

Abstract
Toxoplasma gondii is a globally distributed protozoan parasite that can infect virtually all warm-blooded animals and humans. Despite the existence of a sexual phase in the life cycle, T. gondii has an unusual population structure dominated by three clonal lineages that predominate in North America and Europe, (Types I, II, and III). These lineages were founded by common ancestors ~10,000 yr ago. The recent origin and widespread distribution of the clonal lineages is attributed to the circumvention of the sexual cycle by a new mode of transmission—asexual transmission between intermediate hosts. Asexual transmission appears to be multigenic and although the specific genes mediating this trait are unknown, it is predicted that all members of the clonal lineages should share the same alleles. Genetic mapping studies suggested that chromosome Ia was unusually monomorphic compared with the rest of the genome. To investigate this further, we sequenced chromosome Ia and chromosome Ib in the Type I strain, RH, and the Type II strain, ME49. Comparative genome analyses of the two chromosomal sequences revealed that the same copy of chromosome Ia was inherited in each lineage, whereas chromosome Ib maintained the same high frequency of between-strain polymorphism as the rest of the genome. Sampling of chromosome Ia sequence in seven additional representative strains from the three clonal lineages supports a monomorphic inheritance, which is unique within the genome. Taken together, our observations implicate a specific combination of alleles on chromosome Ia in the recent origin and widespread success of the clonal lineages of T. gondii.
Toxoplasma gondii is a globally distributed protozoan parasite that infects ~25% of the human population and a similar proportion of birds and mammals (Dubey and Beattie 1988; Hall et al. 2001). Acute infection during pregnancy can result in birth defects and the parasite may cause fatal encephalitis in immunocompromised individuals (Hall et al. 2001). However, the vast majority of infections go unnoticed or cause mild flu-like symptoms, resulting in an asymptomatic, chronic infection lasting the lifetime of the host. Importantly, the relatively quiescent tissue cyst form of the parasite in the chronic infection is readily transmitted between intermediate hosts by carnivorous or omnivorous feeding (Dubey and Beattie 1988). This is thought to be the driving force behind the recent and rapid expansion of T. gondii (Su et al. 2003).
By their very nature, parasitic organisms have evolved complex life cycles that exploit their intimate association with their hosts. For apicomplexan species, including the causative agents of malaria (Plasmodium spp.) and avian coccidiosis (Eimeria spp.), the maintenance of a sexual cycle may be vital for generating the diversity necessary to adapt to varying environments by evading host immunity and optimizing transmission and virulence (Kyes et al. 2001; Mackinnon and Read 2004). The cost of this diversity comes at the expense of being reliant on a single definitive host and the more general problem of breaking up advantageous combinations of alleles (Maynard-Smith 1978). T. gondii, arguably the most successful species in the phylum, is a striking exception because, along with the sexual cycle, the parasite can transmit asexually by ingestion of tissue cysts by intermediate hosts (Dubey and Beattie 1988). This property, termed asexual oral transmission, underpins its highly clonal population structure with only three predominant lineages (Types I, II, and III) widespread in North America and Europe, where members within each lineage are virtually identical (Darde et al. 1992; Sibley and Boothroyd 1992; Howe and Sibley 1995). Equally remarkable, when comparing between lineages, there exist only two alleles at each locus. Together, these patterns indicate that the clonal lineages arose very recently from a common set of parental strains (referred to as Adam and Eve or A&E) and subsequently underwent only limited genetic exchange (Grigg et al. 2001a; Su et al. 2003). Thus, the need to define the mechanisms and common genetic elements that underlie asexual transmission and the ubiquitous distribution of T. gondii provides a compelling rationale for genetic and comparative genomic analyses.
Experimental genetic crosses between prototypic members of the three common lineages demonstrate a Mendelian pattern of inheritance (Su et al. 2002; Khan et al. 2005). There are 14 distinct linkage groups that comprise the ~65 Mb haploid genome. The recently compiled genome map relies on the presence of frequently occurring A&E polymorphisms for 229 well-dispersed genetic markers (Su et al. 2002; Khan et al. 2005); polymorphisms occur at a frequency of ~1 in 100 bp on all chromosomes with the exception of chromosome Ia (chrIa). To our surprise, and unlike any other chromosome, genetic polymorphisms proved exceedingly difficult to identify on chrIa. Moreover, the density of polymorphic markers elsewhere in the genome (spaced approximately every 300 kb) suggests that chrIa is unique in its lack of polymorphism compared with the rest of the genome. This lack of polymorphism has been confirmed by a recent global analysis of single nucleotide polymorphisms (SNPs) identified by mapping ESTs against the whole-genome sequence of T. gondii. This analysis showed that the genome-wide polymorphism rate between the three lineages is ~0.65%, while it is only ~0.03% on chrIa (Boyle et al. 2006). These findings prompted us to generate and analyze the finished genomic sequence for chrIa and the similarly sized, but independent chromosome Ib (chrIb) from the RH (Type I) strain. We subsequently compared nucleotide composition, repeats, and genes between the two chromosomes. Analyses of the levels of polymorphisms were used to address directly and accurately the oddity of apparent low polymorphism specific to chrIa, by comparison with the ME49 clone B7 (Type II) strain. Sequencing of several regions at intervals across chrIa for an additional seven representative strains, including Type III strains, was also used to explore the generality of the observation that chrIa appears to be monomorphic.
Results
Comparison of chrIa and chrIb in the Type I strain RH reveals similar features
To provide a basis for initial comparative genomic analysis, we undertook complete sequencing of chrIa and chrIb genomic DNA from the RH strain of T. gondii. The genomic sequence for the RH strain reveals that the 1.92-Mb chrIa and the 2.01-Mb chrIb have an average G+C content of just over 52% with 211 protein-coding sequences (CDS) predicted for chrIa and 252 for chrIb (Table 1). Expression of 47% of the total predicted genes was confirmed by comparison with an extensive set of Expressed Sequence Tag (EST) data available at http://www.cbil.upenn.edu/apidots/downloads/ (Wan et al. 1996; Ajioka et al. 1998), of which 35% were identified completely. Comparative genome analyses revealed that both chrIa and chrIb have similar levels of coding and noncoding regions and contain conventional telomeres (Table 1; Supplemental material). Crucially, chrIa does not appear to be unusual in structure or content compared with chrIb, which we have used to be representative of the rest of the T. gondii genome.
Table 1.
Features of T. gondii chromosome Ia and Ib DNA sequence for the RH strain

Chromosomal comparison between RH and ME49 clone B7 reveal virtually identical chrIa
Having completed the analysis of the sequence of the Type I RH strain, we were able to compare it with the recently completed whole-genome shotgun analysis of the Type II ME49 strain undertaken at TIGR (available at http://www.tigr.org and http://www.toxodb.org/ToxoDB.shmtl). Genomic sequence alignments between the Type I RH and Type II ME49 clone B7 for both chromosomes show that each chromosome pair has the same gene content and structural elements. However, there is a very dramatic difference in the levels of polymorphism between the two chromosomes (Fig. 1; Supplemental Table 2): chrIb has 12,925 SNPs compared with only 332 SNPs on chrIa (Table 1). An analysis of the SNPs in the intron sequences of a sample of genes from different chromosomal locations estimates that about seven SNPs per kilobase are true biallelic polymorphisms (i.e., A&E) due to sequence differences between the original parents (Su et al. 2003). Since this estimate may be biased because it is based solely on intron sequence, 53.6 kb of genomic sequence from a chromosome XII-derived BAC (RH) was generated. The alignment between RH and ME49 for this region estimates 6.60 SNPs per kilo-base and predicts 13,289 SNPs due to the A&E dimorphism for chrIb, again a figure consistent with the observed number of SNPs on chrIb between these two strains. This frequency of A&E SNPs on chrIb also agrees closely with that observed for ~200 genetic markers distributed across the genome (Su et al. 2002; Khan et al. 2005) and a more recent global analysis of strain-specific SNPs (Boyle et al. 2006). In stark contrast, chrIa is virtually identical between RH and ME49, having only 0.17 SNPs per kilobase. This is likely to be an overestimate, because more than half of the apparent polymorphism is likely to be due to sequencing errors, even with the low sequencing error rate of ~1in104 bp achieved in this study. The 332 SNPs on chrIa are not distributed at random across the chromosome (P~0), largely due to the paucity of SNPs between positions 150,000 and 750,000 and at the right-hand end of chrIa (Fig. 1). The genes in the relatively polymorphism-free region in the center of the chromosome can be identified from the start positions given in Supplemental Table 3. There is no obvious bias in the functional categories of genes in these regions that would explain the lower polymorphism, although many of the genes are annotated as “unknown.” The overall low rate of polymorphism argues that chrIa is derived from a single ancestral chromosome that is extremely recent in origin.
Figure 1.

Single Nucleotide Polymorphisms (SNPs) on chromosome Ia vs. chromosome Ib for the RH vs. ME49 comparison. Distribution of SNPs (counts per 2000 bp). chrIa has 332 SNPs, chrIb has 12,925 SNPs.
Table 2.
Summary of clonal strains used for sequencing regions of chromosome Ia

chrIa is shared in all lineages
In order to assess the generality of these observations, we analyzed a Type III strain called VEG, which represents the third predominant lineage. Since whole-genome sequence is not yet available, the EST database (Li 2003) was screened and 26 scattered regions of 800–900 bp that included at least three ESTs were selected for sequencing. There were only two SNPs in 23,654 bp of chrIa sequenced from VEG (Fig. 2). This low rate of polymorphism argues that the Type III lineage also inherited the same version of chrIa without apparent crossover. In order to confirm the uniparental inheritance of chrIa among the three clonal lineages, we sequenced 12 of the 26 regions from chrIa in two additional strains from each clonal type. These strains were selected from human or animal isolates that are representative of the three clonal lineages that predominate in the USA and France (Table 2). No SNPs were found in the sequence from these additional strains (a total of 9646 bp for each strain; see Fig. 2). These data are consistent with a common inheritance of chrIa for all members of the clonal lineages, which predominate in North America and Europe.
Figure 2.

Composition of chrIa for clonal (I, II, III) strains based on sequence analysis of 26 regions. Yellow represents areas not sequenced in the alternative strains, white represents sequenced areas that are identical to RH and ME49, and red represents regions containing a single SNP. Twelve regions are represented in all nine strains. Only two SNPs are seen in these 12 regions, which represents a total of 86,814 bp of sequence across the nine strains.
Time of origin of the common chrIa is coincident with origin of the clonal lineages
The combined data show two SNPs in 128,146 total bp, where 86,814 bp represents sequence from the 12 regions available for all nine strains (Fig. 2;Table 2). This very low rate of polymorphism of 2/86,814 bp for chrIa is strikingly similar to the overall rate of unique SNPs between the three lineages. We have previously shown that while A&E polymorphisms occur across the genome at about 1/100 bp, when these biallelic polymorphisms are removed, the remaining unique polymorphism rate is on the order of 2/90,000 bp (Su et al. 2003). Based on the assumption that each strain evolves independently and these polymorphisms represent random (Poisson) neutral mutations that have occurred since their common ancestry (Rich et al. 1998), we have previously estimated that the three lineages originated ~10,000 yr ago (Su et al. 2003). Using this same model, the simplest explanation of our observations is that chrIa was inherited in common by all three lineages at a time coincident with their recent origin.
Repression of meiotic recombination does not explain the monomorphic chrIa
How might a virtually monomorphic chrIa be established and maintained within an otherwise polymorphic genome? A monomorphic chrIa is currently maintained in the population because the frequency of a sexual encounter with anything other than a member of a clonal lineage is very low (i.e., when crosses do occur, chrIa is effectively “selfing” in the vast majority of crosses). However, this does not explain how the current monomorphic chrIa was generated and initially propagated in a sexually reproducing polymorphic population. In sexually reproducing populations, population genetic theory suggests that chromosomes/chromosomal regions where meiotic recombination is suppressed may have reduced levels of polymorphism (Maynard-Smith and Haigh 1974; Charlesworth et al. 1993). This could be due to a selective sweep or purifying selection as proposed for the small fourth chromosome of Drosophila melanogaster (Berry et al. 1991; Jensen et al. 2002; Bartolome and Maside 2004), and similarly, it is possible that suppression of recombination could have led to the establishment of a monomorphic chrIa in T. gondii. To test whether a similar process might occur in T. gondii, we analyzed the segregation of SNPs on chrIa in the progeny of a genetic cross between the Type I and Type III lineages as previously reported (Khan et al. 2005). Our analysis showed that 12 of the 31 progeny showed one or more crossing-over events in a single meiosis on chrIa (Fig. 3) and, in experimental crosses, it is observed to have the highest frequency of recombination of any T. gondii chromosome (Khan et al. 2005). Thus, there is no physical suppression of recombination on chrIa. Nevertheless, in experimental crosses of T. gondii, individual chromosomes can be inherited uniparentally (i.e., without crossing over). This raises the possibility that it is simply the result of chance that chrIa is the only chromosome/chromosomal region common between the three clonal lineages, particularly if they are related by only a few crosses.
Figure 3.

Progeny from genetic crosses between the Type I and Type III lineage were analyzed using six polymorphic markers on each of chrIa and chrIb. The resulting allele patterns are shown for each of 31 progeny at each locus (solid boxes, Type III lineage; open boxes, Type I lineage). Progeny where recombination has occurred are indicated at the bottom by an asterisk. The maps at the top represent the linear genetic maps for each chromosome with the markers shown on the right and the genetic distances shown on the left in centiMorgans.
Chance inheritance of chrIa is highly unlikely
A study of EST-identified SNPs by Boyle et al. (2006) revealed a surprising pattern of SNP distribution across the genome— haplotype blocks are conserved across large regions of the genome. Further analysis of these patterns indicates that the relationship between the clonal lineages might be explained by as few as two separate crosses. They propose that the existence of three clonal lineages were generated by two crosses, in which one lineage (Type II) is a common parent of the other two, and that the subsequent lineages have undergone little mixing since this recent origin. Given the two-cross model, it is expected that some chromosomes will not recombine and will be shared in their entirety between the three lineages, some chromosomes will have shared regions, and some will have no shared regions. Experimental crosses with T. gondii predict a recombination rate of 0.4 (Khan et al. 2005); hence, the expected frequencies of these three classes are 0.09, 0.36, and 0.55, respectively. The probability of observing a single intact chromosome with no other chromo-some having shared regions can be calculated using the trinomial distribution:
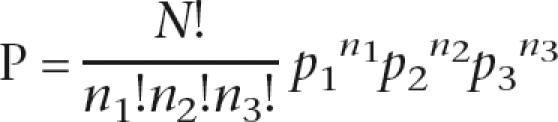 |
where N is the number of chromosomes, n 1, n 2, n 3 are the observed numbers of shared chromosomes, chromosomes with shared sections, and chromosomes with no shared sections, and p 1, p 2, p 3 are the probabilities of observing these three types of chromosomes on the two-cross model.
Therefore:
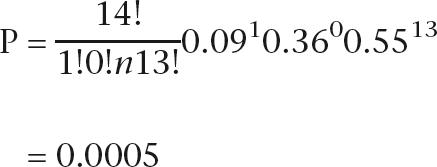 |
Collapsing the first two classes to give a minimum expected number greater than five, the expected numbers of chromosomes with shared material and with no shared material are 7.7 and 6.3 compared with observations of 1 and 13, giving 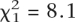 (P = 0.004). Given the two-cross model, the observed pattern is highly unlikely, with fewer shared chromosome/chromosomal regions than expected by chance. If more than two crosses led to the extant clonal lineages, the probability is further reduced, so it is extremely unlikely that this pattern would occur by chance.
(P = 0.004). Given the two-cross model, the observed pattern is highly unlikely, with fewer shared chromosome/chromosomal regions than expected by chance. If more than two crosses led to the extant clonal lineages, the probability is further reduced, so it is extremely unlikely that this pattern would occur by chance.
Discussion
Comparative genome analysis of the two smallest chromosomes of T. gondii, chrIa and chrIb reveals a similar chromosome organization and content of genes and repeats between two of the three common lineages of T. gondii. Strikingly, analysis of the frequency of polymorphisms within these chromosomes reveals a markedly different pattern. chrIb contains the expected frequency of SNPs seen in other regions of the genome of approximately one change per 100 bp. In contrast, the extremely low polymorphism rate on chrIa argues that the Type I and Type II lineages acquired this entire chromosome from a common source. While our sampling of the Type III lineages and other representative strains is more limited, given the relatively low rate of recombination in T. gondii, it is unlikely that the intervening regions differ from this pattern. Hence, our data suggest that all members of the three clonal lineages that predominate in North America and Europe possess the identical chrIa. Moreover, the estimated time of the establishment of this monomorphic chromosome is coincident with the origin of these clonal lineages ~10,000 yr ago (Su et al. 2003). Although the clonal lineages may be related by as few as two crosses, probability estimates indicate that the common inheritance of chrIa alone is extremely unlikely to have arisen by chance. Rather, our analyses support a model where natural selection acting on the products of a recombination event in the wild led to the unusual fixation of chrIa.
Despite the results from the experimental cross that argue that chrIa monomorphism is not due to chromosome-specific repression of meiotic recombination, variations of a positive selective sweep model remain a possibility. Examination of a cross between relatively avirulent Type II and Type III T. gondii strains demonstrates that recombination can generate progeny far more virulent than either parent (Grigg et al. 2001a; Grigg and Suzuki 2003); not surprisingly, virulence involves multiple genes, as shown by linkage mapping studies (Su et al. 2002). The monomorphic chrIa could be the result of just such a recombination process, in which the generation of a particular combination of alleles from different parents led to successful expansion of the clonal lineages bearing this unique chromosome. While the precise mechanism at play is uncertain, one plausible model suggests that this event first allowed or greatly enhanced asexual oral transmission between intermediate hosts (Su et al. 2003). Parasites capable of such asexual oral transmission would be expected to expand rapidly via clonal propagation, thus limiting, but not excluding exposure to sexual, meiotic recombination. The special combination of alleles on chrIa would subsequently be maintained together by asexual growth, while still allowing the rare opportunity for recombination to establish related but distinct lineages (Fig. 4). At present, we cannot predict which gene or genes on chrIa might be responsible for the success of the clonal lineages, due to the fact that this chromosome has been maintained in entirety in all of the strains we have examined (all regions are equally associated with the phenotype; annotated genes are listed in Supplemental Table 3). Successful testing of this model will rely on identifying ancestral or exotic strains that express a wider range of asexual oral transmission phenotypes that we predict will have more variable chrIa genotypes.
Figure 4.

Model of the evolutionary histories of chrIa and chrIb. (A) Original A&E cross. (A) blue; (E) pink; (shorter bar) chrIa; (longer bar) chrIb. (B) Recombinant progeny from a restricted gene pool. A special chrIa (black outline), generated by recombination, carries allele(s) that allow or enhance transmission between intermediate hosts, permitting asexual propagation and suppressing recombination events that might break up the special combination of alleles. (C) Asexual growth expands the lineage in the population. Infrequent sexual events with highly recombinant relatives from the original cross allow parasites with the special chromosome Ia to acquire highly recombinant chromosomes including chromosome Ib. These new recombinant parasites can now expand in the population through asexual growth. (D) Natural selection for asexual growth maintains the special chrIa intact and three different clonal lineages (i.e., Types I, II, and III) expand and become predominant in the population.
Asexual transmission now affords T. gondii with population dynamics more akin to zoonotic viruses, where rare recombinants can spread via direct horizontal transmission (Worobey and Holmes 1999). This has reached a culmination in the example of the three clonal lineages that predominate in North America and Europe, where widely different geographic regions share identical mutilocus genotypes. However, recent studies have revealed that the population structure of T. gondii in South America differs from this pattern and appears genetically more divergent (Ajzenberg et al. 2004; Lehmann et al. 2004; Khan et al. 2006; Su et al. 2006). It is presently uncertain whether a clonal population structure also predominates in these regions, albeit dominated by different genotypes, or whether they are truly panmictic. More severe forms of toxoplasmosis in humans have been associated with so-called atypical strains from such regions (Darde et al. 1998; Grigg et al. 2001b; Khan et al. 2006) and it is uncertain to what extent these strains share the monomorphic chrIa described here. Despite a recently derived and highly clonal population structure, the continued recombination of T. gondii in the wild, while infrequent, may lead to strains that acquire new pathogenic mechanisms and can readily expand in the population. Further investigations into the genetic basis of this and other mechanisms that profoundly alter transmissibility and virulence will provide a greater understanding of the evolution of toxoplasmosis and global parasitic disease in general.
Methods
Parasite culture for DNA preparation
Tachyzoites of the RH (American Type Culture Collection [ATCC] # 50174) strain were propagated in monolayers of human foreskin fibroblasts as described previously (Su et al. 2003). Parasite harvesting, DNA isolation, and shotgun library preparation for chrIa and chrIb are described in the Supplemental material.
Whole-chromosome sequencing and annotation
Whole-chromosome shotgun sequencing (Hall et al. 2003) was used to determine the 1.92 and 2.01 Mb DNA sequences of chrIa and chrIb for T. gondii strain RH. The methods used in annotating the RH sequence are provided in the Supplemental material.
Comparison of strain RH with strain ME49
The manually finished sequence of chrIa and chrIb from the RH strain (http://www.sanger.ac.uk/Projects/T_gondii/) was compared with a whole-genome shotgun assembly of the Type II ME49 (B7 clone) strain generated by TIGR (preliminary sequence data was obtained from The Institute for Genomic Research Web site at http://www.tigr.org). The polymorphisms were identified and classified as SNPs, insertion/ deletions, and replacements (Supplemental material). Su et al. (2003) analyzed the frequency of SNPs in non-coding regions (including 11 introns and the ITS1 region) in four Type I, three Type II, and three Type III strains. The frequency of SNPs between Type I and Type II strains was estimated from Table S3 of their Supplemental online materials as 28 SNPs per 4067 bp or 6.9 SNPs per kilobase compared.
The SNPs are expected to follow a Poisson distribution if they are randomly distributed across chrIa. The chromosome was divided into “bins” and the number of SNPs in each bin were counted. Agreement with the Poisson distribution was assessed using the Poisson dispersion test. The result was not sensitive to the bin size chosen.
Recombination parameters for chrIa and chrIb
To identify genetic markers for chrIa, we analyzed 10-kb sequences spaced ~500 kb apart along the chromosome. We compared the genomes of Type I and Type II strains using CLUSTALX (Higgins et al. 1996) to identify potential SNPs. Once SNPs had been identified, we amplified and sequenced genomic regions flanking SNPs from all three lineages using the type strains—RH (Type I), ME49 (Type II), and VEG (Type III) (Supplemental material). CLUSTALX was used to align the sequences and identify strain-specific polymorphisms. Further analysis of these markers is described in the Supplemental material. In total, nine polymorphic markers were developed on chrIa. However, due to the biallelic pattern of T. gondii, only six of these detect differences between the Type I and Type III lineages, while three of them detect differences between the Type II and Type III lineages. Due to the larger number of markers and progeny available for the IxIII cross, we analyzed the segregation of markers in the 31 progeny from a previous cross (Su et al. 2002). Progeny were analyzed for segregation of polymorphisms on chrIa and chrIb by PCR amplification, restriction enzyme digestion, and gel electrophoresis to score alleles. The resulting genotypes were analyzed using MapMAKER (Lander et al. 1987) to generate genetic linkage maps of the chromosomes.
ACKNOWLEDGMENTS
We acknowledge the support of the Wellcome Trust, the National Institutes of Health, the Wellcome Trust Sanger Institute core sequencing and informatics groups, the Washington University Genome Sequencing Center, and The Institute for Genomic Research. We thank Andrew Berry, Wally Gilks, John Boothroyd, and Jon Boyle for discussion and review of the manuscript. Preliminary sequence data for the Type II ME49 (B7 clone) strain of Toxoplasma gondii was obtained from The Institute for Genomic Research Web site at http://www.tigr.org. Sequencing of Toxoplasma gondii was funded by the National Institute of Allergy and Infectious Disease.
Footnotes
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.5318106.
Supplemental material is available online at www.genome.org. The sequence data from this study have been submitted to EMBL under accession nos. AM055942 and AM055943. Accession numbers for full-length cDNAs associated with chromosomes Ia and Ib are AK223637–AK223774.
REFERENCES
- Ajioka J.W., Boothroyd J.C., Brunk B.P., Hehl A., Hillier L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Boothroyd J.C., Brunk B.P., Hehl A., Hillier L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Brunk B.P., Hehl A., Hillier L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Hehl A., Hillier L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Hillier L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Manger I.D., Marra M., Overton G.C., Roos D.S., Wan K.L., Marra M., Overton G.C., Roos D.S., Wan K.L., Overton G.C., Roos D.S., Wan K.L., Roos D.S., Wan K.L., Wan K.L., et al. Gene discovery by EST sequencing in Toxoplasma gondii reveals sequences restricted to the Apicomplexa. Genome Res. 1998;8:18–28. doi: 10.1101/gr.8.1.18. [DOI] [PubMed] [Google Scholar]
- Ajzenberg D., Banuls A.L., Su C., Dumetre A., Demar M., Carme B., Darde M.L., Banuls A.L., Su C., Dumetre A., Demar M., Carme B., Darde M.L., Su C., Dumetre A., Demar M., Carme B., Darde M.L., Dumetre A., Demar M., Carme B., Darde M.L., Demar M., Carme B., Darde M.L., Carme B., Darde M.L., Darde M.L. Genetic diversity, clonality and sexuality in Toxoplasma gondii . Int. J. Parasitol. 2004;34:1185–1196. doi: 10.1016/j.ijpara.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Bartolome C., Maside X., Maside X. The lack of recombination drives the fixation of transposable elements on the fourth chromosome of Drosophila melanogaster . Genet. Res. 2004;83:91–100. doi: 10.1017/s0016672304006755. [DOI] [PubMed] [Google Scholar]
- Berry A.J., Ajioka J.W., Kreitman M., Ajioka J.W., Kreitman M., Kreitman M. Lack of polymorphism on the Drosophila fourth chromosome resulting from selection. Genetics. 1991;129:1111–1117. doi: 10.1093/genetics/129.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle J.P., Rajasekar B., Saeij J.P., Ajioka J.W., Berriman M., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Rajasekar B., Saeij J.P., Ajioka J.W., Berriman M., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Saeij J.P., Ajioka J.W., Berriman M., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Ajioka J.W., Berriman M., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Berriman M., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Paulsen I., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Roos D.S., Sibley L.D., White M.W., Boothroyd J.C., Sibley L.D., White M.W., Boothroyd J.C., White M.W., Boothroyd J.C., Boothroyd J.C. Just one cross appears capable of dramatically altering the population biology of an eukaryotic pathogen like Taxoplasma gondii . Proc. Natl. Acad. Sci. 2006;103:10514–10519. doi: 10.1073/pnas.0510319103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Morgan M.T., Charlesworth D., Morgan M.T., Charlesworth D., Charlesworth D. The effect of deleterious mutations on neutral molecular variation. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darde M.L., Bouteille B., Pestre-Alexandre M., Bouteille B., Pestre-Alexandre M., Pestre-Alexandre M. Isoenzyme analysis of 35 Toxoplasma gondii isolates and the biological and epidemiological implications. J. Parasitol. 1992;78:786–794. [PubMed] [Google Scholar]
- Darde M.L., Villena I., Pinon J.M., Beguinot I., Villena I., Pinon J.M., Beguinot I., Pinon J.M., Beguinot I., Beguinot I. Severe toxoplasmosis caused by a Toxoplasma gondii strain with a new isoenzyme type acquired in French Guyana. J. Clin. Microbiol. 1998;36 doi: 10.1128/jcm.36.1.324-324.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey J.P., Beattie C.P., Beattie C.P. Toxoplasmosis of animals and man. CRC Press; Boca Raton, FL.: 1988. [Google Scholar]
- Grigg M.E., Suzuki Y., Suzuki Y. Sexual recombination and clonal evolution of virulence in. Toxoplasma. Microbes Infect. 2003;5:685–690. doi: 10.1016/s1286-4579(03)00088-1. [DOI] [PubMed] [Google Scholar]
- Grigg M.E., Bonnefoy S., Hehl A.B., Suzuki Y., Boothroyd J.C., Bonnefoy S., Hehl A.B., Suzuki Y., Boothroyd J.C., Hehl A.B., Suzuki Y., Boothroyd J.C., Suzuki Y., Boothroyd J.C., Boothroyd J.C. Success and virulence in Toxoplasma as the result of sexual recombination between two distinct ancestries. Science. 2001a;294:161–165. doi: 10.1126/science.1061888. [DOI] [PubMed] [Google Scholar]
- Grigg M.E., Ganatra J., Boothroyd J.C., Margolis T.P., Ganatra J., Boothroyd J.C., Margolis T.P., Boothroyd J.C., Margolis T.P., Margolis T.P. Unusual abundance of atypical strains associated with human ocular toxoplasmosis. J. Infect. Dis. 2001b;184:633–639. doi: 10.1086/322800. [DOI] [PubMed] [Google Scholar]
- Hall S.M., Ryan M., Buxton D., Ryan M., Buxton D., Buxton D. Epidemiology. In: Joynson D.H.M., editor; Wreghitt T.G., editor. Toxoplasmosis: A comprehensive clinical guide. Cambridge University Press; Cambridge, UK: 2001. pp. 58–124. [Google Scholar]
- Hall N., Berriman M., Lennard N.J., Harris B.R., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Berriman M., Lennard N.J., Harris B.R., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Lennard N.J., Harris B.R., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Harris B.R., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Hertz-Fowler C., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Bart-Delabesse E.N., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Gerrard C.S., Atkin R.J., Barron A.J., Bowman S., Atkin R.J., Barron A.J., Bowman S., Barron A.J., Bowman S., Bowman S., et al. The DNA sequence of chromosome I of an African trypanosome: Gene content, chromosome organisation, recombination and polymorphism. Nucleic Acids Res. 2003;31:4864–4873. doi: 10.1093/nar/gkg674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins D.G., Thompson J.D., Gibson T.J., Thompson J.D., Gibson T.J., Gibson T.J. Using CLUSTAL for multiple sequence alignments. Methods Enzymol. 1996;266:383–402. doi: 10.1016/s0076-6879(96)66024-8. [DOI] [PubMed] [Google Scholar]
- Howe D.K., Sibley L.D., Sibley L.D. Toxoplasma gondii comprises three clonal lineages: Correlation of parasite genotype with human disease. J. Infect. Dis. 1995;172:1561–1566. doi: 10.1093/infdis/172.6.1561. [DOI] [PubMed] [Google Scholar]
- Jensen M.A., Charlesworth B., Kreitman M., Charlesworth B., Kreitman M., Kreitman M. Patterns of genetic variation at a chromosome 4 locus of Drosophila melanogaster and D. simulans . Genetics. 2002;160:493–507. doi: 10.1093/genetics/160.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Taylor S., Su C., Mackey A.J., Boyle J., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Taylor S., Su C., Mackey A.J., Boyle J., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Su C., Mackey A.J., Boyle J., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Mackey A.J., Boyle J., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Boyle J., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Cole R., Glover D., Tang K., Paulsen I.T., Berriman M., Glover D., Tang K., Paulsen I.T., Berriman M., Tang K., Paulsen I.T., Berriman M., Paulsen I.T., Berriman M., Berriman M., et al. Composite genome map and recombination parameters derived from three archetypal lineages of Toxoplasma gondii . Nucleic Acids Res. 2005;33:2980–2992. doi: 10.1093/nar/gki604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A., Jordan C., Muccioli C., Vallochi A.L., Rizzo L.V., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Jordan C., Muccioli C., Vallochi A.L., Rizzo L.V., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Muccioli C., Vallochi A.L., Rizzo L.V., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Vallochi A.L., Rizzo L.V., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Rizzo L.V., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Belfort R., Jr., Vitor R.W., Silveira C., Sibley L.D., Vitor R.W., Silveira C., Sibley L.D., Silveira C., Sibley L.D., Sibley L.D. Genetic divergence of Toxoplasma gondii strains associated with ocular toxoplasmosis, Brazil. Emerg. Infect. Dis. 2006;12:942–949. doi: 10.3201/eid1206.060025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyes S., Horrocks P., Newbold C., Horrocks P., Newbold C., Newbold C. Antigenic variation at the infected red cell surface in malaria. Annu. Rev. Microbiol. 2001;55:673–707. doi: 10.1146/annurev.micro.55.1.673. [DOI] [PubMed] [Google Scholar]
- Lander E.S., Green P., Abrahamson J., Barlow A., Daly M.J., Lincoln S.E., Newburg L., Green P., Abrahamson J., Barlow A., Daly M.J., Lincoln S.E., Newburg L., Abrahamson J., Barlow A., Daly M.J., Lincoln S.E., Newburg L., Barlow A., Daly M.J., Lincoln S.E., Newburg L., Daly M.J., Lincoln S.E., Newburg L., Lincoln S.E., Newburg L., Newburg L. MAPMAKER: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics. 1987;1:174–181. doi: 10.1016/0888-7543(87)90010-3. [DOI] [PubMed] [Google Scholar]
- Lehmann T., Graham D.H., Dahl E.R., Bahia-Oliveira L.M.G., Gennari S.M., Dubey J.P., Graham D.H., Dahl E.R., Bahia-Oliveira L.M.G., Gennari S.M., Dubey J.P., Dahl E.R., Bahia-Oliveira L.M.G., Gennari S.M., Dubey J.P., Bahia-Oliveira L.M.G., Gennari S.M., Dubey J.P., Gennari S.M., Dubey J.P., Dubey J.P. Variation in the structure of Toxoplasma gondii and the roles of selfing, drift, and epistatic selection in maintaining linkage disequilibria. Infect. Genet. Evol. 2004;4:107–114. doi: 10.1016/j.meegid.2004.01.007. [DOI] [PubMed] [Google Scholar]
- Li L., Brunk B.P., Kissinger J.C., Pape D., Tang K., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Brunk B.P., Kissinger J.C., Pape D., Tang K., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Kissinger J.C., Pape D., Tang K., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Pape D., Tang K., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Tang K., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Cole R.H., Martin J., Wylie T., Dante M., Fogarty S.J., Martin J., Wylie T., Dante M., Fogarty S.J., Wylie T., Dante M., Fogarty S.J., Dante M., Fogarty S.J., Fogarty S.J., et al. Gene discovery in the apicomplexa as revealed by EST sequencing and assembly of a comparative gene database. Genome Res. 2003;13:443–454. doi: 10.1101/gr.693203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon M.J., Read A.F., Read A.F. Virulence in malaria: An evolutionary viewpoint. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:965–986. doi: 10.1098/rstb.2003.1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard-Smith J. The Evolution of Sex. Cambridge University Press; Cambridge, UK: 1978. [Google Scholar]
- Maynard-Smith J., Haigh J., Haigh J. The hitchhiking effect of a favorable gene. Genet. Res. 1974;23:23–35. [Google Scholar]
- Rich S.M., Licht M.C., Hudson R.R., Ayala F.J., Licht M.C., Hudson R.R., Ayala F.J., Hudson R.R., Ayala F.J., Ayala F.J. Malaria's Eve: Evidence of a recent population bottleneck throughout the world populations of Plasmodium falciparum . Proc. Natl. Acad. Sci. 1998;95:4425–4430. doi: 10.1073/pnas.95.8.4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibley L.D., Boothroyd J.C., Boothroyd J.C. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature. 1992;359:82–85. doi: 10.1038/359082a0. [DOI] [PubMed] [Google Scholar]
- Su C., Howe D.K., Dubey J.P., Ajioka J.W., Sibley L.D., Howe D.K., Dubey J.P., Ajioka J.W., Sibley L.D., Dubey J.P., Ajioka J.W., Sibley L.D., Ajioka J.W., Sibley L.D., Sibley L.D. Identification of quantitative trait loci controlling acute virulence in Toxoplasma gondii . Proc. Natl. Acad. Sci. 2002;99:10753–10758. doi: 10.1073/pnas.172117099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C., Evans D., Cole R.H., Kissinger J.C., Ajioka J.W., Sibley L.D., Evans D., Cole R.H., Kissinger J.C., Ajioka J.W., Sibley L.D., Cole R.H., Kissinger J.C., Ajioka J.W., Sibley L.D., Kissinger J.C., Ajioka J.W., Sibley L.D., Ajioka J.W., Sibley L.D., Sibley L.D. Recent expansion of Toxoplasma through enhanced oral transmission. Science. 2003;299:414–416. doi: 10.1126/science.1078035. [DOI] [PubMed] [Google Scholar]
- Su C., Zhang X., Dubey J.P., Zhang X., Dubey J.P., Dubey J.P. Genotyping of Toxoplasma gondii by multilocus PCR-RFLP markers: A high resolution and simple method for identification of parasites. Int. J. Parasitol. 2006;36:841–848. doi: 10.1016/j.ijpara.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Wan K.L., Blackwell J.M., Ajioka J.W., Blackwell J.M., Ajioka J.W., Ajioka J.W. Toxoplasma gondii expressed sequence tags: Insight into tachyzoite gene expression. Mol. Biochem. Parasitol. 1996;75:179–186. doi: 10.1016/0166-6851(95)02524-3. [DOI] [PubMed] [Google Scholar]
- Worobey M., Holmes E.C., Holmes E.C. Evolutionary aspects of recombination in RNA viruses. J. Gen. Virol. 1999;80:2535–2543. doi: 10.1099/0022-1317-80-10-2535. [DOI] [PubMed] [Google Scholar]