

Abstract
Investigation of axonal biology in the central nervous system (CNS) is hindered by a lack of an appropriate in vitro method to probe axons independently from cell bodies. Here we describe a microfluidic culture platform that polarizes the growth of CNS axons into a fluidically isolated environment without the use of targeting neurotrophins. In addition to its compatibility with live cell imaging, the platform can be used to (i) isolate CNS axons without somata or dendrites, facilitating biochemical analyses of pure axonal fractions and (ii) localize physical and chemical treatments to axons or somata. We report the first evidence that presynaptic (Syp) but not postsynaptic (Camk2a) mRNA is localized to developing rat cortical and hippocampal axons. The platform also serves as a straightforward, reproducible method to model CNS axonal injury and regeneration. The results presented here demonstrate several experimental paradigms using the microfluidic platform, which can greatly facilitate future studies in axonal biology.
Neurons in the CNS extend axons over considerable distances and through varying extracellular microenvironments to form synapses, the basis of neuronal connectivity. Axonal damage is critical to the etiology of CNS injuries and neurodegenerative disease (for example, spinal cord injury and Alzheimer disease)1-3; therefore, considerable effort focuses on the molecular and cellular mechanisms that influence axonal plasticity and response to injury. In vitro models facilitate the study of axonal biology in the peripheral nervous system (PNS), but no suitable method has been developed for the study of the CNS because of the challenges associated with culturing CNS neurons.
In vitro studies using compartmentalized ‘Campenot’ chambers have greatly improved the understanding of axonal biology within the PNS4-6. Campenot chambers use a compartmented Teflon divider attached to a collagen-coated petri dish via a thinly applied silicone grease layer; typically nerve growth factor (NGF) promotes neuritic growth through the grease layer. Much of the work involving Campenot chambers focused on the influence and transport of NGF. More recently, Campenot chambers have been used to study the effects of lipoproteins on retinal ganglion axonal growth7 and the effect of Rho antagonists on superior cervical ganglion axons8. To date, all neurons cultured in Campenot chambers require the use of either NGF or brain-derived neurotrophic factor (BDNF).
CNS neurons involved in the pathology of most neurodegenerative diseases and injuries (for example, cortical, hippocampal and spinal cord neurons) have not been successfully cultured in Campenot chambers. These neurons are traditionally more difficult to culture and do not have the same dependency on neurotrophic targets for axonal growth as PNS or retinal ganglion neurons. Chambers to isolate hippocampal axons, which used a thin coverslip and a grease layer to separate hippocampal neurites from somata, have also been developed9. These chambers, however, were extremely challenging to fabricate and assemble, precluding high-throughput experimentation. In addition, these chambers had a tendency to leak owing to an imperfect grease seal, and even slight mechanical disturbances caused lesioning of the neurites. Finally, both of the chambers had several problems that restricted adapting the technique for sophisticated microscopy.
Microfluidics is becoming an increasingly useful tool for cell biologists owing to its ability to precisely control, monitor and manipulate cellular microenvironments10-14. Several biological studies use microfluidic systems fabricated with poly(dimethylsiloxane) (PDMS) as a platform for miniature immunoassays, separation of proteins and DNA, sorting and manipulation of cells, and microscale bioreactors15-19. Development of microfabricated devices for neurons has generally been engineering-oriented, to develop retinal protheses20 and to use neurons for biosensor applications17,21. Here we report the use of a microfluidic device for long-term culture and compartmentalization of primary CNS neurons with potential applications in neuroscience experiments.
The microfluidic platform can be used to isolate and direct the growth of CNS axons without the use of neurotrophins, providing a highly adaptable system to model many aspects of CNS neuro-degeneration and injury. We have successfully cultured and manipulated standard CNS neuronal populations (that is, primary rat cortical and hippocampal neurons) within the microfluidic device. We used the culture platform to isolate axonal mRNA from mammalian CNS neurons, an achievement not possible by either in vitro or in vivo methods22. Further, we investigated the utility of the microfluidic platform as an in vitro model of axonal injury; demonstrating the ability to selectively lesion axons and biochemically analyze their somata for immediate early gene expression. Notably, this technique can be used as a method to screen compounds of interest for regenerative potential. Specifically, we show axonally restricted BDNF- and neurotrophin 3 (NT-3)-enhanced regeneration after axotomy. The platform also permits the establishment of axonally restricted cocultures. We cocultured oligodendrocytes with CNS axons to show the potential use of this method to study myelination as well as demyelinating disease. Finally, we demonstrate that this microfluidic culture platform is ideally suited for high-resolution axonal transport studies using live cell imaging with optical microscopy (for example, phase contrast, differential interference contrast, epifluorescence and confocal microscopy).
RESULTS
Fabrication of the microfluidic culture platform
The microfluidic culture platform consists of a molded elastomeric polymer piece placed against a glass coverslip (Fig. 1a,b)23,24. The design of the device incorporates a physical barrier with embedded microgrooves separating two mirror image compartments. Microgrooves that connect the compartments act as a ‘filter’, allowing passage of neuritic processes into the axonal side but not of cell bodies. Within 4 d in vitro after plating dissociated neurons into one of the compartments (somal side), neuritic processes began to extend into the axonal side. The somal compartment contained approximately 3,000 neurons after 7 d in vitro, as labeled by immunocytochemistry with neuronal-specific marker, MAP2. Notably, neuritic growth of cortical and hippocampal neurons is continuous for over 2 weeks in the axonal side without the use of exogenously supplied neurotrophic support.
Figure 1.

The microfluidic-based culture platform directs axonal growth of CNS neurons and fluidically isolates axons. (a) The culture chamber consists of a PDMS mold containing a relief pattern of somal and axonal compartments (1.5 mm wide, 7 mm long, 100 mm high) connected by microgrooves (10 μm wide, 3 μm high). The optically transparent PDMS adheres to a polylysine-coated coverslip. Rat CNS neurons (green) are added to the somal-side reservoir and are drawn into the somal channel (black) by capillary action. Within 3–4 d, axonal growth is guided into the axonal side (yellow) through the microgrooves. (b) A volume difference between the somal side and axonal side (∼50 μl) allows chemical microenvironments to be isolated to axons for over 20 h owing to the high fluidic resistance of the microgrooves. Similarly, the volume difference can be reversed to isolate a chemical microenvironment to the somal side. (c) Fluidic isolation of Texas red dextran (top panel) to the axonal compartment demonstrates that axonal or somatic microenvironments can be independently manipulated using this culture platform. Axonally restricted application of CellTracker Green (middle panel) backtracked neurons from their isolated axons. The bottom image is the merged figure. Scale bar, 100 μm. (d) Counts of radioactivity in samples from somal and axonal compartments after [35S]methionine was localized to the axonal compartment for over 20 h. Counts in the somal compartment (3.7 c.p.m. ± 1.5 s.e.m.) were similar to background levels. Error bars, s.e.m. (n = 3).
Fluidic isolation within the axonal compartment
The platform allows the fluidic isolation of axonal microenvironments by establishing a minute volume difference between the two compartments. The high fluidic resistance of the microgrooves produces a small but sustained flow between the compartments that counteracts diffusion23,24. To demonstrate fluidic integrity of the compartments, we incubated Texas red dextran (3,000 Da; 20 μM) in the axonal compartment for over 20 h. We detected no trace of fluorescence within the somal compartment by either UV spectroscopy (equivalent to background) or confocal microscopy (Fig. 1c). We also validated the fluidic isolation by restricting [35S]methionine (131 Da) to the axonal side of the chamber for over 20 h (Fig. 1d). Scintillation counts of 35S radioactivity revealed an average of 17,537 c.p.m. on the axonal side versus 3.7 c.p.m. on the somal side. In a similar manner, we can establish fluidic isolation of the somal compartment by reversing the hydrostatic pressure (that is, higher volume on the axonal side).
Directional growth and isolation of axons
Having established the fluidic integrity of the compartment, we wanted to empirically establish a barrier length that would allow axonal isolation. To accomplish this we took advantage of the growth properties of axons, which grow faster and longer than dendrites25. After 7 d in culture, we observed that axons, but not dendrites, extended into the axonal side of the platform, as assessed by immunocytochemistry. At 14 d in vitro, however, a small number of MAP2 immunoreactive dendrites (usually four to five per chamber) began to cross into the axonal compartment when we used 150-μm barriers (Fig. 2). As mature receptor phenotype is typically established between 10–14 d in vitro, we widened the barrier widths to isolate mature axons. Multiple immunocytochemical experiments confirmed that up to 14 d in vitro, no detectable dendrites entered the axonal side of chambers when barrier widths were greater than 450 μm (Fig. 2). In this manner barriers can be modified and lengthened to isolate axons at varying culture ages.
Figure 2.

Axons are isolated without somata or dendrites. When barrier widths of 450 μm or more were used, axons (red; tau) extended past the barrier at 14 d in vitro without detecting dendrites (green; MAP2). Frames including the longest dendrites in each chamber were imaged. Dashed lines indicate the barrier region.
Isolation of axonal mRNA
The neuronal cell body is classically considered the exclusive source of axonal proteins. But recent evidence suggests that axonal protein synthesis occurs and is important in the development, maintenance and plasticity of synapses22. Much of this work uses invertebrate models such as Aplysia and squid giant axon owing to the relative ease of isolating axonal material. Vertebrate studies are limited by the experimental challenge of isolating axons without somal and dendritic contamination. Using the microfluidic platform, we isolated mammalian axonal mRNA from the axonal compartment of 6 d in vitro neurons. The presence or absence of specific mRNAs further validated that pure axon fractions can be isolated to the axonal compartment (Fig. 3). Prior evidence suggests that axonal RNA localization is developmentally regulated and diminishes with maturation22. Therefore, we analyzed mRNA isolated from developing axons (6 d in vitro). We isolated approximately 60 ng of axonal RNA from each chamber. RT-PCR analysis revealed the presence of H1 histone (H1f0), calcium/calmodulin-dependent protein kinase II α (Camk2a), β actin (Actb) and synaptophysin (Syp) mRNAs within the somal compartment. In contrast, axons contained β actin and synaptophysin mRNAs, but did not contain H1 histone or calcium/calmodulin-dependent protein kinase II α mRNA. The absence of H1 histone has previously been used to ensure axonal homogeneity of RNA material from axons of chick retinal explants separated from cell bodies by a microknife26. We did not detect H1 histone mRNA, even after as many as 65 rounds of PCR amplification. In addition, we did not detected dendritically targeted calcium/calmodulin-dependent protein kinase II α mRNA27, confirming that isolated axons do not contain dendritic mRNA. Until now, there have been no suitable methods to analyze for the presence of presynaptic mRNAs. Notably, we provide the first evidence that mRNA coding for the presynaptic vesicle protein, synaptophysin, is present within developing axons.
Figure 3.
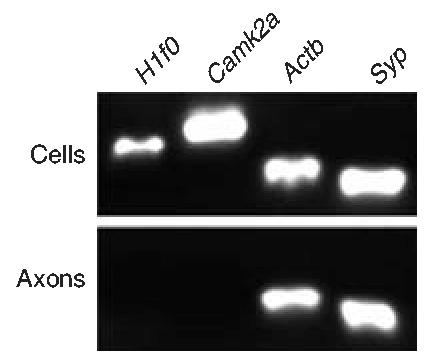
RNA encoding the presynaptic vesicle protein synaptophysin is localized to CNS axons at 6 d in vitro. RT-PCR analysis of the samples from somal and axonal compartments demonstrate that β actin and synaptophysin mRNAs are localized to axons. H1 histone and calcium/calmodulin-dependent protein kinase II α mRNAs were not detected in isolated axons in chambers with 450 μm barriers at 6 d in vitro.
CNS axonal injury and regeneration
The microfluidic platform can serve as an in vitro model for axonal injury and regeneration within the CNS (Fig. 4a). Axotomy induces rapid transcription of immediate early genes within lesioned somata in vivo28. We used the microfluidic platform to measure changes in somal transcription activity in response to selective axotomy. Vacuum aspiration in the axonal compartment (axotomy) was followed by collection of somal fraction. Changes in mRNA expression of the immediate early gene FBJ murine osteosarcoma viral oncogene homolog (Fos; also known as c-fos) were analyzed using RT-PCR (Fig. 4b). Axotomy resulted in a ∼200% increase in induction of c-fos mRNA expression within 15 min of injury, which was further elevated to 320% within 2 h after lesion. These data illustrate the effectiveness of the microfluidic culture platform for analyzing gene expression changes owing to altering axonal environments.
Figure 4.

Axotomy leads to rapid transcription of immediate early genes and regeneration is enhanced by axonal neurotrophin treatment. (a) Axonal side prior to (left), immediately after (middle) and 20 h after (right) axotomy. Arrows indicate axonal regeneration. Scale bar, 50 μm. (b) RNA was isolated from the somal compartment of unlesioned cultures and cultures at 15 min and 2 h after axotomy (9 d in vitro). Semiquantitative RT-PCR was performed for c-fos and GAPDH.PCR products were visualized on a 1% agarose gel and quantified. Error bars, s.e.m. (n = 3; ANOVA, P < 0.0015; Fischer's PLSD P < 0.02). (c) Neurotrophins enhance axonal regeneration following a lesion. Neurofilament immunofluorescence (red) and f actin–labeled growth cones (green, phalloidin), shown approximately 200 μm from the edge of the barrier in the axonal side, reveal extensive axonal arborization and increased ingrowth after axonal BDNF and NT-3 treatment (left), control (right). Data is representative of four experiments. Scale bar, 25 μm.
Next, we validated the use of this platform as a potential method to screen candidate molecules for axonal regeneration. We investigated the influence of axonally localized neurotrophin treatment on regeneration. Lesioned axons (6 d in vitro) were treated with a combination of BDNF (2.4 nM) and NT-3 (6 nM) for 5 d. Controls had an equivalent amount of medium without BDNF and NT-3 added to lesioned axons. Texas red fluorescent dextran (20 μM) was also added to axonal compartments (including controls) to verify axonal localization of neurotrophins. The isolated axons in neurotrophin-treated chambers had a dramatic increase in axonal branching and growth versus control-treated chambers, as assessed by neurofilament immunofluorescence (Fig. 4c). In addition, the neuronal growth cones had a marked increase in density in response to BDNF and NT3 treatment.
DISCUSSION
The microfluidic culture platform provides a new method to direct, isolate, lesion and biochemically analyze CNS axons. Soft lithography uses replica molding to fabricate devices, resulting in reproducible experiments. Notably, we can modify the device design to suit particular experimental applications. For example, proximal or more distal axonal sites can be accessed by varying the barrier length. Another advantage of the microfluidic platform is that it uses hydrostatic pressure to fluidically isolate one side of the chamber; this bypasses the need for external pumps that make implementation more difficult and limit the number of experiments that can be run in parallel. It is also possible to achieve fluidic isolation in both compartments by incorporating constant flow through each compartment.
This microfluidic culture platform provides a unique method to investigate axonal mRNA in developing neurons without detectable somal or dendritic contamination. Future work to characterize cultures of differing maturity will be of interest, as localization of RNA to axons may be developmentally regulated22. Recent evidence suggests that BDNF-induced potentiation of neurotransmitter release requires local axonal protein synthesis29. It is interesting to speculate that the existence of axonal pools of synaptophysin mRNA, which we detected using RT-PCR, may lead to local protein synthesis of this synaptic vesicle protein and thereby mediate this neurotransmitter release.
Spinal cord injury is a challenging field of study, in part because simple and reproducible in vitro models do not exist. Instead, researchers must perform time-consuming and technically demanding studies in vivo that preclude the rapid, high-throughput screening of compounds of interest. Here we demonstrated that the microfluidic platform provides a method to model aspects of CNS axonal injury and can be used to test soluble factors involved in injury and regeneration30,31. Cocultures with other cell types, such as oligodendrocytes or astrocytes will also help to investigate mechanisms of axonal injury and regeneration32,33. Furthermore, somata or axons can be independently exposed, providing an initial screen for new compounds and combinations that may enhance CNS axonal regeneration. In vivo work demonstrated that dorsal-column sensory axons regenerate beyond a lesion site only with combined treatments of cAMP to the neuronal cell bodies and NT-3 to lesioned axons34. With further refinement in design, the microfluidic platform may assess such combinatorial approaches in vitro.
The microfluidic platform provides a tool to study other important areas of axonal biology that have previously proven challenging. For example, studies of axonal transport require establishing transport direction following fixation and tracing. With the microfluidic platform, the embedded microgrooves define transport direction (Supplementary Fig. 1 online). This aspect greatly facilitates time-lapse imaging and quantification of transport owing to the large numbers of axons that can be examined in a given chamber. In addition, investigators can readily assess the influence of localized axonal or somal microenvironments on transport.
The microfluidic platform may also prove invaluable for various coculture applications. For example, we cocultured oligodendrocytes in the axonal compartment to potentially investigate mechanisms of axonal myelination and demyelination (Fig. 5). We also cocultured neurons overexpressing APP (Tg2576) and wild-type neurons in separate compartments (data not shown).
Figure 5.
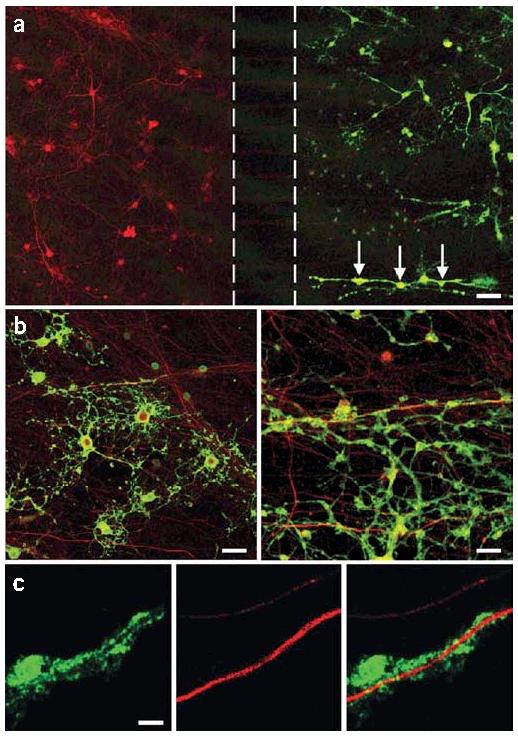
Axons can be cocultured with other cell types such as oligodendrocytes. Oligodendrocytes were added to the axonal compartment of 7 d in vitro neurons. (a) Low-power confocal image demonstrates the coculture of neurons (red, neurofilament) with oligodendrocytes (green, myelin basic protein) in separate compartments for eight additional days. In some cases, oligodendrocytes align themselves along axons (arrows) in a pattern reminiscent of white matter tracts in vivo. Dashed lines indicate the barrier region. Scale bar, 60 μm. (b) Higher-power views demonstrate the association of oligodendrocyte processes (green) with neuronal axons (red). Scale bars, 30 μm(left) and 10 μm (right). (c) A single confocal optical Z-slice demonstrates myelin basic protein immunoreactive processes surround an axon. Scale bar, 4 μm.
The ability to direct growth of neuronal processes and to control the microenvironments of CNS axons offers the chance to overcome several major limitations in the exploration of axonal biology. The results presented here demonstrate a new platform that allows the examination of multiple aspects of axonal biology, including axonal RNA localization, injury and regeneration.
METHODS
Microfluidic culture platform fabrication
We fabricated the chambers in PDMS using soft lithography and replica molding. We used photolithography to pattern two layers of negative photoresist, SU-8, on a silicon wafer, resulting in a master with positive relief patterns of cell culture compartments and microgrooves23,24 (Supplementary Methods online). We cast and cured a prepolymer mixture of Sylgard 184 (Dow Corning) against the positive relief master to obtain a negative replica-molded piece. After curing, we peeled the PDMS away from the master and punched out reservoirs with a sharpened needle. We then sterilized the PDMS pieces with 70% ethanol. We cleaned glass coverslips (24 × 40 mm, No. 1, Corning Inc.) and immersed them in sterile aqueous solution of 1.0 mg/ml poly(l-lysine) (PLL, M.W. 70,000–150,000; Sigma) in borate buffer for 24 h before use. Sealing the PDMS piece to a polylysine-coated glass coverslip by conformal contact formed the enclosed channels (Fig. 1a). This type of reversible contact resulted in both a water-tight seal during use and the separation of the PDMS piece from the glass cell culture surface after the experiment for immunostaining steps.
Cell preparation
In accordance with AAALAC guidelines, the vivarium of the Gillespie Neuroscience Research Laboratories at the University of California, Irvine, housed the rodents. We prepared cortical and hippocampal dissociated neurons from embryonic rat (E18) and mouse (E17) as described previously23,24. We used approximately 3×106 cells/ml, yielding approximately 3,000 cells in the somal side of the chamber.
We isolated oligodendrocytes from postnatal rat pups (P1 or P2) according to previous published procedures35. We mechanically separated the cells by trituration through 18G and 22G needles in cold minimal essential medium (MEM) with 40 mM glucose and 15% fetal bovine serum, then cultured them in a PLL-coated flask. After 10 d, the flask was shaken for 3 h at 250 r.p.m. at 37 °C. We replaced the medium and re-equilibrated the flask in a 5% CO2 incubator for 1 h. The flask was shaken overnight at 37 °C. We collected the oligodendrocytes by passing medium through a 40-μm cell filter.
Isolation of soluble microenvironments
To isolate a soluble microenvironment to the axonal compartment, we established a volume difference of 30 μl between the two compartments with the lesser volume being the axonal side (for molecules greater than 500 Da). For molecules less than 200 Da, we used a volume difference of 50 μl. This small hydrostatic pressure difference resulted in a slow continuous flow through the high-resistance microgrooves and fluidic isolation of the axonal compartment. We then added a soluble compound (1–2 μl) to the axonal compartment. Continuous flow produced by the hydrostatic pressure difference prevented this soluble compound from diffusing into the somal side. We maintained fluidic isolation for more than one day by periodically adding or removing small volumes of medium.
Testing of fluidic isolation
We tested fluidic isolation in triplicate using chambers without cells, using both fluorescein isothiocyanate (FITC)-dextran (3 kDa) and radioactively labeled [35S]methionine (131 Da). We fluidically isolated FITC-dextran to the compartment containing the lesser volume (that is, axonal compartment) to yield a final concentration of 0.2 mg/ml. After more than 20 h, we analyzed 50 μl from each compartment using a UV spectrophotometer and compared these data with varying concentrations of FITC-dextran diluted in medium ranging from 200 p.p.m. to 0.02 p.p.m., and also with a background sample containing medium only. To test fluidic isolation using radioactively labeled [35S]methionine, we added 350 μl of medium to the somal side and then 300 μl to the axonal side. The chambers sat for 10 min after pipetting to minimize potential convective flow from filling chambers. We added concentrated [35S]methionine to the axonal compartment, 0.5 μl in each well, resulting in a final radioactivity of 33 μCi/ml. We gently added the [35S]methionine to the axonal wells far from the entry into the chamber to minimize flow disturbances. After 20 h, we removed the entire volume of the axonal compartment by simultaneously pipetting out the contents of both wells and then repeating the procedure for the somal side with new pipette tips. We added 1 μl of each volume removed from the compartments to the scintillation cocktail and used a liquid scintillation counter to record counts over 1 min.
Fixation and immunocytochemistry
We fixed cultures using 4% paraformaldehyde for 30 min at room temperature. We washed cultures twice with phosphate-buffered saline (PBS) for 5 min, and then permeabilized them using PBS with 0.2% Triton X-100 for 30 min. To block nonspecific binding, we used PBS with 0.2% Triton X-100 and 10% goat serum. We incubated the primary antibodies in PBS with 0.2% Triton X-100 and 5% goat serum at 4 °C overnight. We used the following dilutions of primary antibodies: monoclonal MAP2 1:1000 (Sigma), polyclonal tau 1:500 (gift of L. Binder), neurofilament H 1:200 (Chemicon), Myelin basic protein 1:750 (Sternberger). For MAP2 immunocytochemistry, we used previously developed procedures36. Briefly, we fixed cultures for 20 min, washed them twice in PBS, and then permeabilized them in PBS with 0.2% Triton X-100 for 5 min. We blocked cultures in PBS with 10% BSA for 2 h at 37 °C. We incubated monoclonal MAP2 in PBS with 1% BSA at 4 °C overnight. We rinsed cultures 3× for 10 min, then incubated them with secondary antibody (Alexa Fluor 488 or 568, 1:200; Molecular Probes) in PBS for 1 h. For MBP and CNPase, we used biotinylated mouse secondary antibodies at 1:150, and a streptavidin-conjugated fluorophore. We rinsed cultures 3× then in PBS, 1× water, and mounted them in fluoroMount G in (Southern Biotech). We fluorescently labeled filamentous actin using phalloidin (Molecular Probes). Control experiments with no primary antibody showed no detectable labeling of neuronal cells or processes (data not shown).
Confocal imaging
We captured confocal images on a Bio-Rad Radiance 2100 using lambda-strobing to avoid nonspecific cross-excitation or detection.
RNA isolation and RT-PCR
We isolated RNA using the RNaqueous-Micro kit (Ambion) from both the axonal and somal side of chambers. We isolated the axonal RNA by adding cell lysis solution to the axonal side while continuously aspirating the somal side. To isolate somal RNA, we removed the chamber, scrapped off the cells within the well areas, and then added cell lysis solution to the remaining neurons. We performed RT-PCR on RNA from 6 d in vitro cells and axons isolated from separate chambers to determine presence of marker mRNAs (42 cycles; primers: H1 histone, 5′-ACCCATTGTTCAAGGACAGC-3′ and 5′-ATCAGGTCCCCCAACTTACC-3′; calcium/calmodulin-dependent protein kinase II α, 5′-GCCCGGGAGTATTACAGTGA-3′ and 5′-GGGTTGATGGTCAGCATCTT-3′; β actin, 5′-AGCCATGTACGTAGCCATCC-3′ and 5′-CTCTCAGCTGTGGTGGTGAA-3′; synaptophysin, 5′-CAGTGGGTCTTTGCCATCTT-3′ and 5′-ATCTTGGTAGTGCCCCCTTT-3′). We replicated axonal RT-PCR experiments using separate cultures from different neuronal preparations. We performed RT-PCR for both the immediate early gene c-fos and the house-keeping control gene GAPDH (for normalization) within the same multiplex PCR (40 cycles; primers: GAPDH, 5′-TCCATGACAACTTTGGCATCGTGG-3′ and 5′-GTTGCTGTTGAAGTCACAGGAGAC-3′; c-fos, 5′- TCTTGTTTCCGGCATCATCT-3′ and 5′-GCTCCAGCTCTGTGACCAT-3′). We optimized conditions to produce yields for both the GAPDH and c-fos amplification products within the same reaction, and detected them using the Agilent bioanalyzer system and DNA-500 LabChips. We used Stratview Software (SAS Institute Inc.) to calculate statistics using analysis of variance (ANOVA) and Fischer's PLSD.
Axotomy
We lesioned the axons with vacuum aspiration applied to the axonal compartment for 5 s. Owing to high fluidic resistance of the microgrooves, the aspiration did not disturb the cell bodies in the somal compartment.
Supplementary Material
ACKNOWLEDGMENTS
We thank W. Poon for help with the radioisotope study. We thank A. Anderson and W. Saadi for reviewing the manuscript. This work was partially funded by the National Institutes of Health (AG-000538, AG-20241, NS-40953 and NS-50895) and the CRPF. A.M.T. thanks the National Institutes of Health for a predoctoral fellowship award (F31NS046208-02).
Footnotes
Note: Supplementary information is available on the Nature Methods website.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests (see the Nature Methods website for details).
References
- 1.Terry R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 2.McKerracher L. Spinal cord repair: strategies to promote axon regeneration. Neurobiol. Dis. 2001;8:11–18. doi: 10.1006/nbdi.2000.0359. [DOI] [PubMed] [Google Scholar]
- 3.Medana IM, Esiri MM. Axonal damage: a key predictor of outcome in human CNS diseases. Brain. 2003;126:515–530. doi: 10.1093/brain/awg061. [DOI] [PubMed] [Google Scholar]
- 4.Salehi A, Delcroix JD, Mobley WC. Traffic at the intersection of neurotrophic factor signaling and neurodegeneration. Trends Neurosci. 2003;26:73–80. doi: 10.1016/S0166-2236(02)00038-3. [DOI] [PubMed] [Google Scholar]
- 5.MacInnis BL, Campenot RB. Retrograde support of neuronal survival without retrograde transport of nerve growth factor. Science. 2002;295:1536–1539. doi: 10.1126/science.1064913. [DOI] [PubMed] [Google Scholar]
- 6.Campenot RB. Local control of neurite development by nerve growth factor. Proc. Natl. Acad. Sci. USA. 1977;74:4516–4519. doi: 10.1073/pnas.74.10.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayashi H, Campenot RB, Vance DE, Vance JE. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 2004;279:14009–14015. doi: 10.1074/jbc.M313828200. [DOI] [PubMed] [Google Scholar]
- 8.Bertrand J, Winton MJ, Rodriguez-Hernandez N, Campenot RB, McKerracher L. Application of rho antagonist to neuronal cell bodies promotes neurite growth in compartmented cultures and regeneration of retinal ganglion cell axons in the optic nerve of adult rats. J. Neurosci. 2005;25:1113–1121. doi: 10.1523/JNEUROSCI.3931-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ivins KJ, Bui ET, Cotman CW. β-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol. Dis. 1998;5:365–378. doi: 10.1006/nbdi.1998.0228. [DOI] [PubMed] [Google Scholar]
- 10.Jeon NL, et al. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat. Biotechnol. 2002;20:826–830. doi: 10.1038/nbt712. [DOI] [PubMed] [Google Scholar]
- 11.Tourovskaia A, Figueroa-Masot X, Folch A. Differentiation-on-a-chip: a microfluidic platform for long-term cell culture studies. Lab Chip. 2005;5:14–19. doi: 10.1039/b405719h. [DOI] [PubMed] [Google Scholar]
- 12.Chung BG, et al. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip. 2005;5:401–406. doi: 10.1039/b417651k. [DOI] [PubMed] [Google Scholar]
- 13.Hung PJ, Lee PJ, Sabounchi P, Lin R, Lee LP. Continuous perfusion microfluidic cell culture array for high-throughput cell-based assays. Biotechnol. Bioeng. 2005;89:1–8. doi: 10.1002/bit.20289. [DOI] [PubMed] [Google Scholar]
- 14.Gu W, Zhu X, Futai N, Cho BS, Takayama S. Computerized microfluidic cell culture using elastomeric channels and Braille displays. Proc. Natl. Acad. Sci. USA. 2004;101:15861–15866. doi: 10.1073/pnas.0404353101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 16.Sia SK, Whitesides GM. Microfluidic devices fabricated in poly(dimethylsiloxane) for biological studies. Electrophoresis. 2003;24:3563–3576. doi: 10.1002/elps.200305584. [DOI] [PubMed] [Google Scholar]
- 17.Park TH, Shuler ML. Integration of cell culture and microfabrication technology. Biotechnol. Prog. 2003;19:243–253. doi: 10.1021/bp020143k. [DOI] [PubMed] [Google Scholar]
- 18.Andersson H, van den Berg A. Microfabrication and microfluidics for tissue engineering: state of the art and future opportunities. Lab Chip. 2004;4:98–103. doi: 10.1039/b314469k. [DOI] [PubMed] [Google Scholar]
- 19.Beebe DJ, Mensing GA, Walker GM. Physics and applications of microfluidics in biology. Annu. Rev. Biomed. Eng. 2002;4:261–286. doi: 10.1146/annurev.bioeng.4.112601.125916. [DOI] [PubMed] [Google Scholar]
- 20.Peterman MC, et al. The Artificial Synapse Chip: a flexible retinal interface based on directed retinal cell growth and neurotransmitter stimulation. Artif. Organs. 2003;27:975–985. doi: 10.1046/j.1525-1594.2003.07307.x. [DOI] [PubMed] [Google Scholar]
- 21.Prasad S, Zhang X, Yang M, Ozkan CS, Ozkan M. Neurons as sensors: individual and cascaded chemical sensing. Biosens. Bioelectron. 2004;19:1599–1610. doi: 10.1016/j.bios.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 22.Piper M, Holt C. RNA translation in axons. Annu. Rev. Cell Dev. Biol. 2004;20:505–523. doi: 10.1146/annurev.cellbio.20.010403.111746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor AM, et al. Microfluidic multicompartment device for neuroscience research. Langmuir. 2003;19:1551–1556. doi: 10.1021/la026417v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhee SW, et al. Patterned cell culture inside microfluidic devices. Lab Chip. 2005;5:102–107. doi: 10.1039/b403091e. [DOI] [PubMed] [Google Scholar]
- 25.Dotti CG, Sullivan CA, Banker GA. The establishment of polarity by hippocampal neurons in culture. J. Neurosci. 1988;8:1454–1468. doi: 10.1523/JNEUROSCI.08-04-01454.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brittis PA, Lu Q, Flanagan JG. Axonal protein synthesis provides a mechanism for localized regulation at an intermediate target. Cell. 2002;110:223–235. doi: 10.1016/s0092-8674(02)00813-9. [DOI] [PubMed] [Google Scholar]
- 27.Steward O, Halpain S. Lamina-specific synaptic activation causes domain-specific alterations in dendritic immunostaining for MAP2 and CAM kinase II. J. Neurosci. 1999;19:7834–7845. doi: 10.1523/JNEUROSCI.19-18-07834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiser M, Baker H, Wessel TC, Joh TH. Axotomy-induced differential gene induction in neurons of the locus ceruleus and substantia nigra. Brain Res. Mol. Brain Res. 1993;17:319–327. doi: 10.1016/0169-328x(93)90017-j. [DOI] [PubMed] [Google Scholar]
- 29.Zhang X, Poo MM. Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron. 2002;36:675–688. doi: 10.1016/s0896-6273(02)01023-1. [DOI] [PubMed] [Google Scholar]
- 30.Pearse DD, et al. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat. Med. 2004;10:610–616. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- 31.Filbin MT. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat. Rev. Neurosci. 2003;4:703–713. doi: 10.1038/nrn1195. [DOI] [PubMed] [Google Scholar]
- 32.Chen MS, et al. Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature. 2000;403:434–439. doi: 10.1038/35000219. [DOI] [PubMed] [Google Scholar]
- 33.Silver J, Miller JH. Regeneration beyond the glial scar. Nat. Rev. Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 34.Lu P, Yang H, Jones LL, Filbin MT, Tuszynski MH. Combinatorial therapy with neurotrophins and cAMP promotes axonal regeneration beyond sites of spinal cord injury. J. Neurosci. 2004;24:6402–6409. doi: 10.1523/JNEUROSCI.1492-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J. Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jareb M, Banker G. The polarized sorting of membrane proteins expressed in cultured hippocampal neurons using viral vectors. Neuron. 1998;20:855–867. doi: 10.1016/s0896-6273(00)80468-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.