

Abstract
Synthesis of Tyr in the human body occurs by hydroxylation of the indispensable amino acid Phe. Until now, it was believed that in humans, this process was restricted to the liver, but we provide compelling evidence of production of Tyr from Phe in the kidney. To determine whether the human kidney produces Tyr, we measured Tyr balance, the Tyr appearance rate, and the Phe-to-Tyr conversion in 12 healthy human subjects by using [15N]Phe and [2H4]Tyr as tracers. Renal plasma flow was measured by using paraaminohippurate, and sampling from the femoral artery and renal veins was performed. The results were compared with those obtained in 12 control subjects undergoing hepatic vein catheterization and infusion of identical tracers. In all 12 subjects, there was a net uptake of Phe by the kidney (2.2 ± 1.2 μmol/min), whereas Tyr was released (5.3 ± 1.5 μmol/min). In contrast, there was a net uptake of both Phe (9.5 ± 1.2 μmol/min) and Tyr (14.3 ± 1.3 μmol/min) by the splanchnic bed. Phe conversion to Tyr occurred at a rate of 5.2 ± 1.2 μmol/min in kidney and 3.0 ± 0.7 μmol/min in the splanchnic bed. The kidney contributed a substantial amount of Tyr to the systemic circulation where the splanchnic bed was a net remover of Tyr. Our results demonstrate that the kidney is the major donor of Tyr to the systemic circulation by its conversion of Phe to Tyr. This observation may have important clinical implications for patients with both renal and hepatic disease, who may be at risk of Phe overloading and Tyr deficiency, and it should be considered when parenteral or enteral nutrients are administered rich in Phe and low in Tyr.
In humans, Phe is an indispensable amino acid (1), which may either be used for protein synthesis or be converted to the nonessential amino acid Tyr, the precursor for catecholamine and thyroid hormone synthesis. Tyr is not considered indispensable, because it can be synthesized from Phe. Deficiency of Tyr can, however, adversely affect the remodeling of body proteins. Remodeling of virtually all tissues in the body is a dynamic process involving breakdown and synthesis of proteins, and Tyr is an essential component of body proteins as well as a prerequisite for continuous synthesis of individual proteins. For synthesis of proteins, it is essential to have a full complement of all amino acids.
The initial step of Phe degradation involves hydroxylation to Tyr by molecular oxygen. This reaction is catalyzed by phenylalanine hydroxylase in the presence of the cofactor tetrahydrobiopterin (2). Phenylketonuria is an autosomal recessive disorder characterized by lack of phenylalanine hydroxylase leading to accumulation of Phe in the circulation and mental retardation in childhood (3). Although phenylalanine hydroxylase activity in the adult range has been demonstrated in fetal liver after 8 weeks of gestation (4), a number of studies have suggested that the overall capacity of the enzyme system is limited in the fetus (5–7). Based on these observations in fetal livers, it has been hypothesized that Tyr is a conditionally essential amino acid for preterm infants (8). In this context, it has been generally assumed that human phenylalanine hydroxylase activity and the Phe-to-Tyr conversion are specific to the liver. This concept is based on the belief that in humans and other primates, phenylalanine hydroxylase is expressed exclusively in the liver, although in rodents, additional enzyme activity has been demonstrated in the kidney (9). If tenable, the perception of Phe-to-Tyr conversion as being restricted to the liver may also have important implications for patients with liver failure in whom elevated concentrations of Phe have been reported (10).
Some findings, however, are not entirely consistent with the belief that the sole fate of Phe in humans is intrahepatic degradation. It has, for instance, repeatedly been shown that despite low hepatic in vitro capacity for Phe degradation, premature and normal newborns are fully capable of converting Phe to Tyr (11, 12). Furthermore, patients with hepatic cirrhosis have high Phe turnover, implying that increased Phe production may be the primary cause of increased Phe levels rather than defective hydroxylation, which might have been anticipated in view of the compromised liver function (13, 14).
The present study was therefore designed to test the hypothesis that the human kidney is an important site for Phe-to-Tyr conversion. For this purpose, we investigated 12 normal healthy volunteers combining femoral artery and renal vein catheterization and Phe and Tyr isotopic dilution techniques in vivo. To allow quantification of the hepatic contribution, we compared these subjects to 12 healthy persons undergoing combined hepatic vein catheterization and isotope dilution (15).
Methods
Subjects.
Twelve healthy subjects (6 men and 6 women) aged 28.3 ± 1.8 years with body mass indexes between 18 and 27 kg/m2 and a total body weight of 68.8 ± 6.9 kg participated in the renal study. Twelve other subjects (6 men and 6 women) aged 29.0 ± 1.5 years with body mass indexes between 18–27 kg/m2 and body weights of 70.9 ± 4.7 kg participated in the splanchnic study. Fasting blood glucose levels were normal (70–100 mg per 100 ml) in all participants and all had a normal physical examination, normal ECGs, and normal hematological, renal, and hepatic function as assessed by biochemical screening. One subject received a replacement dose of levothyroxine because of stable primary hypothyroidism. All subjects ingested a standardized weight-maintaining diet (20% protein, 30% fat, and 50% carbohydrate) prescribed by a research dietician for 3 days before the study. Preceding the study, the protocol was submitted to and approved by the Mayo Institutional Review Board and the purpose and potential risks of the study were explained to all subjects. Informed, written consent was obtained from each participant.
Experimental Design.
The studies were conducted after a 12-h overnight fast in the General Clinical Research Center at Mayo Clinic. The evening before the study, an intravenous catheter was inserted into an antebrachial vein and kept patent with saline. At 0800 h the following morning, duplicate baseline samples were drawn and priming doses followed by continuous infusion of [15N]Phe (0.1 mg/kg and 0.1 mg/kg/h) and [2H4]Tyr (0.6 mg/kg and 0.6 mg/kg/h) together with a priming dose of [15N]Tyr (0.05 mg/kg) were given for determination of isotopic enrichment of [15N]Phe, [2H4]Tyr, and [15N]Tyr. To optimize determination of regional renal Phe fluxes, the remaining six subjects received an increased dose of [15N]Phe (0.75 mg/kg and 0.75 mg/kg/h) and a priming dose of [15N]Tyr (0.3 mg/kg). These subjects also received tracer infusions of glucose and Leu as a part of a separate study.
For the renal study, cannulation of the femoral vein, one renal vein, and the femoral artery were performed at 0900 h, as described (16–19). Renal vein catheters were inserted under fluoroscopic guidance and correct positioning was confirmed by contrast injection. The femoral artery line was used for infusion of indocyanine green (0.6 mg/min) to measure blood flow in the leg and for blood sampling. Femoral vein and renal vein catheters were used to collect blood samples. The peripheral antebrachial vein was used for infusion of isotopes and paraaminohippurate for measurements of renal plasma flow. At 1030 h, Cardiogreen was infused at a rate of 30 mg/h for 90 min. Paraaminohippurate was given as a bolus of 10 mg/kg total body weight followed by a constant infusion of 1.5 g/h from 1030 onward (17–19). The study was stopped after 240 min at 1200 h; the catheters were removed and homeostasis obtained.
The 12 subjects undergoing hepatic vein catherization were investigated under similar conditions, with the exception that isotope administration was commenced after catheter insertion and thus maintained for only 150 min. Six of these subjects continued the isotope infusion for another 360 min, which allowed us to determine whether any time-related changes occurred. These studies showed no significant change in whole-body and regional protein dynamics between 120–150 min and 300–360 min. Hepatic catheters were also inserted under fluoroscopic guidance and the position verified by contrast injection (16).
Blood samples were collected at baseline and at 15-min intervals during the last 60 min of the study. Simultaneous samples were collected from the femoral artery, hepatic vein, and renal vein.
Materials.
l-[15N]Phe (99 atom percent excess) and l-[1-13C, 15N]Leu (99 atom percent excess) were purchased from Cambridge Isotope Laboratories (Andover, MA) and l-[ring-2H4]Tyr (99 atom percent excess) and l-[15N]Tyr (99 percent atom excess) were purchased from Isotec. The chemical, isotopic, and optical purity of these compounds was confirmed before use. Solutions were prepared under sterile conditions in the pharmacy and were shown to be free of bacteria and pyrogens before administration. Cardiogreen (indocyanine) was purchased from Becton Dickinson and para-aminohippurate, from Merck.
Analysis of Samples.
Blood glucose was measured by a glucose oxidase method (Beckman Instruments, Fullerton, CA). Hormonal assays were performed as described (20). Plasma insulin and growth hormone were measured by a chemiluminescent sandwich assay (Sanofi Diagnostics, Chaska, MN), and glucagon and cortisol were measured by RIAs (Diagnostic Products, Los Angeles). Indocyanine green concentrations were measured by spectrophotometry and para-aminohippurate colorimetrically.
Plasma enrichment levels of [15N]Phe, [2H4]Tyr, [15N]Tyr, [1-13C,15N]Leu, and [1-13C]Leu were determined by gas chromatography/mass spectrometry as described (15). Fifty microliters of an internal standard solution containing 60 μg/ml of [2H5]phenylalanine hydrochloride (Sigma) and 20 μg/ml of [13C6]Tyr (Isotec) was added to six standards containing Phe and Tyr with concentrations ranging from 0 to 300 μmol/liter (Sigma), and to 100-μl plasma samples. The amino acids from plasma were isolated by ion-exchange chromatography and dried down. All samples were derivatized with N-methyl-N-(t-butyldimethylsilyl)trifluoroacetamide with 1% t-butyl-dimethylchlorosilane in acetonitrile (Regis, Morton Grove, IL) at room temperature overnight. The derivatives were injected onto a 30-m × 0.25-mm × 0.25 μm DB5MS column (J & W Scientific, Folsom, CA) operating with a constant helium flow of 1.1 ml/min under the following temperature conditions: 120°C initially, then ramped to 210°C at 25°C/min, to 225°C at 10°C/min, to 255°C at 25°C/min, to 265°C at 10°C/min, to 300°C at 25°C/min, and finally to 325°C at 10°C/min and held for 2 min. Under electron ionization, fragment ions were monitored at m/z 336, 337 for Phe, 341 for [2H5]Phe, and 466, 467, 470, and 472 for Tyr by using a Hewlett Packard 6890 MSD (Hewlett–Packard) to determine the isotopic enrichment and concentrations of Phe and Tyr.
Calculations.
Plasma flows from the kidney or the splanchnic bed were calculated as described (16, 17) and converted to blood flows by using the equation: Blood flows = Plasma flows/(1 − hematocrit). Isotopic plateau was observed during the last 60 min of the study period when samples were collected. This was assessed based on the observation that when isotopic enrichment values of Phe and Tyr in different sites were plotted against time, the ensuing slopes were not different from zero. Mean values for any isotope at each plateau were used for all calculations of amino acid kinetics.
Whole-body Phe flux (QPhe) was calculated (21) as follows:
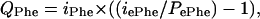 |
in which iPhe is the rate of Phe infusion (μmol/kg body weight/min), iePhe is the isotopic enrichment of the infusate, and PePhe is [15N]Phe enrichment at isotopic plateau. Similar calculations were performed for [2H4]Tyr (QTyr).
Regional Dynamics of Phe and Tyr.
Net amino acid balances across organs were calculated by multiplying arteriovenous differences by blood flows.
For calculation of across-organ isotope kinetics, we used the equations described (16). The rate of appearance of Phe (Ra(Phe)), representing local protein breakdown, was determined as follows:
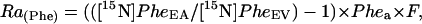 |
where [15N]PheEA and [15N]PheEV are enrichments in artery and vein, Phea is the arterial concentration of Phe, and F is the blood flow. The rate of appearance of Tyr was calculated similarly.
The local conversion of Phe-to-Tyr (IPT) was calculated as follows:
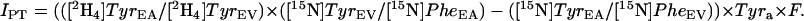 |
Calculations of local Phe incorporation into protein can now be determined after calculation of the Phe rate of disappearance [Rd(Phe)]
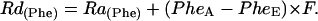 |
Statistics.
All values are given as means ± SEM. Paired t tests were used to assess whether any parameter differed significantly from zero. A P value under 0.05 was considered statistically significant.
Results
Plasma Flows, Hormones, and Glucose.
Renal and splanchnic blood flow values of 1,225 ± 70 and 1,431 ± 153 ml/min were recorded. Circulating concentrations of insulin, growth hormone, glucagon, and glucose were similar and within the normal range in the two study groups (Table 1).
Table 1.
Renal and splanchnic blood flows and plasma concentrations of hormones and glucose in 12 subjects undergoing renal vein catheterization and 12 subjects undergoing hepatic vein catheterization
| Renal group (n = 12) | Splanchnic group (n = 12) | |
|---|---|---|
| Renal blood flow, ml/min | 1225 ± 70 | 1431 ± 153 |
| Insulin, pmol/liter | 33.1 ± 4.8 | 38.0 ± 4.1 |
| Glucagon, pg/ml | 63.2 ± 3.1 | 56.1 ± 3.9 |
| Growth hormone, ng/ml | 0.67 ± 0.30 | 1.5 ± 0.4 |
| Glucose, mmol/liter | 5.0 ± 0.1 | 4.9 ± 0.1 |
Amino Acids and Whole-Body Protein Dynamics.
Arterial Phe and Tyr concentrations were stable and comparable in both groups (Table 2). Likewise, there was no difference in whole-body fluxes of Phe or Tyr, which plateaued around 40 and 30 μmol/kg/h, respectively.
Table 2.
Arterial plasma concentrations of amino acids and whole body phenylalanine and tyrosine fluxes in 12 subjects undergoing renal vein catheterization and 12 subjects undergoing hepatic vein catheterization
| Renal group (n = 12) | Splanchnic group (n = 12) | |
|---|---|---|
| Phenylalanine, μmol/liter | 46.9 ± 6.1 | 51.7 ± 3.9 |
| Tyrosine, μmol/liter | 45.0 ± 6.9 | 41.1 ± 7.8 |
| Phenylalanine flux, μmol/kg/h | 35.8 ± 0.9 | 43.2 ± 0.6 |
| Tyrosine flux, μmol/kg/h | 28.3 ± 1.2 | 32.1 ± 1.0 |
Regional Protein Dynamics.
Regional Phe and Tyr kinetics.
Fig. 1 shows Phe and Tyr kinetics in the kidney, and Fig. 2 shows that of the splanchnic bed. In general, amino acid kinetics in the splanchnic bed occur at a higher level than in the kidney. Ra(Phe) in both the kidney (2.2 ± 0.3 μmol/min) (P < 0.05) and splanchnic bed (19.2 ± 1.5) (P < 0.01) were lower than that of Rd(Phe) (kidney = 4.7 ± 1.3 and splanchnic bed = 28.6 ± 2.3). In the kidney, Rd(Phe) is explained on the basis of Phe conversion to Tyr (5.2 ± 1.2 μmol/min), whereas in the splanchnic bed, only a fraction of Rd(Phe) is caused by conversion to Tyr (3.0 ± 0.7 μmol/min). In the kidney, a large part of Ra(Tyr) (7.0 ± 1.5 μmol/min) is caused by Phe conversion to Tyr, whereas in the splanchnic bed, only a small portion of Ra(Tyr) (16.0 ± 2 μmol/min) is caused by Phe conversion to Tyr.
Figure 1.

Phe and Tyr balance and kinetics across kidney. (A) Phe and Tyr balance. There was a net uptake of Phe (2.2 ± 0.3 μmol/min) across the kidney and a net release of Tyr (−5.2 ± 1.5 μmol/min). (B) The kinetics of Phe and Tyr. Ra(Phe) was lower than Rd(Phe) (P < 0.01). Ra(Tyr) is higher than Rd(Tyr) (P < 0.01). Phe conversion to Tyr (5.2 ± 1.2 μmol/min) also was higher than Rd(Tyr) (1.8 ± 1.0 μmol/min) (P < 0.01).
Figure 2.

Phe and Tyr balance and kinetics across splanchnic bed. (A) Phe and Tyr balance. There was net uptake of both Phe (9.5 ± 1.2 μmol/min) and Tyr (14.3 ± 1.3 μmol/min) across the splanchnic bed. (B) The kinetics of Phe and Tyr. Ra(Phe) was lower than Rd(Phe) (P < 0.01). Ra(Tyr) was also lower than Rd(Tyr) (P < 0.01). Phe conversion to Tyr (3.0 ± 0.7 μmol/min) was also lower than Rd(Tyr) (30.3 ± 3.3 μmol/min) (P < 0.01).
The kidney is a net releaser of Tyr to systemic circulation (−5.3 ± 1.5 μmol/min), whereas the splanchnic bed is a net receiver of Tyr (14.3 ± 1.3 μmol/min).
Discussion
This study demonstrates a metabolic role of the human kidney. Based on labeled Phe and Tyr measurements across the kidney, it was demonstrated that a substantial amount of Phe conversion to Tyr occurs in kidney. The rate at which Tyr is produced in the kidney is greater than the Tyr disposal rate in the kidney. As a result, kidney is a net contributor of Tyr to the systemic circulation. In contrast, the splanchnic bed is a net remover of Tyr from the systemic circulation. Although the splanchnic bed (presumably in liver) also converts Phe to Tyr, it consumes Tyr at a rate higher than the production of Tyr in this tissue bed. As a result, there is a net uptake of Tyr in the splanchnic bed. It is, therefore, apparent from this study that although Phe conversion to Tyr occurs across both the kidney and splanchnic bed, it is kidney that contributes Tyr to the systemic circulation and thus makes Tyr available to other tissues for protein synthesis.
Hitherto, it has been assumed that in primates, the initial enzymatic step in Phe degradation occurs exclusively in the liver (4). There is also considerable experimental evidence to support that the liver converts Phe to Tyr in humans (15, 16, 19). In contrast, no experimental evidence is available in humans demonstrating that any organs other than liver converts Phe to Tyr. It has, however, been demonstrated that phenylalanine hydroxylase is present in the rodent kidney (9) and guinea pig (22). In addition, phenylalanine hydroxylase has also been demonstrated in the human kidney cortex (23), although studies based on biopsy samples from diseased kidney and autopsy samples failed to demonstrate phenylalanine hydroxylase in human kidney (9, 24). It should be emphasized that needle biopsy samples from diseased kidney may not represent the whole kidney, and careful studies based on freshly removed kidney demonstrated this enzyme activity in the human kidney (25). It has also been reported that the enzyme in kidney is less stable than that in liver (24). However, the stability has been shown to be the same after purification, indicating that the kidney enzyme is unstable in kidney extract (25). It is, therefore, likely that the inability to demonstrate the enzyme activity in autopsy samples (9) is related to the instability of this enzyme. Further studies have demonstrated that the same isoenzymes of phenylalanine hydroxylase exist in the liver and kidney (25). The kidney also has been reported to release Tyr on long-term fasting (26) and in subjects renal catheterization is done for diagnostic purpose (19). The current study based on tracer methodology clearly demonstrated that Phe is converted to Tyr in kidney which supports the presence of phenylalanine hydroxylase in the kidney.
In the current study, we measured arteriovenous flux across the kidney of [15N]Phe and [15N]Tyr during a primed continuous infusion of [15N]Tyr enrichment. In addition, we independently measured total Tyr flux across the kidney by using an independent tracer of Tyr ([2H4]Tyr) and demonstrated a decrease in the enrichment across the kidney ([2H4]Tyr enrichment as molar percent excess in the femoral artery = 8.87 ± 0.23 and in the renal vein = 7.96 ± 0.18, P < 0.01). Under steady-state conditions, the observation that the renal vein [15N]Tyr was consistently higher than that of the artery (femoral artery = 1.38 molar percent excess and renal vein = 1.92 ± 0.096, P < 0.01), whereas the renal vein [2H4]Tyr enrichment was consistently lower than that of the artery in all subjects, which can only be explained on the basis of conversion of [15N]Phe to [15N]Tyr in the kidney.
The estimated conversion rate of Phe to Tyr is higher in the kidney (5.2 ± 1.2 μmol/min) than in the splanchnic bed (3.0 ± 0.7 μmol/min). It is, however, likely that splanchnic (liver) conversion of Phe to Tyr is underestimated. The true precursor for Phe conversion to Tyr is intracellular Phe. The enrichment of intracellular [15N]Phe in the liver is likely to be lower than that of the femoral artery (27), presumably because of the appearance of unlabeled Phe from protein breakdown of proteins in the gut and liver. It is, therefore, not possible to accurately estimate the relative contributions of the kidney and splanchnic bed to the total body Phe conversion to Tyr. In addition, it is likely that the calculations of whole-body Phe conversion to Tyr based on plasma Phe and Tyr enrichment values are also underestimations, because intracellular isotopic enrichments are likely to be lower (27, 28) than that of plasma for these amino acids.
It is clear from the current study that the Tyr disposal rate in the splanchnic bed (30.3 ± 3.3 μmol/min) is much greater than its production and appearance from protein breakdown (16.0 ± 2.0 μmol/min). As a result, the splanchnic bed in the postabsorptive state removes Tyr from the circulation. In contrast, in the kidney the Tyr production rate plus appearance from protein breakdown (7.0 ± 1.5 μmol/min) is greater than its disposal rate (1.8 ± 1.0 μmol/min), resulting in a net release of Tyr from the kidney during the postabsorption state. Of note, in patients with cirrhosis, circulating Tyr concentrations are higher than in healthy control subjects (29) because of the greater impact the liver has on the Tyr disposal rate than on Tyr production (Phe conversion to Tyr). Theoretically, renal failure should have little impact on the Tyr disposal rate if the liver is functioning normally but will have relatively higher impact on Tyr production and appearance in the systemic circulation. Renal failure may therefore reduce the availability of Tyr to other tissues such as skeletal muscle, impeding their ability to maintain protein synthesis and thus contributing to the catabolic state in renal failure (30). Lack of kidney involvement in converting Phe in renal failure could become an even greater problem when parenteral nutrition is administered to the renal failure patients. Most parenteral nutrition preparations have very low Tyr because it is assumed that Tyr will be produced in vivo from conversion of Phe and also because Tyr has very low solubility, requiring a large volume of water (31). It is undesirable to infuse a large volume to people with renal failure and water retention. For protein synthesis to take place efficiently, it is essential to have a full complement of all amino acids. It is, therefore, important to determine whether Tyr should be treated as an essential amino acid to replace in people with renal failure.
In conclusion, the current study demonstrated that kidney is a major organ involved in the conversion of Phe to Tyr. Because kidney disposes less Tyr than it produces, the kidney is a net contributor of Tyr to the systemic circulation, unlike liver, which disposes more Tyr than it produces. This observation has important clinical implications in people with renal failure.
Acknowledgments
We thank the skillful technical contributions by G. C. Ford, Lawrence Ward, Mai Persson, and Dawn Morse, and the support by the General Clinical Research Center nursing staff and the volunteers for the study. This work was supported by National Institutes of Health Grants RO1 DK 41973 and RR00585.
Abbreviations
- Ra
rate of appearance
- Rd
rate of disappearance
References
- 1.Womak M, Rose W C. J Biol Chem. 1934;107:449–458. [Google Scholar]
- 2.Hufton S E, Jennings I G, Cotton R G H. Biochem J. 1995;311:353–366. doi: 10.1042/bj3110353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schriver C R. Clin Biochem. 1995;28:137–144. doi: 10.1016/0009-9120(94)00076-8. [DOI] [PubMed] [Google Scholar]
- 4.Raiha N C. Pediatr Res. 1973;7:1–4. doi: 10.1203/00006450-197301000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Kenney F T, Kretchmer N. J Clin Invest. 1959;38:2189–2196. doi: 10.1172/JCI103998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenneman A R, Kaufman S. J Biol Chem. 1965;240:3617–3622. [PubMed] [Google Scholar]
- 7.Ryan W L, Orr W. Arch Biochem Biophys. 1966;113:684–686. doi: 10.1016/0003-9861(66)90248-7. [DOI] [PubMed] [Google Scholar]
- 8.Laidlaw S A, Kopple J D. Am J Clin Nutr. 1987;46:593–605. doi: 10.1093/ajcn/46.4.593. [DOI] [PubMed] [Google Scholar]
- 9.Hsieh M C, Berry H K. J Exp Zool. 1979;208:161–168. doi: 10.1002/jez.1402080204. [DOI] [PubMed] [Google Scholar]
- 10.Morgan M Y, Marshall A W, Milsom J P, Sherlock S. Lancet. 1987;2:1051–1056. [Google Scholar]
- 11.Kilani R A, Cole F S, Bier D M. Am J Clin Nutr. 1995;61:1218–1223. doi: 10.1093/ajcn/61.6.1218. [DOI] [PubMed] [Google Scholar]
- 12.Denne S C, Karn C A, Ahlrics J A, Dorotheo A R, Wang J, Liechty E A. J Clin Invest. 1996;97:746–754. doi: 10.1172/JCI118473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tessari P, Biolo G, Inchiostro S, Orlando R, Vettore M, Sergi G. Gastroenterology. 1993;104:1712–1721. doi: 10.1016/0016-5085(93)90650-2. [DOI] [PubMed] [Google Scholar]
- 14.Tessari P, Zanetti M, Barazzoni R, Biolo G, Orlando R, Vettore M, Inchiostro S, Perini P, Tiengo A. Gastroenterology. 1996;111:127–137. doi: 10.1053/gast.1996.v111.pm8698191. [DOI] [PubMed] [Google Scholar]
- 15.Meek S E, Persson M, Ford G C, Nair K S. Diabetes. 1998;47:1824–1835. doi: 10.2337/diabetes.47.12.1824. [DOI] [PubMed] [Google Scholar]
- 16.Nair K S, Ford G C, Ekberg K, Fernquist-Forbes E, Wahren J. J Clin Invest. 1995;95:2926–2937. doi: 10.1172/JCI118000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wahren J, Felig P. Diabetes. 1975;24:730–734. doi: 10.2337/diab.24.8.730. [DOI] [PubMed] [Google Scholar]
- 18.Stumvoll M, Chintalapudi U, Periello G, Welle S, Gerich J. J Clin Invest. 1995;96:2528–2533. doi: 10.1172/JCI118314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tessari P, Garibotto G, Inchiostro S, Robaudo C, Saffioti S, Vettore M, Zanetti M, Russo R, Deferrari G. J Clin Invest. 1996;98:1481–1492. doi: 10.1172/JCI118937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charlton M R, Adey D B, Nair K S. J Clin Invest. 1996;98:90–99. doi: 10.1172/JCI118782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson G N, Pacy P J, Meritt H, Ford G C, Read M A, Cheng K N, Halliday D. Am J Physiol. 1989;256:E631–E639. doi: 10.1152/ajpendo.1989.256.5.E631. [DOI] [PubMed] [Google Scholar]
- 22.Berry H K, Cripps R, Nicholls K, McCandless D, Harper C. Biochim Biophys Acta. 1971;261:315–320. doi: 10.1016/0304-4165(72)90053-0. [DOI] [PubMed] [Google Scholar]
- 23.Ayling J E, Boehn G R, Textor S C, Pirson R A. Biochemistry. 1973;12:2045–2051. doi: 10.1021/bi00735a004. [DOI] [PubMed] [Google Scholar]
- 24.Murthy L J, Berry H K. Biochem Med. 1975;12:392–397. doi: 10.1016/0006-2944(75)90072-1. [DOI] [PubMed] [Google Scholar]
- 25.Ayling J E, Pierson W D, Al-Janabi J M, Helfand G D. Biochemistry. 1974;13:78–85. doi: 10.1021/bi00698a013. [DOI] [PubMed] [Google Scholar]
- 26.Felig P, Owen O, Wahren J, Cahill G F. J Clin Invest. 1969;48:584–594. doi: 10.1172/JCI106017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baumann P Q, Stirewalt W S, O'Rourke B D, Howard D, Nair K S. Am J Physiol. 1994;267:E203–E209. doi: 10.1152/ajpendo.1994.267.2.E203. [DOI] [PubMed] [Google Scholar]
- 28.Ljungqvist O, Persson M, Ford G C, Nair K S. Am J Physiol. 1997;273:E564–E570. doi: 10.1152/ajpendo.1997.273.3.E564. [DOI] [PubMed] [Google Scholar]
- 29.Marchesini G, Bianchi G P, Vilstrup H, Checchia G A, Patrono D, Zoll M. J Hepatol. 1987;4:108–117. doi: 10.1016/s0168-8278(87)80017-x. [DOI] [PubMed] [Google Scholar]
- 30.Greiber S, Mitch W E. Miner Electrolyte Metab. 1992;18:233–236. [PubMed] [Google Scholar]
- 31.American Hospital Formulary Service. AHFS Drug Information. Bethesda, MD: Am. Soc. Hospital Pharmacists; 1999. pp. 2287–2293. [Google Scholar]