


Abstract
Here we show that regions upstream of first transcribed genes have oligonucleotide signatures that distinguish them from regions upstream of genes in the middle of operons. Databases of experimentally confirmed transcription units do not exist for most genomes. Thus, to expand the analyses into genomes with no experimentally confirmed data, we used genes conserved adjacent in evolutionarily distant genomes as representatives of genes inside operons. Likewise, we used divergently transcribed genes as representative examples of first transcribed genes. In model organisms, the trinucleotide signatures of regions upstream of these representative genes allow for operon predictions with accuracies close to those obtained with known operon data (0.8). Signature-based operon predictions have more similar phylogenetic profiles and higher proportions of genes in the same pathways than predicted transcription unit boundaries (TUBs). These results confirm that we are separating genes with related functions, as expected for operons, from genes not necessarily related, as expected for genes in different transcription units. We also test the quality of the predictions using microarray data in six genomes and show that the signature-predicted operons tend to have high correlations of expression. Oligonucleotide signatures should expand the number of tools available to identify operons even in poorly characterized genomes.
INTRODUCTION
In Escherichia coli, regions upstream of first transcribed genes contain higher densities of sigma-70 promoter-like signals than both coding regions and intergenic regions downstream of convergently-transcribed genes (1). Thus, differences in promoter-like signals of upstream regions might help predict operons (2,3), stretches of genes in the same-strand transcribed into a single messenger RNA. Sigma-70 is not the only sigma factor in Prokaryotes and examples of promoters for other sigma factors are scarce. Still, a consequence of the high concentration of sigma-70 or other promoter-like signals within promoter regions (PRs) might be a bias in oligonucleotide signatures. Oligonucleotide signatures might also be different at regions upstream of genes inside operons (see Figure 1). Furthermore, differences in oligonucleotide signatures might result from other characteristics of PRs, such as increased curvature (4–12), higher stacking energies (higher stacking energies mean the regions are easier to melt) (11,13), and higher AT content (9,11,14). Signatures for upstream regions are available as soon as the genome annotation is ready. Thus, signatures might constitute an alternative method for overall operon predictions across Prokaryotes.
Figure 1.
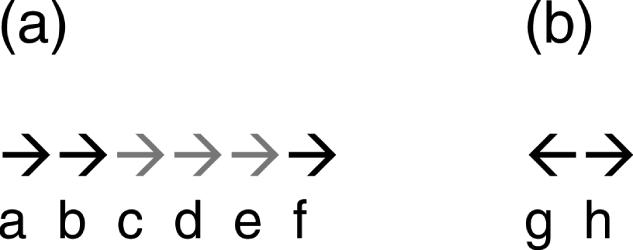
Adjacent genes and upstream regions. (a) The arrows represent a directon, a stretch of adjacent genes in the same-strand with no intervening gene in the opposite strand. The gray arrows represent operon ‘cde’. Pairs of genes within operons (WO pairs) would be pairs ‘cd’ and ‘de’. OJs would be the regions upstream of genes ‘d’ and ‘e’. Pairs ‘bc’ and ‘ef’ would be same-strand TUBs. PRs would be those upstream of genes ‘c’ and ‘f’. (b) Genes ‘g’ and ‘h’ are divergently transcribed. The regions upstream of genes ‘g’ and ‘h’ are PRs of divergently transcribed genes (dPRs).
In this work we show that densities of sigma-70 promoter-like signals distinguish co-directional transcription unit boundaries (TUBs) from operon junctions (OJs) in the genomes of E.coli and Bacillus subtilis. Then we show that oligonucleotide signatures have improved accuracies in operon predictions over those obtained with promoter-like signals. We expand the work to genomes with no experimentally characterized operons using regions upstream of divergently transcribed genes, forcefully TUBs, and regions between highly conserved co-directional genes, most probably in operons (15–17) as training sets to learn oligonucleotide signatures. We evaluate the genome-wide predictions obtained by this approach using diverse functional genomics data and demonstrate the capability of this method to produce high-quality operon predictions across genomes. (See Figure 1).
MATERIALS AND METHODS
Promoter-signal densities
We calculated the density of promoter-signals using a strategy published elsewhere (1). Briefly, with CONSENSUS (18) we obtained weight matrices for both the −10 and the −35 boxes of the Sigma-70 promoters of E.coli K12 reported in RegulonDB (19,20). With these matrices, we used PATSER (18) to calculate the average scores and SDs of the set of known promoters. Then, we scanned the −200 to −10 regions upstream of all gene starts in both E.coli and B.subtilis accepting promoter-signals formed of −10 and −35 boxes with scores equal or higher than the average minus 2.5 standard deviations of the boxes in known promoters. We obtained promoter-signal density as number of signals divided by number of bases (the data is available at http://tikal.ccg.unam.mx/sarath/sig_predictions/).
Upstream regions (Figure 1)
Known OJs are regions −200 to −10 bp from the start codon of genes inside experimentally verified operons. Known PRs consist of regions upstream of first transcribed genes (also −200 to −10). To find both of these kinds of regions we used the current dataset of transcription units of E.coli K12 (21) found in RegulonDB (19,20), and a dataset of experimentally verified transcription units of B.subtilis (http://odb.kuicr.kyoto-u.ac.jp/). With these data we found adjacent co-directional TUB pairs, consisting of the last gene in one transcription unit and the first in the next (the latter would be a first transcribed gene by definition), and we also found adjacent pairs of genes in operons (WO pairs), as described previously (22,23).
Conserved operon junctions (cOJs) are regions upstream of genes predicted to be inside operons by their conservation of adjacency in evolutionarily distant genomes as described elsewhere (17). We used predictions obtained at a confidence value of 0.95 (17). Regions upstream of divergently transcribed genes were representatives of regions upstream of first transcribed genes. To avoid overrepresentation we used the upstream regions of only one of the genes chosen randomly when the separation between the genes was <300 bp. If the space between the divergent genes was ≥300 bp we used the upstream regions of both genes as promoter regions (dPR).
Operon predictions based on intergenic distances
We predicted operons by intergenic distances using a method described previously (22,23). The method is based on log-likelihoods (dist-LLHs) calculated at different distance intervals measured in base pairs (23), and it has been shown to be successful in most Prokaryotes (22).
Oligonucleotide signatures
We built oligonucleotide signatures for each region upstream of all annotated genes in the set of about 330 Prokaryotic genomes available at the NCBI Reference Sequence (RefSeq) Database (24,25) (rsync://rsync.ncbi.nih.gov/genomes/Bacteria/) on April 2006. For individual signatures, we counted the occurrence of each possible, overlapping, oligonucleotide along the −200 to −10 regions of each gene. The size of regions were chosen on the basis of preliminary tests showing that the sequence from −10 to 0 made the OJs and PRs signatures look more alike, probably due to the presence of the ribosome-binding site (data not shown). The overall signatures are defined as the average occurrences of each oligonucleotide within each dataset (e.g all OJs or all PRs). We calculated dinucleotide and trinucleotide signatures. Longer oligonucleotides make the statistics problematic. For instance, if we were to use tetranucleotides, therewould be 44 = 256 possible combinations. A region 190 bp in length would contain 190 − 4 = 186 overlapping tetranucleotides, much less than the number of possible combinations.
The distance of the signature of each upstream region to the overall OJ-signature or to the overall PR-signature was estimated using χ2, which is a common method to test if a sample belongs to a given population.
where Oi and Ei are the observed and expected oligonucleotides frequencies, respectively. Ei is the frequency of the oligonucleotide in the overall signatures. The number of oligonucleotide types in the signature is k (64 for trinucleotide signatures). Since the OJ and PR oligonucleotide signatures have the same degrees of freedom, the X2 of each of them can be directly compared. Thus, we can calculate the log-likelihood of a co-directional pair to belong to the OJ sample as follows.
where and correspond to the χ2 values calculated with respect to the oligonucleotide signatures of PRs and of OJs, respectively.
Phylogenetic profile analyses
The phylogenetic profiles used here consist of vectors where each item represents either the presence (number 1) or the absence (number 0) of an ortholog to the gene in a given genome (26). To compare the phylogenetic profiles of any pair of genes we calculated mutual information (MI) in bits. The MI for two vectors I and J is defined as (27):
where Pij is the proportion of a given pair ij in the alignment of vectors I and J, Pi is the proportion of the value i in vector I and Pj the proportion of j in vector J.
To build the phylogenetic profiles we found orthologs in a non-redundant dataset of genomes built as explained elsewhere (17,22). Our working definition of orthology consisted of reciprocal-best hits and fusions as described elsewhere (22).
RESULTS
We used four main kinds of upstream regions (−200 to −10 from start codon, see also Figure 1): (i) known OJs are regions upstream of genes inside experimentally verified operons; (ii) known PRs consist of regions upstream of first transcribed genes; (iii) cOJs are regions upstream of genes predicted to be inside operons by their conservation of adjacency in evolutionarily distant genomes (17); (iv) regions upstream of divergently transcribed genes (dPRs) are representatives of regions upstream of first transcribed genes. Oligonucleotide signatures consist of the counts of overlapping oligonucleotides along these regions. See Materials and Methods for further details. We applied the signatures of these different kinds of regions to predict whether agiven pair of adjacent genes in the same-strand (co-directional pair), is in an operon (WO pair) or at a TUB pair. To measure the quality of any predictions we calculated sensitivity [true positives/(true positives + false negatives)], specificity [true negatives/(true negatives + false positives)] and accuracy (here the average of sensitivity and specificity). In all cases, we cross validated the predictions using a leave-one-out procedure.
Densities of sigma-70 promoter-like signals distinguish known PRs from OJs
To test promoter-signals in the distinction of PRs and OJs we calculated log-likelihoods to be an OJ at 0.01 intervals of promoter-signals per base pair using the formula log10(fOJ/fPR), where fOJ is the fraction of OJs with such promoter-signal density (see Materials and Methods) and fPR is be the fraction of PRs with the same promoter-signal density (Figure 2). We were able to distinguish OJs from PRs with maximum accuracies of 0.70 in E.coli and of 0.65 in B.subtilis. Sigma-70 promoters are not the only kind of promoter in any of these organisms. E.coli K12 has 6 more annotated sigma factors, while B.subtilis has 15. The difference in number of sigma factors might be partly responsible for the lower success rate in B.subtilis. Alternatively, the results might reflect how prevalent are sigma-70 promoters in these two bacteria, or at least the particular prevalence of sigma-70 promoters within the datasets of known transcription units.
Figure 2.

Density of sigma-70 promoter-like signals in PRs and OJs. Signals consist on predicted sigma-70 promoters, which have been found to abound in PRs (1).
Signatures of cOJs and dPRs distinguish experimentally known OJs and PRs with high accuracy
To test signatures in the distinction of experimentally known WO pairs and TUB pairs we calculated log-likelihoods (sig-LLH) for each gene to be in the same operon with the previous (upstream) gene as described in Materials and Methods. The maximum prediction accuracies were 0.78 for E.coli K12 and 0.72 for B.subtilis (Figure 3). The values are very close to those obtained using the overall signatures of known OJs and PRs as training datasets (0.78 and 0.76, respectively). Thus, despite co-directional PRs might have different requirements than divergent PRs, like the presence of terminator signals, divergent PRs produce signatures useful for discriminating co-directional PRs. Conserved OJs also seem to be a good sample representing known OJs and thus equivalent datasets might help discriminate OJs from co-directional PRs in other genomes.
Figure 3.

Statistics of signature-based predictions. The maximum accuracy for E.coli K12 was 0.78 and that for B.subtilis was 0.72.
To test whether it was necessary to use trinucleotide signatures we also checked the statistical values of predictions obtained using dinucleotide signatures. Dinucleotide signatures are very common in genomic analyses (28). The maximum accuracies attained using dinucleotide-derived sig-LLHs were 0.76 for E.coli K12 and 0.71 for B.subtilis. Both numbers are below the results obtained with trinucleotides. Thus, at least in these model organisms, trinucleotide signatures might contain more information than dinucleotide signatures, making them more suitable for the task of distinguishing PRs from OJs (see also next section).
Proper separation of cOJs and dPRs vary across Prokaryotes
The sig-LLH threshold giving the highest accuracy using experimentally confirmed OJs and PRs is 0.00 sig-LLH in both E.coli K12 and B.subtilis, the same value that gives the highest accuracy of separation of the training data itself (cOJs and dPRs). Thus, the threshold for best discrimination of the training data might help decide a proper threshold for operon predictions in other Prokaryotes. In most cases the threshold giving the highest accuracy was 0.00 sig-LLH. The maximum accuracies of separation of training data range from 0.66 in Prochlorococcus marinus MIT 9313 to 0.97 in Mesoplasma florum L1. The maximum accuracies of separation of training data in E.coli K12 and B.subtilis are 0.88 and 0.87, respectively. Sixty-seven of the set of 219 non-redundant genomes had accuracies of separation of training data ≥0.87 (see website for accuracies obtained in each of the complete genomes analyzed).
To further test whether trinucleotide signatures provide better predictions than dinucleotide signatures we also calculated the accuracy of separation of training data using dinucleotide signatures. In 207 of 219 (0.95) non-redundant genomes the trinucleotide signatures show higher accuracies of separation of training data. The differences in accuracy (trinucleotide–dinucleotide) range from −0.008 to 0.18, with a mean of 0.03 and a median of 0.02. This result indicates, again, that trinucleotide signatures provide more information than dinucleotide signatures for differentiation of PRs from operon juctions.
We performed statistical analyses with several variables in an attempt to find out if any of them affects the maximum accuracies attained. The variables tested were: (i) number of sigma factors. Since our inspiration came from promoter-signal densities, we thought that the presence of several sigma factors might reduce the discrimination power because PRs recognized by different sigma factors might have different oligonucleotide signatures. We counted sigma factors based on Gene Ontology (GO) annotations in HAMAP (29,30) (ftp://ca.expasy.org/databases/complete_proteomes). To be considered a sigma factor the protein had to be associated to GO:0003700: Transcription factor (TF) activity, and either of GO:0016987: sigma factor activity; and GO:0003899: DNA-directed RNA polymerase activity. (ii) Number of TFs. The number and/or the proportion of TFs should also have an effect on the variability of oligonucleotide signatures because of the presence of binding sites for TFs at PRs. We also found TFs using GO annotation in HAMAP. The proteins had to be annotated as pertaining to GO:0003677: DNA-binding, and any of the following: GO:0003700: transcription factor activity; GO:0000156: two-component response regulator activity; GO:0030528: transcription regulator activity; GO:0016563: transcriptional activator activity and GO:0016564: transcriptional repressor activity. We also included TFs as predicted by Perez-Rueda et al. (31). (iii) Genome size and number of genes. There is evidence that as the genome size increases so does the regulatory complexity [see for instance (32–35)]. Thus this factor is related to (i) and (ii) above. (iv) Size of training samples. Despite we eliminated any genome when any of the samples (cOJs and dPRs) was less than 50, small training samples might still bias the oligonucleotide signatures and result in poor predictions. (v) Finally, genome overannotation (the annotation of genes that do not exist). We used the ‘SwissProt match’ method of Skovgaard et al. (36) to account for overannotation. The effect should be that signatures would be contaminated with the wrong information.
We used the ‘R’ package (37) (http://www.R-project.org/) to compute the correlations between accuracy and each variable. The two most important variables were overannotation and the size of the dPR sample (Table 1). We were expecting the number of sigma factors and of other transcription factors to have the largest effect, because complex regulation might result in more variable signatures at upstream regions. However, these were less correlated to accuracy than other variables (Table 1). Large genomes are expected to have more complex regulation. Accordingly, the third and fourth most important variables were genome size and number of genes (Table 1). The failure of the number of TF to represent the complexity of regulation might be due to: (a) the lack of knowledge of sigma and other transcription factors in organisms other than model organisms; (b) to the possibility that even if the number of TFs and sigma factors is higher, the proportion of genes regulated by them might be very small and (c) to the presence of forms of regulation other than DNA-binding regulatory proteins, like ribo-switches and attenuation.
Table 1.
Correlations of several variables versus accuracy of separation of training data
| Variable | Correlation | Significance |
|---|---|---|
| Overannotation | −0.32 | 9.06e−07 |
| Number of dPR | −0.30 | 7.736e−06 |
| Genome size | −0.25 | 0.0002 |
| Number of genes | −0.23 | 0.0006 |
| Number of cOJ | −0.14 | 0.0430 |
| Sigma factors | −0.04 | 0.5484 |
| Transcription factors | −0.01 | 0.8697 |
dPRs in training set; cOJs in training set; TFs (including sigma factors).
A multiple regression analysis, with accuracy as the dependent variable and all the variables in Table 1 as independent variables, results in an R-square of 0.26 (P = 7.662e−11). R-square is a measure of the amount of variation in accuracy that is explained by the variables in the model. Thus, the variables in Table 1 only explain 0.26 of the total variation in accuracy. The accuracies for each genome can be found at the web page: http://tikal.ccg.unam.mx/sarath/sig_predictions/.
Signature-predicted operons in any Prokaryote qualify as functionally related by phylogenetic profile analyses
We used phylogenetic profile analyses to test whether we are separating functionally related pairs of genes (genes in operons) from less related genes (genes at TUBs) across all genomes. Phylogenetic profiles constitute a genomic context tool used to predict functional interactions among gene products (26). We calculated the MI, measured in bits, of the phylogenetic profiles for all same-strand pairs of genes (see Materials and Methods). The expectation for functionally related pairs of genes is that they will exhibit a higher average MI than other pairs of genes. Thus, we compared the MI of pairs of genes predicted to be in the same operon (predicted WO pairs) against the MI of pairs of co-directionally transcribed genes at predicted TUBs. We found that the average MI for predicted WO pairs is higher than that for predicted TUB pairs in all genomes. The ratio of these averages (average MI of predicted WO pairs divided by the average MI of predicted TUB pairs) seems to increase with the maximum accuracy attained (Figure 4a). This result indicates that we are effectively separating functionally related genes from less related genes in any of the genomes analyzed. The minimum ratio was 1.20 in Tropheryma whipplei TW08/27 and the maximum was 7.80 in Mycoplasma penetrans. The ratios in E.coli K12 and in B.subtilis were 4.43 and 3.05, respectively.
Figure 4.

Quality of signature-based predictions. This figure shows ratios of average MI (a) and of proportions of same-KEGG map pairs of genes, with predicted pairs of genes in the same operon (WO pairs) in the numerator and predicted TUB pairs in the denominator. The expectation is that WO pairs will have higher MI than genes at TUB pairs, and that the proportions of genes in the same pathway will be higher among WO pairs than among TUB pairs. Thus, the ratios should be higher than 1 (dotted lines).
Signature-predicted operons contain higher proportions of genes in the same metabolic maps than predicted TUBs
In order to further test whether signature-based operon predictions separate functionally related genes from less related genes we downloaded the current KEGG metabolic map dataset (38,39). These metabolic maps are often used to show how predictions of functionally related genes result in higher proportions of genes whose products are in the same metabolic map [see for instance (27,40)]. We found that the proportions of same-KEGG pairs in predicted WO pairs are higher than those in predicted TUB pairs in 93% of the genomes. As in the phylogenetic profile analysis above, the ratio of same-KEGG WO pairs to same-KEGG TUB pairs tends to increase with the accuracy (Figure 4b). The minimum ratio was 0.58 in Methanospirillum hungatei JF-1, and the maximum was 7.66 in Methanococcus maripaludis S2. The ratios for predictions in E.coli K12 and in B.subtilis were 2.35 and 1.60, respectively.
Signature-predicted operons have high correlations of expression
Another source for confirmation on the quality of predictions comes from the analysis of correlation of expression of each pair of genes in microarrays. We used the data for six genomes published elsewhere (41). The signature-based predictions produce gene pairs whose correlation of expression follows that of highly conserved genes (cWO pairs) (Figure 5). The cases of E.coli K12, and B.subtilis are clear. Both genomes display a good contrast in correlation of expression between predicted WO pairs and pairs of genes in opposite strands. Other cases are not as clear. All the datasets of Helicobacter pylori J99 seem to have the same tendencies towards correlated expression. Such behavior might be due to this organism being a parasite with a reduced genome. It is plausible that in this kind of organism the reduced genome contains a higher proportion of functionally related genes, and/or of genes that require little regulation due to a simpler lifestyle, thus resulting in high correlations of expression. Chlamydia trachomatis, another organism with a reduced genome, has a similar, though not as evident, tendency.
Figure 5.

Correlation of expression in microarray data. Abbreviations as in Figure 4. Conserved pairs represent true WO pairs, while divergent pairs represent how true TUB should behave. Note that the tendencies for predicted WO pairs tend to follow those in conserved pairs, and that the quality seems to be at least as good as that for distance-based predictions produced as described elsewhere (22,23).
The cyanobacterium Synechocystis PCC6803 has been a challenge for operon predictions before (22), mainly due to gene annotation problems and unusually high spacing between genes. In this organism the signature-based WO pairs exhibit higher correlations of expression than genes in opposite strands, but not as high as those of conserved pairs. The correlations somewhat follow those of our distance-based predictions (Figure 5), and those of the distance-based predictions presented by Price et al. (41). If conserved gene-pairs truly represent operons in this organism, then neither method has been very accurate to predict operons in this genome. Accordingly, the accuracy of discrimination of training data in this organism was the second to lowest at 0.68. An alternative explanation to genome annotation problems might be that operons in this organism abound in secondary signals between genes. If so, operons in Synechocystis will not be easy to predict by means other than precise finding of promoters, termination of transcription and other signals.
Signatures provide information complementary to intergenic distances
Since the first publication of a successful method to predict operons (23), most authors have found that the most informative feature is the distance between genes (41–48). The current maximum prediction accuracies using dist-LLHs as evaluated against experimentally known WO pairs and TUB pairs are 0.83 in E.coli K12 and 0.87 in B.subtilis, both higher than the accuracies obtained using sig-LLHs. The purpose of this report is to show that other features remain to be explored, and to measure what can be attained using oligonucleotide signatures in particular. One salient feature of signature-based predictions is that samples for training the method do not have to be as large as the training data necessary to calculate dist-LLHs at different intergenic distance intervals. This is especially important if the distances between genes in the same operon and/or the distances between same-strand TUBs in a genome of interest do not follow those of model organisms.
One more goal to explore other features for operon predictions is to increase the accuracies over those produced previously. In this regard, adding sig-LLHs to dist-LLHs produces maximum accuracies of 0.84 in E.coli K12 and 0.88 in B.subtilis (0.01 above dist-LLHs alone). Fitting to a linear model to combine the log-likelihoods, following Price et al. (41), result in the very same maximum accuracies.
CONCLUDING REMARKS
We have shown that trinucleotide signatures can help distinguish regions upstream of genes inside operons from those upstream of first transcribed genes, thus allowing for operon predictions. We have also shown that such signatures can be derived from pairs of adjacent same-strand genes conserved in evolutionarily distant genomes as examples of genes in operons, and from divergently transcribed genes as examples of genes at TUBs. This makes overall signature-based operon predictions possible for most Prokaryotic genomes.
Further data and predictions for each genome can be found at http://tikal.ccg.unam.mx/sarath/sig_predictions/.
Acknowledgments
G.M.H. acknowledges funds from WLU, Gary Molenkamp for computer assistance, and SHARCNET for computer cluster usage as well as for support as chair in Biocomputing. WLU has provided research funds. The authors thank Ernesto Pérez-Rueda for providing predicted transcription factors for all genomes, and Morgan N. Price for providing the microarray data. SCJ has been supported by grants given to Julio Collado-Vides. Funding to pay the Open Access publication charges for this article was provided by Wilfrid Laurier University.
Conflict of interest statement. None declared.
REFERENCES
- 1.Huerta A.M., Collado-Vides J. Sigma70 promoters in Escherichia coli: specific transcription in dense regions of overlapping promoter-like signals. J. Mol. Biol. 2003;333:261–278. doi: 10.1016/j.jmb.2003.07.017. [DOI] [PubMed] [Google Scholar]
- 2.Jacob F., Perrin D., Sanchez C., Monod J. [Operon: a group of genes with the expression coordinated by an operator.] C R Hebd. Seances Acad. Sci. 1960;250:1727–1729. [PubMed] [Google Scholar]
- 3.Jacob F., Perrin D., Sanchez C., Monod J., Edelstein S. [The operon: a group of genes with expression coordinated by an operator. C.R.Acad. Sci. Paris 250 (1960) 1727–1729] C. R. Biol. 2005;328:514–520. doi: 10.1016/j.crvi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Plaskon R.R., Wartell R.M. Sequence distributions associated with DNA curvature are found upstream of strong E. coli promoters. Nucleic Acids Res. 1987;15:785–796. doi: 10.1093/nar/15.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Espinosa-Urgel M., Tormo A. Sigma s-dependent promoters in Escherichia coli are located in DNA regions with intrinsic curvature. Nucleic Acids Res. 1993;21:3667–3670. doi: 10.1093/nar/21.16.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carmona M., Magasanik B. Activation of transcription at sigma 54-dependent promoters on linear templates requires intrinsic or induced bending of the DNA. J. Mol. Biol. 1996;261:348–356. doi: 10.1006/jmbi.1996.0468. [DOI] [PubMed] [Google Scholar]
- 7.Gabrielian A.E., Landsman D., Bolshoy A. Curved DNA in promoter sequences. In Silico Biol. 1999;1:183–196. [PubMed] [Google Scholar]
- 8.Bolshoy A., Nevo E. Ecologic genomics of DNA: upstream bending in prokaryotic promoters. Genome Res. 2000;10:1185–1193. doi: 10.1101/gr.10.8.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ussery D., Larsen T.S., Wilkes K.T., Friis C., Worning P., Krogh A., Brunak S. Genome organisation and chromatin structure in Escherichia coli. Biochimie. 2001;83:201–212. doi: 10.1016/s0300-9084(00)01225-6. [DOI] [PubMed] [Google Scholar]
- 10.Jauregui R., Abreu-Goodger C., Moreno-Hagelsieb G., Collado-Vides J., Merino E. Conservation of DNA curvature signals in regulatory regions of prokaryotic genes. Nucleic Acids Res. 2003;31:6770–6777. doi: 10.1093/nar/gkg882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ussery D.W., Tindbaek N., Hallin P.F. Genome update: promoter profiles. Microbiology. 2004;150:2791–2793. doi: 10.1099/mic.0.27504-0. [DOI] [PubMed] [Google Scholar]
- 12.Olivares-Zavaleta N., Jauregui R., Merino E. Genome analysis of Escherichia coli promoter sequences evidences that DNA static curvature plays a more important role in gene transcription than has previously been anticipated. Genomics. 2006;87:329–337. doi: 10.1016/j.ygeno.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen A.G., Jensen L.J., Brunak S., Staerfeldt H.H., Ussery D.W. A DNA structural atlas for Escherichia coli. J. Mol. Biol. 2000;299:907–930. doi: 10.1006/jmbi.2000.3787. [DOI] [PubMed] [Google Scholar]
- 14.Mitchison G. The regional rule for bacterial base composition. Trends Genet. 2005;21:440–443. doi: 10.1016/j.tig.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 15.Moreno-Hagelsieb G., Trevino V., Perez-Rueda E., Smith T.F., Collado-Vides J. Transcription unit conservation in the three domains of life: a perspective from Escherichia coli. Trends Genet. 2001;17:175–177. doi: 10.1016/s0168-9525(01)02241-7. [DOI] [PubMed] [Google Scholar]
- 16.Ermolaeva M.D., White O., Salzberg S.L. Prediction of operons in microbial genomes. Nucleic Acids Res. 2001;29:1216–1221. doi: 10.1093/nar/29.5.1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janga S.C., Moreno-Hagelsieb G. Conservation of adjacency as evidence of paralogous operons. Nucleic Acids Res. 2004;32:5392–5397. doi: 10.1093/nar/gkh882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hertz G.Z., Stormo G.D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences. Bioinformatics. 1999;15:563–577. doi: 10.1093/bioinformatics/15.7.563. [DOI] [PubMed] [Google Scholar]
- 19.Huerta A.M., Salgado H., Thieffry D., Collado-Vides J. RegulonDB: a database on transcriptional regulation in Escherichia coli. Nucleic Acids Res. 1998;26:55–59. doi: 10.1093/nar/26.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salgado H., Gama-Castro S., Peralta-Gil M., Diaz-Peredo E., Sanchez-Solano F., Santos-Zavaleta A., Martinez-Flores I., Jimenez-Jacinto V., Bonavides-Martinez C., Segura-Salazar J., et al. RegulonDB (version 5.0): Escherichia coli K-12 transcriptional regulatory network, operon organization, and growth conditions. Nucleic Acids Res. 2006;34:D394–D397. doi: 10.1093/nar/gkj156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blattner F.R., Plunkett G., 3rd, Bloch C.A., Perna N.T., Burland V., Riley M., Collado-Vides J., Glasner J.D., Rode C.K., Mayhew G.F., et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- 22.Moreno-Hagelsieb G., Collado-Vides J. A powerful non-homology method for the prediction of operons in prokaryotes. Bioinformatics. 2002;18:S329–S336. doi: 10.1093/bioinformatics/18.suppl_1.s329. [DOI] [PubMed] [Google Scholar]
- 23.Salgado H., Moreno-Hagelsieb G., Smith T.F., Collado-Vides J. Operons in Escherichia coli: genomic analyses and predictions. Proc. Natl Acad. Sci. USA. 2000;97:6652–6657. doi: 10.1073/pnas.110147297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maglott D.R., Katz K.S., Sicotte H., Pruitt K.D. NCBI's LocusLink and RefSeq. Nucleic Acids Res. 2000;28:126–128. doi: 10.1093/nar/28.1.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pruitt K.D., Tatusova T., Maglott D.R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005;33:D501–D504. doi: 10.1093/nar/gki025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pellegrini M., Marcotte E.M., Thompson M.J., Eisenberg D., Yeates T.O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl Acad. Sci. USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huynen M., Snel B., Lathe W., 3rd, Bork P. Predicting protein function by genomic context: quantitative evaluation and qualitative inferences. Genome Res. 2000;10:1204–1210. doi: 10.1101/gr.10.8.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karlin S., Campbell A.M., Mrazek J. Comparative DNA analysis across diverse genomes. Annu. Rev. Genet. 1998;32:185–225. doi: 10.1146/annurev.genet.32.1.185. [DOI] [PubMed] [Google Scholar]
- 29.Boeckmann B., Bairoch A., Apweiler R., Blatter M.C., Estreicher A., Gasteiger E., Martin M.J., Michoud K., O'Donovan C., Phan I., et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gattiker A., Michoud K., Rivoire C., Auchincloss A.H., Coudert E., Lima T., Kersey P., Pagni M., Sigrist C.J., Lachaize C., et al. Automated annotation of microbial proteomes in SWISS-PROT. Comput. Biol. Chem. 2003;27:49–58. doi: 10.1016/s1476-9271(02)00094-4. [DOI] [PubMed] [Google Scholar]
- 31.Perez-Rueda E., Collado-Vides J., Segovia L. Phylogenetic distribution of DNA-binding transcription factors in bacteria and archaea. Comput. Biol. Chem. 2004;28:341–350. doi: 10.1016/j.compbiolchem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Cherry J.L. Genome size and operon content. J. Theor. Biol. 2003;221:401–410. doi: 10.1006/jtbi.2003.3194. [DOI] [PubMed] [Google Scholar]
- 33.van Nimwegen E. Scaling laws in the functional content of genomes. Trends Genet. 2003;19:479–484. doi: 10.1016/S0168-9525(03)00203-8. [DOI] [PubMed] [Google Scholar]
- 34.Babu M.M., Luscombe N.M., Aravind L., Gerstein M., Teichmann S.A. Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol. 2004;14:283–291. doi: 10.1016/j.sbi.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Ranea J.A., Buchan D.W., Thornton J.M., Orengo C.A. Evolution of protein superfamilies and bacterial genome size. J. Mol. Biol. 2004;336:871–887. doi: 10.1016/j.jmb.2003.12.044. [DOI] [PubMed] [Google Scholar]
- 36.Skovgaard M., Jensen L.J., Brunak S., Ussery D., Krogh A. On the total number of genes and their length distribution in complete microbial genomes. Trends Genet. 2001;17:425–428. doi: 10.1016/s0168-9525(01)02372-1. [DOI] [PubMed] [Google Scholar]
- 37.R_Development_Core_Team. Vienna, Austria: 2006. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing. ISBN 3-900051-07-0. [Google Scholar]
- 38.Ogata H., Goto S., Fujibuchi W., Kanehisa M. Computation with the KEGG pathway database. Biosystems. 1998;47:119–128. doi: 10.1016/s0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 39.Kanehisa M., Goto S., Kawashima S., Okuno Y., Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32:D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snel B., Lehmann G., Bork P., Huynen M.A. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28:3442–3444. doi: 10.1093/nar/28.18.3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price M.N., Huang K.H., Alm E.J., Arkin A.P. A novel method for accurate operon predictions in all sequenced prokaryotes. Nucleic Acids Res. 2005;33:880–892. doi: 10.1093/nar/gki232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craven M., Page D., Shavlik J., Bockhorst J., Glasner J. A probabilistic learning approach to whole-genome operon prediction. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2000;8:116–127. [PubMed] [Google Scholar]
- 43.Bockhorst J., Qiu Y., Glasner J., Liu M., Blattner F., Craven M. Predicting bacterial transcription units using sequence and expression data. Bioinformatics. 2003;19:I34–I43. doi: 10.1093/bioinformatics/btg1003. [DOI] [PubMed] [Google Scholar]
- 44.Chen X., Su Z., Xu Y., Jiang T. Computational prediction of operons in Synechococcus sp. WH8102. Genome Inform Ser. Workshop Genome Inform. 2004;15:211–222. [PubMed] [Google Scholar]
- 45.De Hoon M.J., Imoto S., Kobayashi K., Ogasawara N., Miyano S. Predicting the operon structure of Bacillus subtilis using operon length, intergene distance, and gene expression information. Pac. Symp. Biocomp. 2004;9:276–287. doi: 10.1142/9789812704856_0027. [DOI] [PubMed] [Google Scholar]
- 46.Romero P.R., Karp P.D. Using functional and organizational information to improve genome-wide computational prediction of transcription units on pathway-genome databases. Bioinformatics. 2004;20:709–717. doi: 10.1093/bioinformatics/btg471. [DOI] [PubMed] [Google Scholar]
- 47.Jacob E., Sasikumar R., Nair K.N. A fuzzy guided genetic algorithm for operon prediction. Bioinformatics. 2005;21:1403–1407. doi: 10.1093/bioinformatics/bti156. [DOI] [PubMed] [Google Scholar]
- 48.Westover B.P., Buhler J.D., Sonnenburg J.L., Gordon J.I. Operon prediction without a training set. Bioinformatics. 2005;21:880–888. doi: 10.1093/bioinformatics/bti123. [DOI] [PubMed] [Google Scholar]