleation of a blind eye is required for pain control or cosmesis.1,6
The etiology of PHPV is not established. Analogous to retinoblastoma, it is usually sporadic and unilateral, but bilateral disease has been described in 2% to 30% of patients.6,8,9 Familial cases10-12 and PHPV associated with congenital syndromes13,14 or other eye anomalies15 further imply that it may have a genetic basis. Mouse studies have shed some light on potential candidate genes for PHPV and provide clues to mechanisms promoting the normal regression of the mammalian HVS. For example, vascular endothelial growth factor (Vegf) stabilizes developing blood vessels16,17 and is essential for mouse development.18 Anatomic studies in the mouse suggest that physical separation of Vegf-expressing lens cells from the TVL may drive its involution.19 Vitreous hyaloid vessels persist in mice lacking angiopoietin-2 (Ang-2),20 a vascular growth factor with anti-angiogenic properties.16,17 This is reminiscent of patients with mild posterior PHPV, but secondary features such as retinal detachment or lens degeneration were not reported in these mice.20 In some lines of mice lacking the proapoptotic p53 gene a PHPV-like disease can develop.21,22 The phenotype has variable penetrance and severity in pure BALB/c p53−/− mice22 and is suppressed in p53−/− mice of a mixed C57Bl/6 × 129/Sv background.21 Together, these studies support the concept that PHPV may have a genetic basis. The variable severity and penetrance and the mouse-strain dependence of the phenotype, though, suggest that additional genetic pathways are involved.
We have reported that Arf−/− mice may provide a reproducible model for studying PHPV pathogenesis.23 The Arf gene partially overlaps with the Ink4a gene in the mouse and human genomes.24-26 Arf encodes p19Arf (p14ARF in humans) which stabilizes the tumor suppressor p53 by interacting with Mdm2 (Hdm2 in humans).27-29 We showed that Arf−/− mice in a mixed C57Bl/6 × 129/Sv background have certain manifestations of PHPV, whereas p53−/− mice in a similar genetic background have only subtle evidence of incomplete HVS involution.23 The present manuscript extends our previous findings by identifying other clinical and pathologic manifestations of PHPV evident in Arf−/− mice and by exploring cellular and molecular mechanisms by which p19Arf normally functions in perivascular cells in the vitreous to prevent severe combined PHPV.
Methods
Mice and Genotyping
All procedures using animals were approved by the St. Jude Children's Research Hospital Animal Care and Use Committee and conform to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Mice in which the Arf exon 1β was inactivated30 or replaced by a reporter gene encoding green fluorescence protein (GFP),31 maintained in a mixed C57BL/6 × 129/SvJ background, were provided by Martine F. Roussel and Charles J. Sherr (St. Jude Children's Research Hospital). Ink4a−/− mice (exon 1α deleted)32 and Ink4a/Arf−/− mice (exon 2 deleted)33 were provided by R. A. DePinho (Dana Farber Cancer Institute, Boston, MA). Mouse genotype was determined by polymerase chain reaction (PCR), using tail DNA as previously described.23,32
Ophthalmoscopic Evaluation
Three-month old Arf+/− or Arf−/− mice were lightly anesthetized with Avertin (1.25% 2,2,2-tribromoethanol and 0.8% tert-pentyl alcohol in water, 0.3 mL intraperitoneally). Pupils were dilated with 1% ophthalmic drops (Cyclomydril; Alcon Pharmaceuticals, Fort Worth, TX). Slit lamp imaging took place using a slit lamp biomicroscope (Carl Zeiss Meditec, Dublin, CA) fitted with a digital video camera (GL1; Canon, Lake Success, NY) for making QuickTime movies. Anterior segment images were selected from the digital video captured during the examination. A small-animal fundus camera (Genesis; Kowa, Torrance, CA) in conjunction with a 90-D condensing lens (Volk Optical, Mentor, OH) was used to produce images of the retina. After ophthalmoscopic evaluation, mice were euthanatized, and the eyes were processed for histology studies.
RT-PCR and Northern Analyses
RNA was isolated from whole postnatal day (P)4 wild-type or Arf−/− eyes and primary cultures of wild-type mouse embryo fibroblasts, using an extraction reagent (TRIzol; Invitrogen, San Diego, CA). Reverse-transcriptase polymerase chain reaction (RT-PCR) for Arf, Ink4a, and Gapdh was performed as described.23,24 Northern blot analysis was performed using 10 μg of RNA extracted from P4 Arf+/− and Arf−/− eyes and 32P-labeled cDNA probes for Arf exon 1β and β-actin in buffer (NorthernMax; Ambion, Austin, TX), according to the manufacturer's recommendations.
Wholemounts of the Hyaloid Vascular System
Wholemounts of the HVS were made essentially as previously described.34 Briefly, eyes were removed from euthanatized P4 Arf+/GFP mice and fixed in 4% paraformaldehyde (PF) in PBS from 4 hours to overnight. After they were rinsed in PBS, eyes were incised adjacent to the optic nerve and at two opposing sites along the equator. A 1.5% solution of low-melting-point agarose (Invitrogen-Gibco) was injected through the posterior incision with a 30-gauge needle until the solution flowed from an equatorial incision. After solidifying at room temperature for 15 minutes, equatorial incisions were extended to remove the anterior half of the globe. The posterior optic cup was inverted to release the lens with the attached agarose cast of the vitreous cavity. The agarose cast was heated in a drop of PBS to 65°C on a glass slide allowing the agarose to melt and the VHP vessels to adhere. Immunofluorescence (IF) staining for GFP was immediately performed.
To assess PM and TVL regression, eyes were removed from P12 wild-type and Arf−/− mice, partially dissected, and stained with hematoxylin. PM and TVL vessels were enumerated as described.2
Histologic Studies
Eyes were removed from euthanatized mice, fixed in 4% PF as described earlier and either processed for paraffin-embedded sections with a tissue processor (Shandon Citadel 1000; Thermo Electron Corp., Waltham, MA) or saturated in 20% sucrose, embedded (Tissue Freezing Medium; Triangle Biomedical Sciences, Durham, NC), and stored at −80°C for cryostat sections. Five- and 12 μm-thick sections were used for histologic studies from paraffin-embedded and frozen sections, respectively.
Antibody-Based Staining of Tissue Sections
For IF staining of GFP expression in Arf+/GFP and ArfGFP/GFP mouse eyes, cryostat sections or wholemount HVSs were blocked in 10 mM Tris (pH 7.4), 100 mM MgCl2, 5% fetal calf serum, 1% bovine serum albumin, and 0.5% Tween-20 and stained with rabbit α-GFP primary antibody (1:750; A6455; Molecular Probes, Eugene, OR). Primary antibodies were detected using Cy3- or rhodamine red-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Flk-1 and HMB-45 were detected by histochemical staining of paraffin sections using goat α-Flk1 (AF644; R&D Systems, Minneapolis, MN) and mouse α-HMB-45 (M0634; Dako, Carpinteria, CA). Dual IF staining was performed with rabbit α-GFP and rat α-CD31 (557355; BD-PharMingen, San Diego, CA) or mouse anti-smooth muscle α-actin (SMA) (M0851; Dako) and species-specific secondary antibodies. Colabeling for GFP and the pericyte marker NG235 was accomplished with rabbit anti-NG2 (provided by William B. Stallcup; The Burnham Institute, La Jolla, CA) and direct evaluation of green fluorescence. To document specificity, control slides were stained using an unrelated primary antibody.
In Situ Hybridization
Digoxigenin (DIG)-labeled riboprobes were prepared with a commercial system (Riboprobe Combination System; Promega, Madison, WI) and 10 mM DIG RNA labeling mix (Roche, Indianapolis, IN) from pBSIIKS-based plasmids containing mouse Arf36 or Vegf (exons 1–5)37 (provided by Patricia A. d'Amore, Schepens Eye Research Institute, Boston, MA). DIG-labeled 713-bp Arf sense and antisense riboprobes were precipitated with LiCl/EtOH, rinsed in 70% EtOH, and resus-pended in RNase-free water. For Vegf, sense and antisense riboprobes were synthesized by using T7 or T3 RNA polymerase (Roche) with 33P-UTP.
ISH for Arf was performed using wholemounts of eyes removed from euthanatized P5 wild-type mice, processed as described.38 Just before staining, two opposing windows were cut through the lateral walls of the optic cup, to facilitate solution penetration. Wholemount ISH was performed using DIG-labeled Arf sense and antisense riboprobes (1 μg/mL), essentially as previously described.38 Hybridization was performed overnight at 70°C in 50% formamide, 5× SSC (pH 4.5), 1% SCS containing yeast RNA (50 μg/mL), and heparin (50 μg/mL). After posthybridization washes, DIG was detected with an alkaline phosphatase-coupled α-DIG antibody (1093274; Roche), according to the manufacturer's protocol. Stained eyes were embedded in a solution of gelatin (0.5%), albumin (30%), and sucrose (20%) and mixed with 2.5% glutaraldehyde. Vibratome sections (100-μm-thick) were mounted on glass slides using with antifade medium (Mowiol; Hoechst, Frankfurt, Germany).
ISH for Vegf was performed using cryostat sections from P4 Arf+/− and Arf−/− eyes. Sense and antisense probes were hybridized overnight at 55°C. Afterward, slides were rinsed in 2× SSC, treated with RNase A, and sequentially rinsed in RNase buffer and 2× and 0.2× SSC. Sections were then dehydrated in a graded series of ethanol solutions, air dried, and exposed to autoradiograph film (β-max Hyperfilm; Eastman Kodak, Rochester, NY) overnight. Dried slides were dipped in NTB2 emulsion (Eastman Kodak) and stored at 4°C for ∼6 days. Slides were mounted under glass coverslips (Permount; Sigma-Aldrich, St. Louis, MO) and visualized by dark-field microscopy.
Results
Clinical and Pathologic Features of PHPV Recapitulated in Arf−/− Mice
We determined whether Arf−/− mice had clinical features of PHPV by comparing the appearance of 2- to 4-month-old Arf+/+ and Arf+/− mice with that of Arf−/− mice (n = 10 in each group). All the eyes in 10 Arf−/− mice and no eyes from the heterozygous or wild-type mice appeared microphthalmic (Fig. 1A). Eyes of several normal-appearing Arf+/+ or Arf+/− mice and Arf−/− mice were assessed by ophthalmoscopy. Dense lens opacity prevented fundoscopic analysis in the Arf−/− eyes, whereas the Arf+/+ and Arf+/− eyes exhibited normal retinal pigmentation patterns, vascularization, and optic nerve head morphology (Fig. 1B, top). Slit lamp examination demonstrated dense lens opacity in the Arf−/− eyes, whereas Arf+/− mice had normal lens surface and structure (Fig. 1B). To correlate ophthalmoscopic findings with pathology, we obtained midline sagittal sections through the eyes of these mice. Lens architecture, posterior lens capsule, vitreous space, and neuroretina were normal in eyes taken from heterozygous and wild-type mice (Fig. 1C, left; additional data not shown). Arf−/− eyes had the expected abnormalities of severely misshapen lens, complete detachment of the neuroretina from the RPE, rosettes, and other dysplastic changes in the retina (Fig. 1C, right).23 Higher magnification showed a fibrovascular mass eroding through the posterior lens capsule, allowing extrusion of lens material into the mass (Fig. 1C, inset). These abnormalities have been consistently observed in several lines of Arf−/− mice derived independently23 (Martin AC, Skapek SX, unpublished observations, 2003). The manifestations of the disease in the Arf−/− mouse evolve over time: initially, the lens and neuroretina are largely normal in appearance and in the expression of lens structural genes.23 The presence of the retrolental mass before other pathologic changes in the lens and retina is consistent with its principal role in the pathogenesis of the disease.
Figure 1.
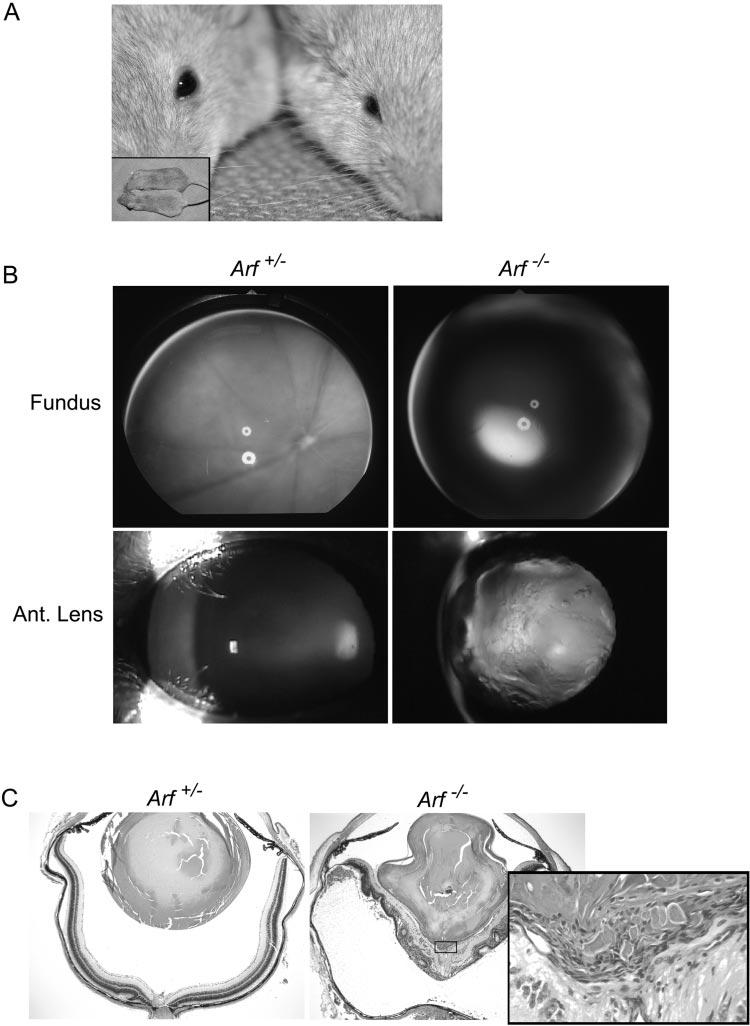
Clinical and pathologic features of severe PHPV in Arf−/− mouse. (A) Photograph of representative Arf+/+ (left) and Arf−/− (right) mice showing microphthalmic appearance without apparent difference in overall body size (inset). (B) Fundoscopic (top) and slit lamp images (bottom) are normal in Arf+/− eye (left). Dens lens opacity obscures fundoscopic view in Arf−/− eye (right). (C) Photomicrographs of hematoxylin and eosin–stained sections of eyes from Arf+/− (left) and Arf−/− (right) mice used for (B). Note misshapen lens, dysplastic neuroretina detached from pigment epithelium in Arf−/− mouse. Inset: high magnification of fibrovascular retrolental mass (box) directly apposed to posterior lens and inner neuroretina. Original magnification: ×40; inset: ×400.
We previously noticed that the retrolental tissue could contain pigmented cells,23 a finding also observed in pathologic specimens from patients with PHPV, in whom they are assumed to represent reactive RPE cells.8 Consistent with this notion, cells in the retrolental tissue of P10 Arf−/− albino mice were detected by the HMB45 antibody (Fig. 2A), which also labels RPE cells adjacent to photoreceptors (Fig. 2A, inset). The RPE-like cells existed in a gradient concentrated around the largest hyaloid vessels (Fig. 2A, arrow). This suggests that they may migrate along remnant vessels and gradually accumulate. Indeed, fewer pigmented cells were observed in the retrolental tissue at P1 than at P8 (Fig. 2B) and RPE-like cells accumulated next to hyaloid vessels close to where they traverse the neuroretina in Arf−/− eyes (Fig. 2C). Although formally possible that the pigmented cells arise directly from the retrolental cells, their gradual accumulation, localization around blood vessels, and antigenic similarity to RPE cells support the notion that they are reactive RPE cells accumulating in response to the vitreous disease.
Figure 2.
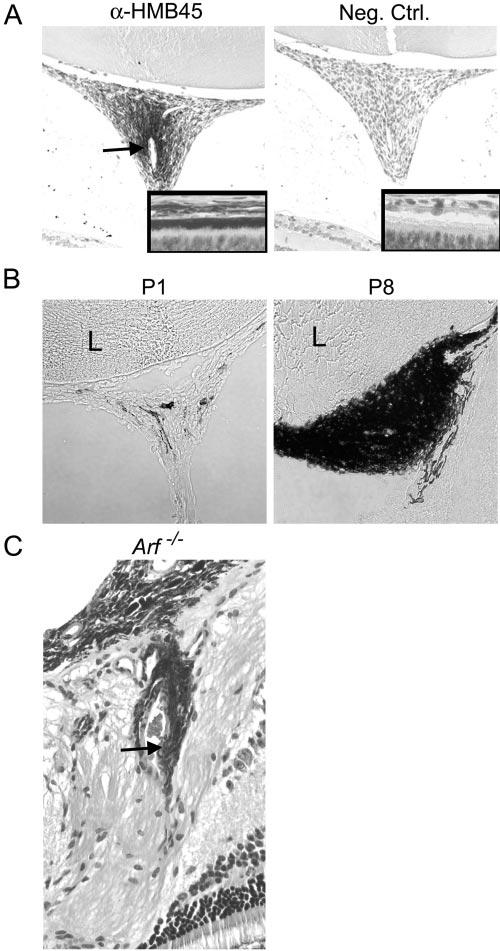
RPE-like cells accumulate in retrolental fibrovascular mass in Arf−/− mouse. (A) Representative photomicrographs of retrolental tissue from P10 albino Arf−/− mouse stained with α-HMB45 antibody (left) and unrelated primary antibody (right). HMB45-positive cells in retrolental tissue localize around large blood vessel (arrow). Insets: antibody staining of RPE as control. (B) Phase contrast photomicrographs of cryostat sections through retrolental tissue showing accumulation of pigmented cells between P1 and P8 in Arf−/− mice. (C) Photomicrograph of hematoxylin and eosin–stained section shows pigmented cells adjacent to vascular structures (arrow) within retina near the optic nerve/hyaloid artery stalk in 3-month-old Arf−/− mouse. Original magnification: (A, B) ×200 and ×400 (insets in A; C).
To assess whether the Arf−/− mice had defects in PM or TVL regression in addition to persistence of the VHP/HA,23 we evaluated hematoxylin-stained wholemounts of eyes of wild-type and Arf−/− littermates at P12, when much of the PM and TVL has already regressed.2 Vessels in the PM were similar in wild-type and Arf−/− eyes (Figs. 3A, top; 3B). TVL vessels were slightly more numerous in Arf−/− mice than wild-type (Fig. 3A, bottom; 3B), but the difference was not statistically significant (P = 0.06). The slight increase may be due to additional TVL vessels extending from the retrolental mass (Fig. 3A, bottom). Together, these findings are compatible with the concept that cells accumulating in the retrolental mass within the VHP/HA may prevent the involution of the adjacent vessels.
Figure 3.
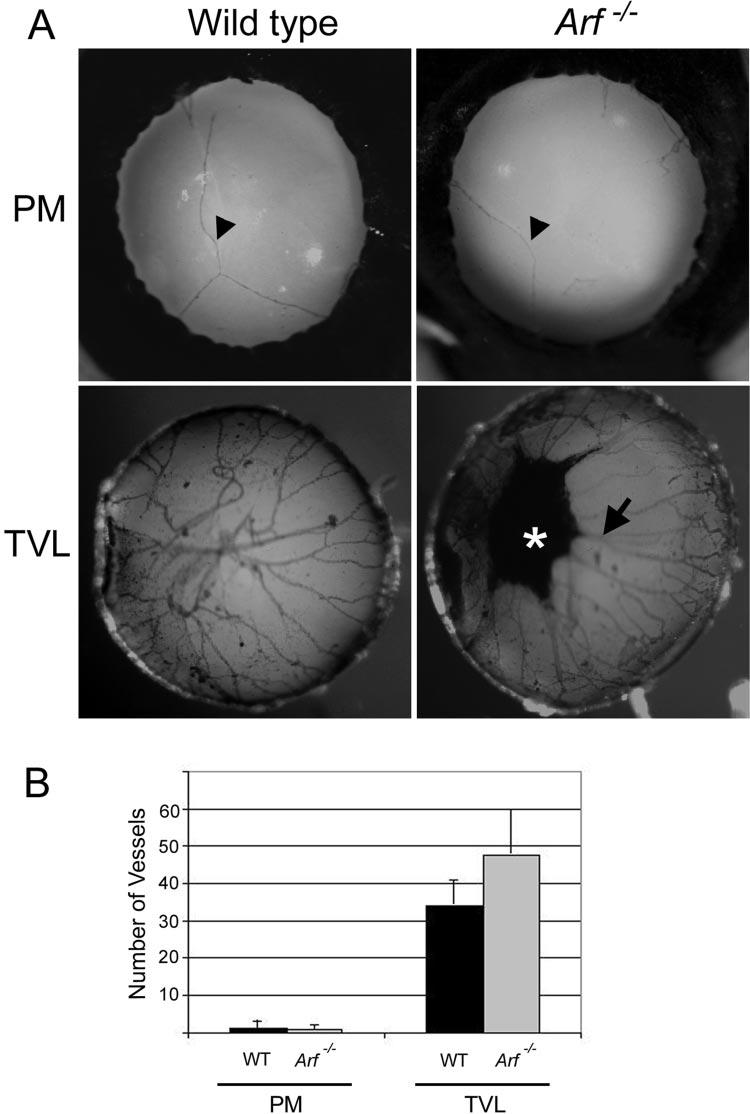
PM and TVL regression in the absence of Arf. (A) Photographs of PM and TVL on posterior lens from representative hematoxylin-stained wholemounts of eyes from wild-type (left) and Arf−/− (right) littermatesharvested at P12. Pigmented mass on posterior lens (⋆) and apparent vascular extension to TVL (arrow) in P12 Arf−/− mouse. (B) Quantitation of vessel number (as in Ref. 2) in PM and TVL present at P12 in wild-type and Arf−/− mice. Differences are not statistically significant (t-test).
No Contribution of Ink4a to Arf−/− Eye Phenotype
Mouse and human forms of the Arf gene reside at a genetic locus that also encodes the Ink4a gene (Fig. 4A; reviewed in Ref. 39). Unlike p19Arf, the Ink4a gene product, p16Ink4a, is a cyclin-dependent kinase inhibitor that regulates the retinoblastoma gene product, Rb. Because of the nature of the shared Arf/Ink4a locus, we asked whether the Arf−/− eye phenotype might be due to secondarily altered Ink4a expression.
Figure 4.
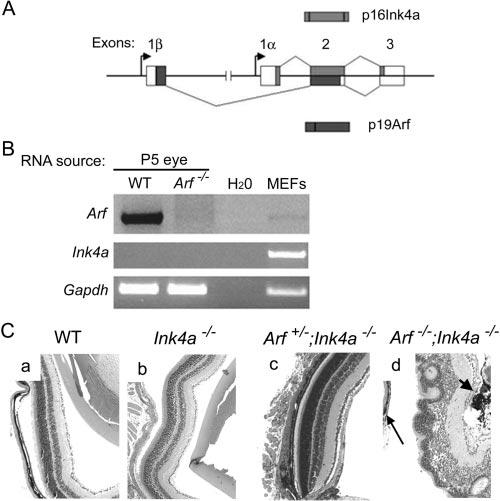
Loss of the Ink4a gene product did not contribute to PHPV in the Arf−/− mouse. (A) Schematic diagram showing overlapping mouse Arf and Ink4a gene products. Exon 1β is disrupted but Ink4a is intact in Arf-deficient mice used in the present study. (B) Ethidium-bromide stained products of RT-PCR for the indicated gene products. (C) Eyes from 2- to 3-month-old wild-type (WT) mice or mice of the indicated genotype. Note retrolental pigmented cells (d, short arrow) and dysplastic retina detached from RPE (d, long arrow) in Arf−/−; Ink4a−/− mice. Original magnification: ×100.
Ink4a could be amplified by RT-PCR from cultured wild-type fibroblasts, but not from RNA from P1 and P5 eyes, at which times Arf expression was detected (Fig. 4B and data not shown). To examine the role of Ink4a more formally, we evaluated the eyes of mice lacking exon 1α or 2 of the Arf/Ink4a locus (Fig. 4A), as they are deficient in p16Ink4 or both p19Arf and p16Ink4 protein expression, respectively (reviewed in Ref. 40). PHPV-like abnormalities were not present in eyes taken from mature Ink4a−/− mice (exon 1α-deficient; Arf+/+) or Ink4a−/−, Arf+/− mice (heterozygous deletions of exons 1α and 2; exon 1β intact) (Figs. 4Ca-Cc). Eyes taken from mice lacking exon 2 (hence, Ink4a−/−, Arf−/−) had lens degeneration, retrolental tissue, and retina dysplasia and detachment as in Arf−/− mice (Fig. 4Cd). Taken together, these data indicate that Ink4a deficiency does not contribute to the development of PHPV in Arf−/− mice.
Arf Expression in Perivascular Cells in the Vitreous
Our previous RT-PCR analyses indicated that Arf was expressed from P1 to P5 and the Arf-expressing cells were localized to the vitreous.23 To identify the cells, DIG-labeled ribo-probes derived from mouse Arf exons 1β and 2 were applied to wholemounts of P5 wild-type eyes for in situ hybridization. The antisense- but not the sense-riboprobe–labeled cells that were adjacent to blood vessels within the vitreous (Fig. 5A). Potential cross-hybridization of the probe to Ink4a mRNA was not deemed relevant because Ink4a mRNA was not detectable by RT-PCR in the postnatal eye (Fig. 4B). To verify this result, we used mice in which a GFP reporter replaced Arf exon 1β. GFP expression in this mouse mimics that of Arf and can be used as a surrogate marker for p19Arf 31 . ArfGFP/GFP mice (effectively Arf−/−) have a phenotype identical with Arf−/− mice. Arf+/GFP mice, like Arf+/− mice, appear to have normal eyes31 (Martin AC, Skapek SX, unpublished data, 2003). Hyaloid vessel preparations were obtained from P4 Arf+/GFP heterozygous eyes (Fig. 5Ba). Direct visualization of GFP fluorescence was confounded by weak signal, probably due to the tissue fixation and relatively high background green autofluorescence. Immunofluorescence staining of the HVS preparations with α-GFP antibody indicated that GFP-expressing cells were adjacent to blood vessels (Figs. 5Bb, 5Bc). To characterize the perivascular GFP-expressing cells further, we performed dual immunofluorescence staining using midline sections through hyaloid vessels in P1 Arf+/GFP and ArfGFP/GFP eyes. GFP-expressing cells were adjacent to CD31-positive endothelial cells and SMA– expressing vascular smooth muscle cells (VSMCs) in the heterozygous Arf+/GFP and ArfGFP/GFP eyes, but there was no apparent coexpression (Figs. 5Bd, 5C; some data not shown). Most cells in the retrolental mass expressed NG2 and some also expressed desmin (Fig. 5Ca, 5Cb and Thornton JD, Skapek SX, unpublished data, 2003), both of which are consistent with their pericyte-like qualities. The significance of the higher and lower levels of NG2 expression (Figs. 5Ca, 5Cb) is not known, but it is possible that the Arf promoter may be regulated during pericyte maturation and p19Arf may influence the process.
Figure 5.
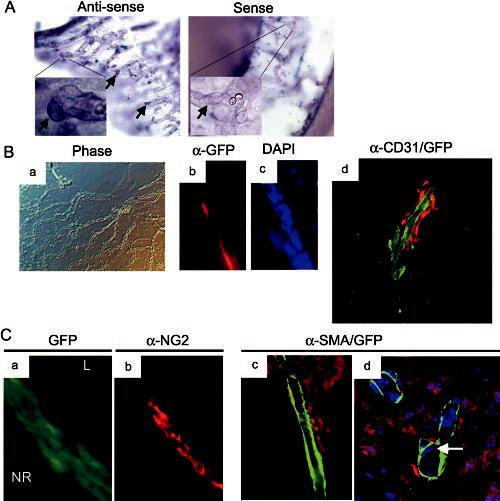
Perivascular cells adjacent to VHP/HA express Arf. (A) Representative photomicrographs of wholemounts of P5 wild-type eyes after in situ hybridization with digoxigenin-coupled antisense and sense riboprobes derived from Arf exons 1β and 2. Perivascular cells (arrows) are labeled by antisense riboprobe. (B) Representative phase contrast (Ba) and immunofluorescence (Bb–Bd) photomicrographs of P4 hyaloid vessel preparations (Ba–Bc) or P1 cryosection (Bd) from Arf+/GFP mice. Note GFP-expressing perivascular cells (red fluorescence in Bb and Bd) flanking CD31-expressing endothelial cells (green fluorescence in Bd). (C) Immunofluorescence (Ca–Cc) and confocal (Cd) photomicrographs of cryostat sections of eyes from P1 ArfGFP/GFP mice. GFP-expressing cells (green in Ca; red in Cc, Cd) between lens (L) and neuroretina (NR) are labeled with α-NG2 antibody (Cb) but not with α-SMA antibody (green in Cc, Cd). DAPI-stained nuclei (presumed to be endothelial cells) (d, arrow) observed internal to SMA-expressing cells in vessel wall. Original magnification: (A) ×200; ×1000 (insets in A, Ba) ×100; (Bb–Bd; Ca–Cc) ×400; (Cd) ×600.
Flk1 and Vegf Expression in Arf−/− Eyes
Because pericytes support the underlying vasculature, we addressed whether a key angiogenic factor may contribute to the abnormal persistence of the VHP/HA in Arf−/− mice, by characterizing the expression of Flk1 protein and Vegf mRNA in the postnatal eye. Both large and small vessels within the retrolental mass in Arf−/− mice at P1 expressed Flk1 (Fig. 6), implying that Vegf signaling is involved in the formation or maintenance of the vessels. Northern blot of RNA taken from Arf+/− and Arf−/− eyes at P4 revealed no difference in total Vegf mRNA (Fig. 7A). Because the pathologic abnormalities in the P4 eye were localized to the retrolental tissue (Figs. 7Ba, 7Bb), we evaluated Vegf expression specifically in that region by in situ hybridization. The antisense probe detected Vegf mRNA throughout the central and peripheral regions of the neuro-retina and within the retrolental mass (Figs. 7Bc, long arrow). In addition to Vegf expression in the retrolental tissue, another difference between Arf+/− and Arf−/− eyes was a strong signal emanating from the dysplastic area of the Arf−/− neuroretina adjacent to the retrolental tissue (Figs. 7Bc, short arrow). Identification of Vegf-expressing cells in the retrolental tissue and dysplastic neuroretina provides a potential mechanism by which the Flk1-positive VHP/HA vessels fail to involute in Arf−/− mice.
Figure 6.
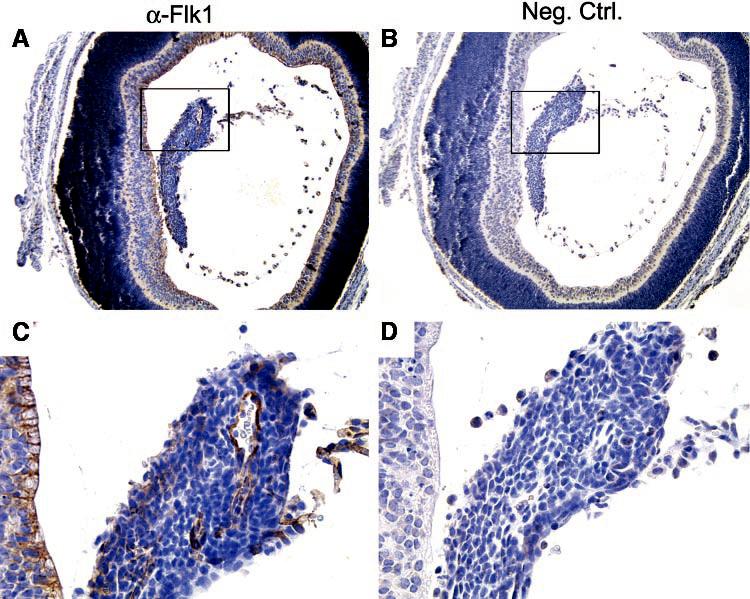
Flk1 is expressed in vascular structures within retrolental tissue in Arf−/− eye. Sections of P1 Arf−/− eye stained with α-Flk1 antibody (A, C) or unrelated primary antibody (B, D). Boxed area (A, B) shown at higher magnification (C, D). Original magnification: (A, B) ×100; (C, D) ×400.
Figure 7.
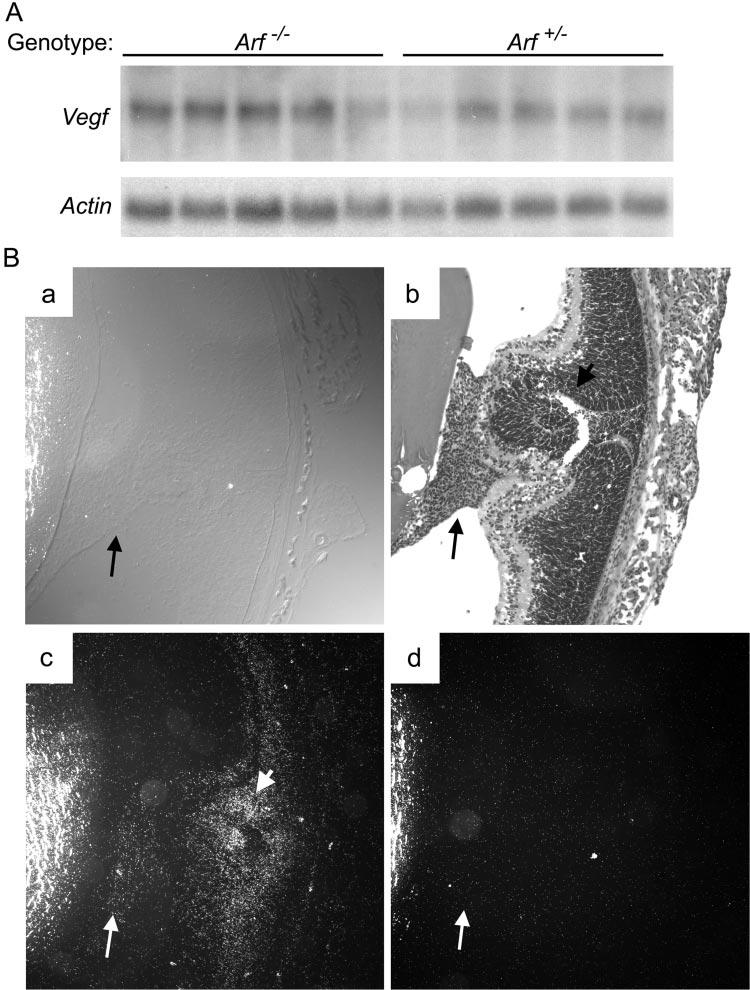
Vegf mRNA was detected in retrolental tissue and dysplastic neuroretina in Arf−/− eye. (A) Northern blot of mRNA from P4 whole eyes of indicated genotype shows equivalent Vegf and Actin expression. Individual lanes represent RNA from eyes of individual mice. (B) Cryostat sections of P4 Arf−/− eye. Phase-contrast (Ba) and hematoxylin and eosin–stained (Bb) images show retrolental tiss ue (long arrow) and adjacent dysplastic neuroretina (Bb, short arrow). Dark-field images after staining with 33P-labeled antisense (Bc) and sense (Bd) riboprobes show Vegf expression in retrolental tissue (Bc, long arrow) and dysplastic neuroretina (Bc, short arrow). Original magnification: ×100.
Discussion
Our findings support the following conclusions: (1) Arf is normally expressed in perivascular cells within the VHP/HA in the developing vitreous. (2) In the absence of Arf, pericyte-like cells that express the Arf promoter accumulate in direct proximity to the VHP/HA, forming a retrolental mass typical of PHPV. (3) The Ink4a gene product, which overlaps with both the mouse and human Arf gene, does not regulate vitreous cell accumulation in the mouse. (4) The retrolental mass of pericytes infiltrates the VHP and HA, which fail to regress in the first postnatal week; other components of the HVS are essentially normal. Hence, the retrolental tissue probably plays a primary role in disease pathogenesis in this mouse model. (5) Vegf is expressed in the retrolental mass and adjacent dysplastic neuroretina and may directly contribute to the failed VHP/HA regression. (6) In response to the retrolental tissue, secondary processes cause other clinical and pathologic features of severe PHPV, including microphthalmia, posterior lens capsule disruption with lens degeneration, dense lens opacity, reactive RPE cell accumulation in the retrolental tissue, and marked retina dysplasia/detachment. We propose that further studies of the Arf gene product and the Arf−/− mouse will provide valuable insight into molecular and cellular mechanisms regulating perivascular cell accumulation and VHP/HA regression as well as mechanisms underlying secondary manifestations of PHPV, like RPE cell accumulation, retinal folding and detachment, and posterior lens capsule destruction.
Potential discrepancies between our mouse model and the human disease are that most cases of PHPV are not bilateral nor are they uniformly severe.1,6 Indeed, in some respects the Arf−/− mouse seems also to mimic X-linked Norrie disease (ND), which is uniformly bilateral and severe and variably associated with cognitive and hearing problems.41,42 Our studies show that the pathogenesis of the Arf−/− phenotype, however, is distinct from that of ND. Most cases of ND are linked to abnormalities in the ND protein (NDP) gene.43,44 In situ hybridization studies in mouse, rabbit, and human show NDP mRNA expression in the outer and inner nuclear and the ganglion cell layers of the retina.45,46 Analysis of Ndp-deficient mice indicates the principal abnormalities to be retrolental structures in the vitreous body and disorganization of the ganglion cell layer in the retina.46 Of note, the published retinal abnormalities are mild compared with what occurs in Arf−/− mice at similar ages. The NDP gene encodes a protein, norrin, that functions as a signaling protein with the frizzled-4 (Fzd4) Wnt receptor and the Lrp5 coreceptor.47 Some propose that norrin may play a role in late retinal cell differentiation and that its loss may secondarily alter other aspects of eye development.46 Alternatively, disruption of norrin–Fzd4 signaling may primarily alter retinal vascular development and secondarily increase retinal Vegf production to prevent HVS involution.47 Either possibility is distinct from the pathogenesis of the Arf−/− phenotype where the chief defect is the excess accumulation of perivascular cells as early as P1. The overlapping clinical features of PHPV and ND support the use of molecular genetic testing as a diagnostic tool.48,49 Up to one third of patients with familial Norrie disease do not have identifiable mutations in NDP.43 Our findings suggest that some of these patients, as well as those with a diagnosis of severe PHPV, may have abnormalities in ARF or ARF-dependent biochemical pathways.
Clinical series stress the wide degree of disease manifestations in PHPV.1,6 In its least severe forms, simple persistence of vascular elements of the VHP or HA—even trace remnants of the VHP (Mittendorf dot) or HA stalk (Bergmeister's papilla)—meet the definition of the disease.1 At the other end of the spectrum, a fibrovascular mass within the VHP/HA, associated with microphthalmia, lens opacity, retinal dysplasia, or detachment represents a severe form of PHPV. Such broad disease manifestations suggest that fundamentally different processes are operative at different ends of the spectrum. Based on our studies and other published mouse models, we propose two distinct cellular mechanisms for PHPV pathogenesis (Fig. 8A). In the first place, it is clear that defects in the machinery directly involved in vascular regression can allow persistence of the components of the HVS (Figs. 8Aa, 8Ab). Examples include p53-deficient21,22 and Ang2-deficient mice.20 The primary defect in p53−/− mice seems to be decreased apoptosis at P7 and P8,22 during which time the VHP normally regresses.2 Similarly, Ang2 more or less directly destabilizes blood vessels,16 as evidenced by its peak expression around atretic ovarian follicles.50 Finally, hyalocytes are thought to transmit signals directly to endothelial cells to promote their apoptosis (reviewed in Ref. 51). Targeting attenuated diphtheria toxin to hyalocytes in a transgenic mouse prevents complete involution of the VHP/HA and pupillary membrane.52 In these examples, the main defect is incomplete regression of the vasculature. The full manifestations of PHPV occur variably or not at all. We propose that the remnants of the HVS serve as a “scaffolding” of sorts around which perivascular cells can accumulate (Fig. 8Ab). Depending on the degree of perivascular cell accumulation, additional manifestations of the disease may occur. Multiple factors may regulate the secondary accumulation of these cells and potentially account for the variable severity and progression in untreated eyes with PHPV.
Figure 8.
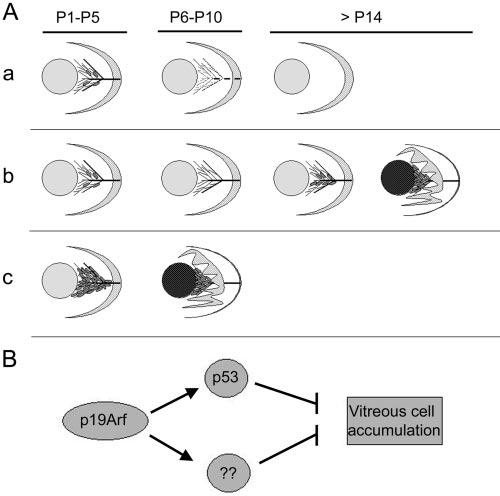
Schematic model for the pathogenesis of PHPV. (Aa) VHP/HA vessels with few Arf-expressing perivascular cells (gray ovals) present between the lens (gray circle) and retina (crescent) at P1 to P5. Between P6 and P10, Arf-expressing cells are absent, and the VHP/HA regresses, leaving an avascular secondary vitreous beyond P14. (Ab) Primary defects in proapoptotic machinery allow VHP/HA to persist beyond P10. Remnant vessels provide a matrix for vitreous cell accumulation (gray ovals) causing secondary PHPV manifestations of lens opacity and retinal folding/detachment. Severity of disease determined by degree of vitreous cell accumulation beyond P14. (Ac) Primary defects controlling perivascular cell accumulation in Arf−/− mice cause secondary manifestations of PHPV. (B) p19Arf may have both p53-dependent and -independent mechanisms to block vitreous cell accumulation.
The Arf−/− mouse provides an alternative pathogenetic mechanism for PHPV. In this case, the principal defect is the accumulation of perivascular cells within the VHP/HA before the vessels begin to regress (Fig. 8Ac). By virtue of their proximity to endothelial cells, the perivascular cells are likely to be a type of pericyte which provides angiogenic factors to support vessels they encase and block their involution.16,53,54 The mass itself secondarily leads to the lens and neuroretina abnormalities, which uniformly occur in the Arf−/− mouse. Although depicted as such (Fig. 8Ac), this proposed pathogenetic mechanism may not always lead to severe PHPV. For example, it is conceivable that hypomorphic mutations in Arf, its regulators, or its effectors lead to a lesser degrees of perivascular cell accumulation and milder disease.
How p19Arf normally prevents the accumulation of perivascular cells is not clear. It seems to be critically important, at least in the mouse, because the phenotype has complete penetrance and is uniformly severe. Its best-characterized function is the activation of p53 by interaction with Mdm2.27,28,55 This does not seem to be its sole activity in the vitreous, because p53−/− mice in a similar mixed genetic background have only subtle disease manifestations.23,21 Conceivably, p19Arf controls vitreous maturation in both p53-dependent and -independent mechanisms (Fig. 8B). One attractive possibility is that Arf expression in developing pericytes may influence platelet-derived growth factor (PDGF)-B–dependent signaling, which is essential for pericyte recruitment by developing vessels.56 Other p53-independent activities for p19Arf include its ability to arrest cell proliferation,57 interfere with ribosomal RNA processing,58 and block the activation of the transcription factor NF-κB.59 Further studies at earlier points in vitreous development are needed to elucidate how these or other Arf-dependent mechanisms prevent perivascular cell accumulation.
Other fundamental questions raised by our studies are by what mechanism does p19Arf normally promote VHP/HA regression and for what reasons do the vessels persist in the absence of Arf. Our findings point to two general possibilities. First, p19Arf may directly or indirectly alter pericyte biology to destabilize the underlying vessels. The temporal correlation of Arf mRNA expression through P5 followed by VHP involution from P6 to P10 is consistent with such a model.2,23 Perivascular cells are known to support endothelial cells by providing angiogenic factors such as Vegf and angiopoietin-1, and they can be a source of such destabilizing factors as Ang-2.16 Vegf expression in the retrolental tissue may contribute to the persistence of the underlying vessels. That p19Arf may repress Vegf expression in pericytes to destabilize the VHP/HA is consistent with others' findings that human p14ARF can repress a hypoxia-responsive element from the human VEGF promoter.60 A second plausible mechanism is that p19Arf primarily functions to check the accumulation of pericytes or to promote their “pruning” from the VHP/HA. This could remove a source of angiogenic factors without invoking a mechanism for p19Arf-mediated modulation of angiogenic factor expression. In addition, such an effect could also predispose the underlying vessels to direct interactions with hyalocytes as direct effectors promoting VHP/HA involution.51,52 Further studies of this mouse should reveal exact cellular and molecular mechanisms driving vascular involution in the developing vitreous and how they go awry in the absence of Arf.
Acknowledgments
The authors thank Dorothy Bush (Fig. 6; Histology Laboratory, St. Jude Children's Research Hospital [SJCRH]), Jennifer Bills (mouse genotyping), and Tie Wei (Fig. 7A) (Skapek Laboratory) for expert technical assistance; Richard A. Lang for technical advice on HVS wholemounts; Michael B. Kastan, Martine F. Roussel, and Charles J. Sherr for helpful discussions; and Martine F. Roussel and Charles J. Sherr (SJCRH) for providing access to the Arf+/GFP mice before publication31 and thus greatly facilitating the work.
Footnotes
Supported by Grants R01-EY012950 (JZ) and R01-EY014368 (SXS) from the National Eye Institute, U01-MH61971 (MMJ), Grant RSG-04-036-01-DDC (SXS) from the American Cancer Society, a grant from Research to Prevent Blindness to the Department of Ophthalmology at the University of Tennessee Health Science Center, and funding from the American Lebanese Syrian Associated Charities (ALSAC) at St. Jude Children's Research Hospital. MMJ is a Research to Prevent Blindness William and Mary Greve Scholar.
Disclosure: A.C. Martin, None; J.D. Thornton, None; J. Liu, None; X.F. Wang, None; J. Zuo, None; M.M. Jablonski, None; E. Chaum, None; F. Zindy, None; S.X. Skapek, None
The publication costs of this article were defrayed in part by page charge payment. This article must therefore be marked “advertisement” in accordance with 18 U.S.C. §1734 solely to indicate this fact.
References
- 1.Goldberg MF. Persistent fetal vasculature (PFV): an integrated interpretation of signs and symptoms associated with persistent hyperplastic primary vitreous (PHPV) LIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 1997;124:587–626. doi: 10.1016/s0002-9394(14)70899-2. [DOI] [PubMed] [Google Scholar]
- 2.Ito M, Yoshioka M. Regression of the hyaloid vessels and pupillary membrane of the mouse. Anat Embryol. 1999;200:403–411. doi: 10.1007/s004290050289. [DOI] [PubMed] [Google Scholar]
- 3.Bishop PN. Structural macromolecules and supramolecular organisation of the vitreous gel. Prog Retin Eye Res. 2000;19:323–344. doi: 10.1016/s1350-9462(99)00016-6. [DOI] [PubMed] [Google Scholar]
- 4.Reese AB. Persistent hyperplastic primary vitreous. Am J Ophthalmol. 1955;40:317–331. doi: 10.1016/0002-9394(55)91866-3. [DOI] [PubMed] [Google Scholar]
- 5.Manschot WA. Persistent hyperplastic primary vitreous. AMA Arch Ophthalmol. 1958;59:188–203. [PubMed] [Google Scholar]
- 6.Pollard ZF. Persistent hyperplastic primary vitreous: diagnosis, treatment and results. Trans Am Ophthalmol Soc. 1997;95:487–549. [PMC free article] [PubMed] [Google Scholar]
- 7.Pruett RC. The pleomorphism and complications of posterior hyperplastic primary vitreous. Am J Ophthalmol. 1975;80:625–629. doi: 10.1016/0002-9394(75)90392-x. [DOI] [PubMed] [Google Scholar]
- 8.Haddad R, Font RL, Reeser F. Persistent hyperplastic primary vitreous: a clinicopathologic study of 62 cases and review of the literature. Surv Ophthalmo. 1978;23:123–134. doi: 10.1016/0039-6257(78)90091-7. [DOI] [PubMed] [Google Scholar]
- 9.Dass AB, Trese MT. Surgical results of persistent hyperplastic primary vitreous. Ophthalmology. 1999;106:280–284. doi: 10.1016/S0161-6420(99)90066-0. [DOI] [PubMed] [Google Scholar]
- 10.Lin AE, Biglan AW, Garver KL. Persistent hyperplastic primary vitreous with vertical transmission. Ophthalmol Paediatr Genet. 1990;11:121–122. doi: 10.3109/13816819009012956. [DOI] [PubMed] [Google Scholar]
- 11.Wang MK, Phillips CI. Persistent hyperplastic primary vitreous in non-identical twins. Acta Ophthalmol. 1973;51:434–437. doi: 10.1111/j.1755-3768.1973.tb06022.x. [DOI] [PubMed] [Google Scholar]
- 12.Yu YS, Chang BL. Persistent hyperplastic primary vitreous in male twins. Kor J Ophthalmol. 1997;11:123–125. doi: 10.3341/kjo.1997.11.2.123. [DOI] [PubMed] [Google Scholar]
- 13.Frydman M, Kauschansky A, Leshem I, Savir H. Oculo-palato-cerebral dwarfism: a new syndrome. Clin Genet. 1985;27:414–419. doi: 10.1111/j.1399-0004.1985.tb02286.x. [DOI] [PubMed] [Google Scholar]
- 14.Milot J, Michaud J, Lemieus N, Allaire G, Gagnon M-M. Persistent hyperplastic primary vitreous with retinal tumor in tuberous sclerosis. Ophthalmology. 1999;106:630–634. doi: 10.1016/S0161-6420(99)90128-8. [DOI] [PubMed] [Google Scholar]
- 15.Storimans CWJM, Van Schooneveld MJ. Rieger's eye anomaly and persistent hyperplastic primary vitreous. Ophthalmic Paediatr Genet. 1989;10:257–262. doi: 10.3109/13816818909009880. [DOI] [PubMed] [Google Scholar]
- 16.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 17.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Ferreira V, Breier G, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell CA, Risau W, Drexler HCA. Regression of vessels in the tunica vasculosa lentis is initiated by coordinated endothelial apoptosis: a role for vascular endothelial growth factor as a survival factor for endothelium. Dev Dyn. 1998;213:322–333. doi: 10.1002/(SICI)1097-0177(199811)213:3<322::AID-AJA8>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 20.Hackett SF, Wiegand S, Yancopoulos G, Campohiaro PA. Angiopoietin-2 plays an important role in retinal angiogenesis. J Cell Physiol. 2002;192:182–187. doi: 10.1002/jcp.10128. [DOI] [PubMed] [Google Scholar]
- 21.Ikeda S, Hawes NL, Chang B, Avery CS, Smith RS, Nishina PM. Severe ocular abnormalities in C57BL/6 but not in 129/Sv p53-deficient mice. Invest Ophthalmol Vis Sci. 1999;40:1874–1878. [PubMed] [Google Scholar]
- 22.Reichel MB, Ali RR, D'Esposito F, et al. High frequency of persistent hyperplastic primary vitreous and cataracts in p53-deficient mice. Cell Death Differ. 1998;5:156–162. doi: 10.1038/sj.cdd.4400326. [DOI] [PubMed] [Google Scholar]
- 23.McKeller RN, Fowler JL, Cunningham JJ, et al. The Arf tumor suppressor gene promotes hyaloid vascular regression during mouse eye development. Proc Natl Acad Sci USA. 2002;99:3848–3853. doi: 10.1073/pnas.052484199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 25.Duro D, Bernard O, Della-Valle V, Berger R, Larsen CJ. A new type of p16Ink4/MTS1 gene transcript expressed in B-cell malignancies. Oncogene. 1995;11:21–29. [PubMed] [Google Scholar]
- 26.Mao L, Merlo A, Bedi G, et al. A novel p16Ink4A transcript. Cancer Res. 1995;55:2995–2997. [PubMed] [Google Scholar]
- 27.Pomerantz J, Schreiber-Agus N, Liegeois NJ, et al. The INK4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell. 1998;92:713–723. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 28.Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARK tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA. 1998;95:8292–8297. doi: 10.1073/pnas.95.14.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Xiong Y, Yarbrough WG. ARF Promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–734. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
- 30.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 31.Zindy F, Williams RT, Baudino TA, et al. Arf tumor suppressor promoter monitors latent oncogenic signals in vivo. Proc Natl Acad Sci USA. 2003;100:15930–15935. doi: 10.1073/pnas.2536808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharpless NE, Bardeesy N, Lee K-H, et al. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature. 2001;413:86–91. doi: 10.1038/35092592. [DOI] [PubMed] [Google Scholar]
- 33.Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D, DePinto RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 34.Kato M, Patel MS, Levasseur R, et al. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient for Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB. NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn. 2001;222:218–227. doi: 10.1002/dvdy.1200. [DOI] [PubMed] [Google Scholar]
- 36.Weber JD, Kuo M-L, Bothner B, et al. Cooperative signals governing Arf-Mdm2 interaction and nucleolar localization of the complex. Mol Cell Biol. 2000;20:2517–2528. doi: 10.1128/mcb.20.7.2517-2528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng YS, Rohan R, Sunday ME, Demello DE, D'Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 38.Conlon RA, Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
- 39.Sherr CJ. The Ink4a/Arf network in tumour suppression. Nat Rev Mol Cell Biol. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
- 40.Sherr CJ. Parsing Ink4a/Arf: “Pure” p16-null mice. Cell. 2001;106:531–534. doi: 10.1016/s0092-8674(01)00486-x. [DOI] [PubMed] [Google Scholar]
- 41.Warburg M. Norrie's disease: A new hereditary bilateral pseudotumour of the retina. Acta Ophthalmol. 1961;39:757–772. [Google Scholar]
- 42.Warburg M. Norrie's disease: differential diagnosis and treatment. Acta Ophthalmol. 1975;53:217–236. doi: 10.1111/j.1755-3768.1975.tb01156.x. [DOI] [PubMed] [Google Scholar]
- 43.Royer G, Hanein S, Raclin V, et al. NDP gene mutations in 14 French families with Norrie disease. Hum Mutat. 2003;22:499. doi: 10.1002/humu.9204. [DOI] [PubMed] [Google Scholar]
- 44.Schuback DE, Chen ZY, Craig IW, Breakefield XO, Sims KB. Mutations in the Norrie disease gene. Hum Mutat. 1995;5:285–292. doi: 10.1002/humu.1380050403. [DOI] [PubMed] [Google Scholar]
- 45.Hartzer MKl, Cheng M, Liu X, Shastry BS. Localization of the Norrie disease gene mRNA by in situ hybridization. Brain Res Bull. 1999;49:355–358. doi: 10.1016/s0361-9230(99)00071-4. [DOI] [PubMed] [Google Scholar]
- 46.Berger W, van de Pol D, Bachner D, et al. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum Mol Genet. 1996;5:59. doi: 10.1093/hmg/5.1.51. [DOI] [PubMed] [Google Scholar]
- 47.Xu Q, Wang Y, Dabdoub A, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 48.Chynn EW, Walton DS, Hahn LB, Dryja TP. Norrie disease. Arch Ophthalmol. 1996;114:1136–1138. doi: 10.1001/archopht.1996.01100140338018. [DOI] [PubMed] [Google Scholar]
- 49.Pendergast SD, Trese MT, Liu X, Shastry BS. Study of the Norrie disease gene in 2 patients with bilateral persistent hyperplastic primary vitreous. Arch Ophthalmol. 1998;116:381–382. [PubMed] [Google Scholar]
- 50.Maisonpierre PC, Suri C, Jones PF, et al. Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis. Science. 1997;277:55–60. doi: 10.1126/science.277.5322.55. [DOI] [PubMed] [Google Scholar]
- 51.Lang RA. Apoptosis in mammalian eye development: lens morphogenesis, vascular regression and immune privilege. Cell Death Differ. 1997;4:12–20. doi: 10.1038/sj.cdd.4400211. [DOI] [PubMed] [Google Scholar]
- 52.Lang RA, Bishop JM. Macrophages are required for cell death and tissue remodeling in the developing mouse eye. Cell. 1993;74:453–462. doi: 10.1016/0092-8674(93)80047-i. [DOI] [PubMed] [Google Scholar]
- 53.Sims DE. The pericyte: a review. Tissue Cell. 1986;18:153–174. doi: 10.1016/0040-8166(86)90026-1. [DOI] [PubMed] [Google Scholar]
- 54.Beck L, Jr, D'Amore PA. Vascular development: cellular and molecular regulation. FASEB J. 1997;11:365–373. [PubMed] [Google Scholar]
- 55.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat Cell Biol. 1999;1:20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 56.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 57.Weber JD, Jeffers JR, Rehg JE, et al. p53-independent functions of the p19Arf tumor suppressor. Genes Dev. 2000;14:2358–2365. doi: 10.1101/gad.827300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugimoto M, Kuo M-L, Roussel MF, Sherr CJ. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol Cell. 2003;11:415–424. doi: 10.1016/s1097-2765(03)00057-1. [DOI] [PubMed] [Google Scholar]
- 59.Rocha S, Campbell KJ, Perkins ND. p53- and Mdm2-independent repression of NF-kB transactivation by the ARF tumor suppressor. Mol Cell. 2003;12:15–25. doi: 10.1016/s1097-2765(03)00223-5. [DOI] [PubMed] [Google Scholar]
- 60.Fatyol K, Szalay AA. The p14ARF tumor suppressor protein facilitates nucleolar sequestration of hypoxia-inducible factor-1a (HIF-1a) and inhibits HIF-1-mediated transcription. J Biol Chem. 2001;276:28421–28429. doi: 10.1074/jbc.M102847200. [DOI] [PubMed] [Google Scholar]