

Abstract
The Id (inhibitor of DNA binding or inhibitor of differentiation) helix–loop–helix proteins are involved in the regulation of cell growth, differentiation and cancer. The fact that the molecular mechanisms of liver regeneration are not completely understood prompted us to study the fate of Id2 in proliferating liver. Id2 increases in liver regeneration after partial hepatectomy, following the early induction of its gene. Co-immunoprecipitation shows that Id2 forms a complex with E2F4, p130 and mSin3A in quiescent liver and all these components are present at the c-myc promoter as shown using ChIP (chromatin immunoprecipitation). Activation of c-myc during hepatocyte priming (G0–G1 transition) correlates with the dissociation of Id2 and HDAC (histone deacetylase), albeit p130 remains bound at least until 6 h. Moreover, as the G0–G1 transition progresses, Id2 and HDAC again bind the c-myc promoter concomitantly with the repression of this gene. The time course of c-myc binding to the Id2 promoter, as determined by ChIP assays is compatible with a role of the oncoprotein as a transcriptional inducer of Id2 in liver regeneration. Immunohistochemical analysis shows that Id2 also increases in proliferating hepatocytes after bile duct ligation. In this case, the pattern of Id2 presence in the c-myc promoter parallels that found in regenerating liver. Our results may suggest a control role for Id2 in hepatocyte priming, through a p130 dissociation-independent regulation of c-myc.
Keywords: cell cycle, c-myc, E2F, histone deacetylase, Id2, liver regeneration
Abbreviations: BDL, bile duct ligation; ChIP, chromatin immunoprecipitation; Ets, E twenty-six; HCC, hepatocellular carcinoma; HDAC, histone deacetylase; Id, inhibitor of DNA binding or inhibitor of differentiation; METS, mitogenic Ets transcriptional suppressor; PH, partial hepatectomy; pRb, retinoblastoma protein; RT, reverse transcription
INTRODUCTION
The Id (inhibitor of DNA binding or inhibitor of differentiation) helix–loop–helix protein family plays a central role in the regulation of cell growth and differentiation in many mammalian tissues [1,2]. Four members of this family, Id1–Id4, have been identified. The first role described for them was their binding to another group of basic helix–loop–helix transcription factors, blocking their association with DNA [3,4]. Although this association may result in the inhibition of cell differentiation (reviewed in [5,6]), the roles of Id proteins in regulating cell growth are far more complicated than originally thought. Id proteins also associate with other factors involved in cell-cycle regulation, for instance the members of the pRb (retinoblastoma protein) family, key factors in the control of the G1–S-phase transition.
Several tumour cell lines, as well as some primary human tumours, have been shown to express Id genes in a deregulated manner [7]. However, Id2, but not Id1 or Id3, associate with the hypophosphorylated forms of the pocket proteins and disrupt their antiproliferative effects [8].
To investigate further the role of Id2 proteins in cell proliferation, the selection of an appropriate biological model is of crucial importance. Liver regeneration is a highly regulated process of tissue repair and replacement involving proliferation of different liver cell populations [9,10]. This process, involved in the response and defence against toxic insults or viral infections, is of outstanding importance because of the capacity of the liver to regulate its own growth and mass, and it plays a crucial role in degenerative processes such as liver cirrhosis [9,11]. Liver regeneration can be experimentally induced by PH (partial hepatectomy), the surgical removal of ∼70% of the liver [12]. This triggers the proliferation of the remaining parenchyma cells that divide once or twice, to rapidly restore the liver mass before returning to quiescence [9,10]. This behaviour is possible because the hepatocytes have a very high replication capacity, comparable with or exceeding that of precursor cells of proliferating tissues [13]. Much effort has been dedicated to understanding the molecular events that trigger liver regeneration. The role of growth factors in the mitogenic response of hepatocytes after PH has been clearly established [9,14]. Nevertheless, quiescent hepatocytes in normal liver do not respond to proliferating stimuli unless primed [15]. It is accepted that priming corresponds to the G0–G1 transition [9]. The ultimate molecular mechanisms responsible for this process are not yet fully understood, although using a microarray methodology, Locker et al. [16] have identified 54 immediate-early genes up-regulated during priming.
In the present study, we analysed the expression of Id2 gene in regenerating rat liver after PH. As one of the genes that is under the control of pocket proteins is c-myc, we also investigated the possible relation between Id2 and hepatic c-myc induction. The fact that c-myc, an essential regulator of the G0–G1 transition [17,18], is one of the immediate-early genes induced after PH [9,16] adds an additional interest to the present study. Our results suggest a role for Id2 protein in the control of hepatocyte priming and provide new insights into the chromatin-associated mechanisms involved in the induction of c-myc promoter during liver regeneration.
EXPERIMENTAL
Animals
Male pathogen-free Wistar rats (220–260 g) were held in groups of two in cages at 22 °C with a 12 h light/12 h dark cycle and fed ad libitum with free access to water. Animals were cared for and handled in conformance with EEC guidelines [18a]. For PH, the two anterior lobes were removed under anaesthesia using the method of Higgins and Anderson [12] and for BDL (bile duct ligation) model, animals were bile duct ligated according to Kontouras et al. [19]. Animals were killed 28 days after surgery under anaesthesia and the liver was quickly removed. The study was approved by the Research Committee of the University of Valencia.
RT (reverse transcriptase)–PCR studies
Total RNA from rat liver was isolated using the guanidinium thiocyanate method [20]. Aliquots of 2 μg were reverse-transcribed using SuperScript II (GibcoBRL) and subsequently amplified by PCR using AmpliTaq DNA polymerase (Applied Biosystems). The primers used for the different genes studied were 5′-ACGAGCAGCATGAAAGCCTT-3′ and 5′-AGGATGCTGATGTCCGTGTTC-3′ for Id2, and 5′-ACCTCGACTACGACTCGGTG-3′ and 5′-AGAAGCCGCTCCACATACAG-3′ for c-myc. 18 S rRNA was simultaneously amplified using the QuantumRNA 18 S Internal Standards (Ambion) as internal loading control. Reactions were resolved using 2% agarose gel electrophoresis, stained with ethidium bromide and quantified using the GeneGenius System and the Gene Tools analysis software (Syngene).
Real-time quantitative PCR was performed using double-stranded DNAbinding dye Syber Green PCR Master Mix in an ABI GeneAmp 7000 Sequence Detection System (Applied Biosystems). Each reaction was carried out in triplicate for two series of animals, and melting curves were constructed, using Dissociation Curves Software (Applied Biosystems), to ensure that only a single product was amplified. As a quantitative control, 18 S rRNA was also analysed.
Immunoblot analysis
Liver was homogenized in 10 ml of ice-cold RIPA buffer (1.85 mM NaH2PO4, 8.4 mM Na2HPO4, pH 7.9, 0.1 M NaCl, 12 mM deoxycholate, 0.1% SDS, 1% Triton X-100 and 0.5% NaN3) per gram of tissue in the presence of protease inhibitors (Sigma). The homogenate was ultracentrifuged at 50000 rev./min for 45 min in an Optima™ Beckman–Coulter centrifuge using an MLA-130 fixed-angle rotor. Equal amounts of protein were subjected to SDS/10% PAGE and transferred on to nitrocellulose membranes. Id2 was detected by using the corresponding antibody (Santa Cruz sc-489) and secondary horseradish-peroxidase-conjugated antibody. Blots were developed by enhanced chemiluminescence (DuPont). The loading of the gels was checked with an antibody specific for β-actin.
Immunohistochemical detection of Id2
Sections (5 μm thick) of paraffin-embedded liver were hydrated and pre-treated with EDTA pH 8. Sections were incubated with 3% H2O2 for 20 min to inhibit endogenous peroxidase and then with normal pig serum (10% in PBS and 0.1% BSA) for 30 min. Incubation with primary antibody (diluted 1:500) (Zymed Laboratories) was performed overnight at 4 °C. Sections were incubated with secondary antibody, pig anti-rabbit (DakoCytomation), diluted 1:200 for 1 h at room temperature (25 °C), and then with a visualization reagent (ABComplex/horseradish peroxidase; DakoCytomation) for 25 min. Positive reactions were detected with diaminobenzidine.
Co-immunoprecipitation
Liver tissues (2 g) were washed twice with 10 ml of cold PBS, pH 7.4, and immersed in 8 ml of PBS supplemented with 2 μl/ml protease inhibitor and 2 μl/ml each of phosphatase inhibitor cocktails I and II (Sigma). Tissues were disrupted with a Dounce homogenizer and centrifuged at 1500 g for 5 min. The pellets were resuspended in 3 ml of cell lysis buffer (5 mM Hepes, pH 8.0, 85 mM KCl and 0.5% Nonidet P40) supplemented with protease and phosphatase inhibitors as above, incubated on ice for 15 min and centrifuged at 3500 g for 5 min to pellet the nuclei. The nuclei were resuspended in 3 ml of nuclear lysis buffer (1×PBS, pH 7.4, 1% Nonidet P40, 0.5% sodium deoxycholate and 0.1% SDS) supplemented with the abovementioned protease and phosphatase inhibitor cocktails. The nuclear extract was fractionated into aliquots containing 1 mg of protein each.
Co-immunoprecipitation was performed by adding 4 μg of the corresponding antibodies against Id2 (sc-489) E2F4 (sc-866), p130 (sc-317) and mSin3A (sc-994) (Santa Cruz) to each aliquot and incubating overnight at 4 °C in a rotating plate. Controls with a non-related antibody (against Arabidopsis thaliana amidase, provided by Dr G. Ríos, Departamento de Bioquímica y Biología Molecular, Universidad de València) and without antibody were carried out.
Immunocomplexes were captured with protein immunoprecipitation matrix (ExactaCruz F, sc-45043; Santa Cruz) for 4 h, and the pellets were washed three times in protease- and phosphatase-inhibitor-supplemented nuclear lysis buffer. Protein complexes were eluted in Laemmli's loading buffer and subjected to Western blotting analysis using horseradish-peroxidase-conjugated antibody probe (ExactaCruz F sc-45043) and ECL® (enhanced chemiluminescence) advance Western blotting detection kit (Amersham Biosciences).
ChIP (chromatin immunoprecipitation) assay and RNA pol-ChIP
Cross-linking of chromatin, ChIP and RNA pol-ChIP procedures were performed using the method of Sandoval et al. [21]. Briefly, isolated nuclei from formaldehyde-cross-linked livers were lysed and cross-linked chromatin was sonicated to yield fragments of ∼500 bp. Diluted soluble chromatin fragments were pre-cleared with blocked Protein A/G–Sepharose, to discard non-specifically bound chromatin fragments. Immunofractionation of complexes was carried out by adding 2 μg of the corresponding antibodies against Id2, E2F4, P130, mSin3A, RNA pol II (sc-899) and c-myc (sc-764) (Santa Cruz) to aliquots containing 50 μg of DNA each. The immunocomplexes were recovered by centrifugation at 13500 g for 1 min after adding blocked Protein A/G–Sepharose and washing extensively. Immunoselected chromatin was eluted and the formaldehyde cross-linking was reverted. The DNA from all samples was purified with a PCR purification kit (Qiagen), and used for PCR analysis, which was carried out with the following primers: c-myc (promoter) 5′-GTGCCCCTCCCGAGTTCC-3′ and 5′-GCGACTCCGGATCCCTCC-3′; c-myc (coding region) 5′-ACAACCGCAAATGCTCCAGC-3′ and 5′-TCCGGTCAGTTTATGCACCAAG-3′; Id2 (promoter) 5′-ACGGGCATTGGCTGCGAACG-3′ and 5′-GAGGAAAGGCCGGGAGGGAG-3′; Id2 (coding region) 5′-ACAAGAAGGTGACCAAGATGGAA-3′ and 5′-GCGATCTGCAGGTCCAAGAT-3′; α-actin (promoter) 5′-AGGGACTCTAGTGCCCAACACC-3′ and 5′-CCCACCTCCACCCTACCTGC-3′; α-actin (coding region) 5′-AGGATTCCTACGTGGGCGAC-3′ and 5′-TAGAGAGACAGCACCGCCTG-3′; β-actin (coding region) 5′-TTCAACACCCCAGCCATGT-3′ and 5′-GTGGTACGACCAGAGGCATACA-3′.
RESULTS
Id2 gene is up-regulated during liver regeneration
The expression of the Id2 gene was first evaluated by both semiquantitative and quantitative RT–PCR analyses. The steady-state levels of Id2 mRNA were significantly increased early after PH, reaching a maximum at approx. 1 h and returning to control values by 6 h (Figure 1A). This peak of Id2 expression occurs during the hepatocyte priming phase (G0–G1 transition). A second, yet flatter, peak of Id2 expression occurs later during regeneration. In accordance with the results of other workers (see, for instance, [22]), c-myc, apart from being an immediate-early gene [9], displays an expression pattern similar to that of Id2 and its expression is resumed during G1–S transition (Figure 1B). Western blot experiments demonstrate that the level of Id2, which is low in quiescent cells, increases soon after PH, following the activation of Id2 gene transcription (results not shown).
Figure 1. Expression of Id2 and c-myc genes during liver regeneration.

Steady-state levels of Id2 (A) and c-myc (B) mRNA over time after PH, as shown by semi-quantitative and quantitative RT–PCR. In the latter instance, histograms give the data for controls (t=0) and after PH, corrected relative to the level of 18 S rRNA. The value of control was arbitrarily set to 1. Samples were run in triplicate for two independent series of animals. Results are means±S.D. The melting curves are also shown to check that a single specific amplicon is being generated.
Id2 is present in the c-myc promoter in quiescent cells
Id2 is able to bind to pocket proteins regulating their function [23,24]. Specifically, the low amount of Id2 found in quiescent fibroblasts is mainly bound to p130 [25]. The co-immunoprecipitation experiment shown in Figure 2 demonstrated that this binding also occur in quiescent liver cells, where p130 and Id2 form a complex with E2F4 and mSin3A [an important component of a HDAC (histone deacetylase) repressor complex]. Massagué and colleagues have found that complexes between E2F4/5 and the pocket protein p107 are essential for c-myc repression in keratinocytes [26]. As the pocket proteins inhibit the action of E2F by recruiting HDACs [27–29], a mechanism that is also involved in placing keratinocytes in a quiescent state [30], the hypothesis that Id2 is involved in liver proliferation by regulating c-myc gene expression through a histone acetylation-dependent mechanism was a plausible one. To check the potential role of these factors in the regulation of c-myc promoter, we carried out the ChIP assay shown in Figure 3. Id2 is present in the c-myc promoter in quiescent cells, together with E2F4, p130 and mSin3A (Figure 3A). It is worth noting that, to the best of our knowledge, this is the first time that Id2 has been shown to be bound to DNA, although, most likely, on the basis of the current knowledge, the binding was not a direct one. The same samples employed for the ChIP assay were used for RNA pol-ChIP [21]. Briefly, this technique consists of using the chromatin immunoprecipitated with an anti-(RNA pol II) antibody for the PCR detection of the coding region of the desired gene. Therefore it provides an excellent internal control of real-time transcription. At 30 min after PH, c-myc is actively transcribed (Figure 3C) and, at this time, Id2 and mSin3A dissociated from the c-myc promoter (Figure 3A). A further comparison of Figures 3(A) and 3(C) reveals that, at 6 h after PH, when the transcription of c-myc passes through a minimum, Id2 and mSin3A again appear to be bound to the promoter. The p130–E2F4 complex remained attached along the whole priming period, independently of the transcriptional state of c-myc.
Figure 2. Complex-associated forms of Id2 in the nuclei of quiescent liver.

Nuclear extracts were immunoprecipitated with antibodies against p130 (α-p130), Id2 (α-Id2), mSin3A (α-sin3A) and E2F4 (α-E2F4). The proteins in the immunoprecipitates were subjected to electrophoresis (8% PAGE for p130 and 15% PAGE for Id2) and analysed by Western blotting using the antibodies against p130 (B, upper panel) and Id2 (B, lower panel). To assess the specificity of p130 and Id2 antibodies, the input sample (5% of total nuclear aliquot) was also analysed by Western blotting (A). Molecular masses (MW) are given in kDa. As a control, a sample was treated in the immunoprecipitation protocol with a non-related antibody (NR Ab). A further control was carried out in the experiment without added antibody (No Ab).
Figure 3. In vivo binding of several factors to the c-myc gene at different times after PH.

(A) Analysis of the c-myc promoter by ChIP assay with antibodies against E2F4 (α-E2F4), p130 (α-p130), mSin3A (α-mSin3A) and Id2 (α-Id2). For each time, a sample of total chromatin (input) was included in the PCR. No Ab, no antibody. (B) PCR products obtained with oligonucleotides specific for α-actin gene promoter, included as a negative control. (C) RNA pol-ChIP analysis of the c-myc transcription. The same cross-linked chromatin samples used in (A) and (B) were immunoprecipitated with an anti-(RNA pol II) antibody, and the immunoprecipitates were analysed by PCR using specific primers from the c-myc coding region. The presence of RNA polymerase in these regions is taken as a measure of actual transcription. As negative and positive controls, the PCR analysis of the immunoprecipitates with primers from the α- and β-actin gene coding regions respectively were also added.
The c-myc oncoprotein is present in the rat Id2 promoter during hepatocyte priming
Lasorella et al. [25] reported that the overexpression of Id2 in human neuroblastoma cells is associated with the presence of extra copies of c-myc and that c-myc and N-myc activate the expression of Id2 in fibroblasts and neuroblastoma cells respectively. Moreover, c-myc directly induces the expression of Id2 in fibroblasts by binding to its consensus sequences in the promoter of the human gene. To ascertain whether c-myc could also up-regulate Id2 during liver regeneration, we searched for c-myc consensus binding sequences in the rat Id2 promoter to find that it contains a cluster of E-boxes susceptible to binding c-myc, as the human and mouse promoters do [25]. In quiescent liver, c-myc is not substantially bound to the Id2 promoter, but the oncoprotein is recruited soon after PH (Figure 4A). Its presence is clear at 30 min and passes through a maximum at 1 h, concomitant with the early activation of Id2 gene expression (compare Figures 4A and 4C). Interestingly, RNA pol II is bound to Id2 promoter even in control liver, when active transcription has not yet started (Figure 4B). This shows that Id2 gene ranks among those in which RNA polymerase is paused at the promoter, waiting for the signal to initiate transcription. On the other hand, comparison of the RNA pol-ChIP analyses of Figures 3(C) and 4(C) shows that transcription of Id2 is somewhat delayed relative to the transcription of c-myc.
Figure 4. Analysis of the Id2 gene for binding of c-myc and RNA polymerase in vivo to the promoter and for transcription after PH.

Immunoprecipitation of formaldehyde-cross-linked chromatin was carried out in control rat liver and at several times after PH, by using antibodies against c-myc (α-c-myc) (A) or RNA pol II (α-RNA pol II) (B) and (C). Immunoprecipitates were analysed by PCR with primers specific for the Id2 promoter (A and B) or for the Id2 coding region, to show actual transcription of the gene (RNA pol-ChIP assay; C). In the right-hand panel of (A), the PCR products obtained with oligonucleotides specific for α-actin gene promoter were included as a negative control. No Ab, no antibody.
Id2 is up-regulated in BDL
Cirrhosis, which is often regarded as a pre-cancerous condition [31–33], shares some mechanistic aspects with liver regeneration. This fact, together with the Id2 overexpression described in different primary human tumours [8], led us to evaluate the expression of the Id2 gene in another model of hepatocyte proliferation, namely BDL rats, which are often regarded as an experimental model of cirrhosis [19]. After 28 days of BDL, the steady-state levels of Id2 mRNA, determined by RT–PCR, were significantly increased in BDL rat liver (Figure 5A), as those of c-myc are (Figure 5B). The Id2 mRNA was actually translated in liver, as confirmed by Western blotting (Figure 5C). Moreover, immunohistochemical analysis allowed us to confirm that Id2 actually accumulates not only in bile duct cells, but also in hepatocytes (Figures 5D and 5E), where it localizes both to nuclei and cytoplasm. PCNA (proliferating-cell nuclear antigen) analyses showed that a significant proportion of hepatocytes are proliferating in BDL rats (results not shown). These results validate the model used in our experiments.
Figure 5. Expression of the Id2 gene in rat liver 28 days after BDL.

(A) RT–PCR analyses of Id2 mRNA for three representative liver samples obtained 28 days after BDL. C, control. (B) RT–PCR analyses of c-myc mRNA in the same samples as in (A). (C) Western blot analysis of Id2 protein at 28 days after BDL, β-actin was used as loading control. (D) Immunohistochemical localization of Id2 in control rat liver. See the Experimental section for experimental details. (E) Immunohistochemical localization of Id2 in rat liver 28 days after BDL.
Finally, we studied the binding of factors to the c-myc promoter in BDL-rats. Figure 6(A) shows that Id2 and mSin3A are absent from the c-myc promoter, whereas E2F4 and p130 are present, in a manner similar to that observed in hepatocyte priming. This situation correlated with the presence of RNA pol II in the coding region of c-myc (Figure 6B). Therefore many Id2-related characteristics are shared by proliferating hepatocytes, irrespective of the cause that triggers proliferation.
Figure 6. mSin3A and Id2 are released in vivo from the c-myc promoter in cirrhotic rats when the gene is being transcribed.
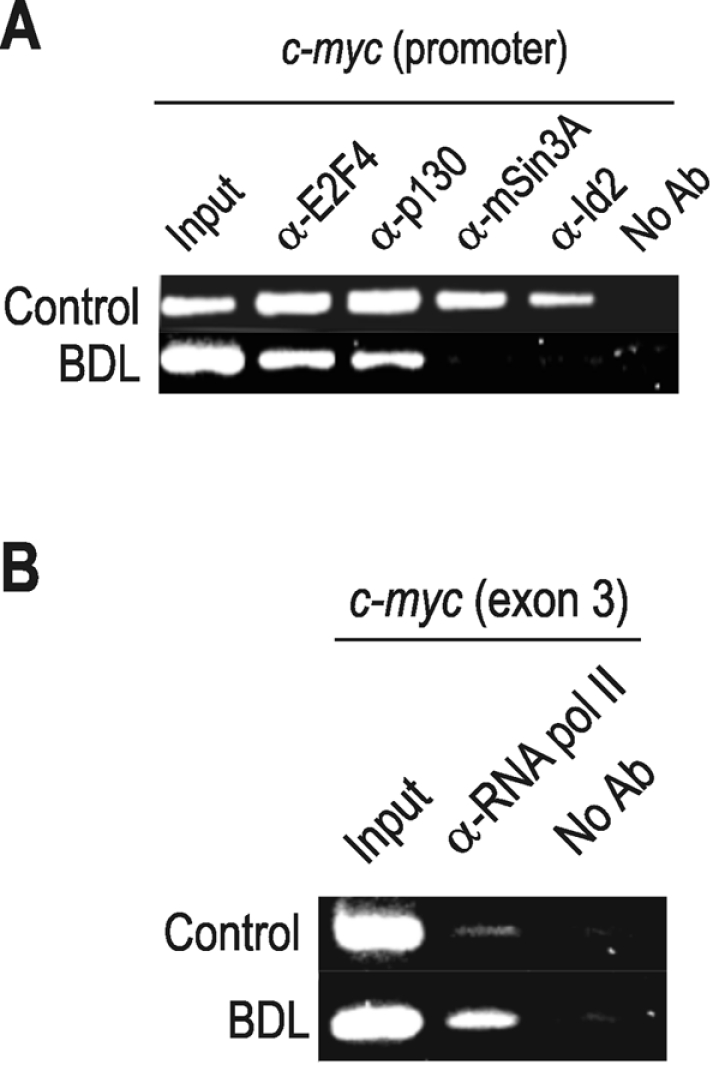
The experiment was conducted as in Figure 3, with samples from rat cirrhotic liver at 28 days after BDL. (A) ChIP analysis was carried out with the antibodies indicated over the PCR results in control and cirrhotic rats. The primers were specific for c-myc promoter. (B) Real-time RNA pol-ChIP analysis of c-myc transcription in normal and cirrhotic rats. An antibody against RNA pol II (α-RNA pol II) was used for immunoprecipitation of chromatin, and the bound DNA was analysed with primers from the third exon of the c-myc gene. No Ab, no antibody.
DISCUSSION
The events that trigger the proliferation of the quiescent hepatocytes after PH are complex and remain unknown in many aspects. The simple infusion of growth factors such as TGFα (transforming growth factor α) or EGF (epidermal growth factor) into the normal liver does not result in increased DNA synthesis, which actually takes place if the infusion is preceded by PH [34]. Therefore hepatocyte proliferation in vivo requires an initial priming stimulus [9], such as PH, which allows the cells to exit from G0. Although their molecular aspects are not yet fully understood, it is known that hepatocyte priming is marked by the induction of c-myc gene expression [35].
Our results show for the first time that Id2 gene expression is up-regulated in rats immediately after PH. Togo et al. [36], working with DNA microarrays, have reported a discrete increase of Id2 transcripts 6 h after PH in mice. Nevertheless, both the quantitative PCR of Figure 1(A) and the RNA pol-ChIP of Figure 4(C) clearly show that the level of Id2 transcription in rats reaches a maximum at approx. 1 h after PH. To date, the central role assigned to Id2 in promoting dedifferentiation and cell proliferation consists in the reversal of the pRb-mediated cell-cycle arrest [23], which in our model would allow the hepatocyte to progress from G1- to S-phase. However, the data presented here suggest another earlier role for Id2 in liver regeneration, namely in the hepatocyte priming, or G0–G1 transition.
The promoter of Id2 is a target for c-myc in many cell types [37–40] and Lasorella et al. [25] have shown that c-myc binding activates Id2 in fibroblasts. The timing of c-myc binding to the Id2 promoter (Figure 4) also agrees with this activating role in regenerating liver, especially during hepatocyte priming. Interestingly, the ChIP experiments of Figure 3 show that Id2 protein, bound to the c-myc promoter in quiescent hepatocytes, leaves it when the gene is expressed. This removal also occurs during hepatocyte priming, concomitantly with the parting of the HDAC complex, and does not result in the displacement of p130 (Figure 3). It is well established that the sequestering of the non-phosphorylated form of the pocket proteins by Id2 results in the progression from G1 to S-phase [41]. This action of Id2 is possible because a second peak of Id2 expression occurs at late G1, for instance, in fibroblasts [42]. During liver regeneration, there is also a second peak of Id2 expression at 24 h (Figures 1A and 4C), which may be involved in the G1–S progression, but the earlier events observed here are markedly different. In a recent work, Russell et al. [43] have shown that Id2 prevents tumour formation in the intestinal epithelium. This result would be explained if Id2 actually were inhibitory of the expression of c-myc in vivo, as our results might indicate. If it were, this unexpected link between Id2 and c-myc would establish an autoregulatory loop between both proteins that may contribute to the exquisite regulation of liver proliferation.
The binding of Id2 to the c-myc promoter is probably indirect as it seems to be recruited to the E2F site through p130. The simultaneous presence of all these factors (Figure 3) shows that binding of E2F and Id2 to pocket proteins is, most likely, not mutually exclusive. On the other hand, the binding of Id2 seems to depend on factors other than its concentration, as the pattern of promoter binding (Figure 3A) does not follow Id2 expression (Figure 1A). The lack of correlation between Id2 concentration and binding to the c-myc promoter also occurs in BDL rats (compare Figures 5D and 6A). It should be kept in mind that the low amounts of Id2 present in quiescent liver are complexed with p130 (Figure 2), as it occurs in quiescent fibroblasts [25], and that the complex containing these components, together with E2F4 and mSin3A, is bound to the c-myc promoter (Figure 3). Interestingly, the early induction of c-myc during hepatocyte priming occurs concomitantly with the removal of both Id2 and the HDAC component mSin3A, while E2F4 and p130 remain bound to the promoter (Figure 3). So, the role of Id2 protein in hepatocyte priming seems to be independent of p130 removal, in contrast with its later role in the G1–S transition [41].
Therefore the control of c-myc transcription during priming is independent of pocket protein displacement, although it may depend on chromatin structure, because activation is concomitant with the removal of the deacetylase complex. The mechanism by which the deacetylases are released during hepatocyte priming remains obscure and may involve other factors, such as NF-κB (nuclear factor κB), a key factor in the activation of c-myc and other immediate-early genes during liver regeneration [44]. On the other hand, the persistence of pocket proteins does not mean that they are ineffective in the priming process. For example, in the growth arrest during macrophage terminal differentiation, i.e. in the exit from the cell cycle to enter G0, Ets (E twenty-six) factors are replaced in the c-myc promoter by the repressor METS (mitogenic Ets transcriptional suppressor), which, in turn, recruits a Sin3A–deacetylase–corepressor complex and the interplay between METS and E2F–pRb complexes can provide a mechanism to exit from the cell cycle [45]. The mechanisms for interaction between METS and E2F–pRb are not known, but Id2 protein might be involved in them owing to its capability of binding pocket proteins as well as repressor complexes. Obviously, more research is needed to ascertain the existence of any of these mechanisms. The use of Id2-knockout mice, in which some aspects of the mechanisms of lactation have been studied [46], may be of great interest to clear up the exact role of Id2 in c-myc control and in hepatocyte priming.
Finally, we wish to emphasize that the occupancy of the c-myc promoter shows an obvious correlation between cirrhotic liver and the priming phase after PH. Cirrhosis constitutes a risk factor for HCC (hepatocellular carcinoma) [47] although the mechanisms for the cirrhosis–HCC progression are still poorly understood. The expression of c-myc in human and animal cirrhotic livers is generally considered to precede the formation of HCC [48–50], so, in view of our results, it could be hypothesized that cirrhotic hepatocytes are in a certain sense ‘primed’ for proliferation. It is desirable to explore this question further, as the feasibility of the ChIP assays might make them a useful technique of prognostic value in evaluating the risks of cirrhosis to HCC transition.
Acknowledgments
This work was supported by grants from The Plan Nacional (BFU2004-01965) and Instituto de Salud Carlos III, (RCMN, Madrid, Spain; C03/08) to J. R. V. and L. T.; BMC2001-2868 from the Ministerio de Ciencia y Tecnología (Spain) to L. F.; BFU2004-03616 from the Ministerio de Educación y Ciencia (Spain) to G. L. R.; Grupos 03/211 from Consellería de Educación, Ciencia y Cultura (Generalitat Valenciana) to L. F.; Instituto de Salud Carlos III (C03/02 and G03/015) to J. M. M. and M. A.; SAFT 2003-05 from the Ministerio de Ciencia y Tecnología (Spain) to J. Sastre.; and Fondazione CRT (Torino, Italy) to M. G. P. J. Sandoval is a recipient of a predoctoral grant from the Consellería de Educación, Ciencia y Cultura (Generalitat Valenciana). We are very indebted to Dr G. Ríos for his helpful assistance in coimmunoprecipitation experiments, and valuable comments on the manuscript.
References
- 1.Norton J. D. Id helix–loop–helix proteins in cell growth, differentiation, tumorigenesis. J. Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 2.Yokota Y., Mori S. Role of Id family proteins in growth control. J. Cell. Physiol. 2002;190:21–28. doi: 10.1002/jcp.10042. [DOI] [PubMed] [Google Scholar]
- 3.Sun X. H., Copeland N. G., Jenkins N. A., Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix–loop–helix proteins. Mol. Cell. Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pesce S., Benezra R. The loop region of the helix–loop–helix protein Id1 is critical for its dominant negative activity. Mol. Cell. Biol. 1993;13:7874–7880. doi: 10.1128/mcb.13.12.7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benezra R. The Id proteins targets for inhibiting tumor cells and their blood supply. Biochim. Biophys. Acta. 2001;1551:F39–F47. doi: 10.1016/s0304-419x(01)00028-2. [DOI] [PubMed] [Google Scholar]
- 6.Benezra R. Regulation by Id. Oncogene. 2001;20:8288–8289. [Google Scholar]
- 7.Lasorella A., Uo T., Iavarone A. Id proteins at the cross-road of development and cancer. Oncogene. 2001;20:8326–8333. doi: 10.1038/sj.onc.1205093. [DOI] [PubMed] [Google Scholar]
- 8.Lasorella A., Iavarone A., Israel M. A. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol. Cell. Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fausto N. Liver regeneration. J. Hepatol. 2000;3:19–31. doi: 10.1016/s0168-8278(00)80412-2. [DOI] [PubMed] [Google Scholar]
- 10.Koniaris L. G., McKillop I. H., Schwartz S. I., Zimmers T. A. Liver regeneration. J. Am. Coll. Surg. 2003;197:634–659. doi: 10.1016/S1072-7515(03)00374-0. [DOI] [PubMed] [Google Scholar]
- 11.Garcia-Trevijano E. R., Martinez-Chantar M. L., Latasa M. U., Mato J. M., Avila M. A. NO sensitizes rat hepatocytes to proliferation by modifying S-adenosylmethionine levels. Gastroenterology. 2002;122:1355–1363. doi: 10.1053/gast.2002.33020. [DOI] [PubMed] [Google Scholar]
- 12.Higgins G. M., Anderson R. M. Experimental pathology of the liver. I: Restoration of the liver of the white rat following surgical removal. Arch. Pathol. 1931;12:186–202. [Google Scholar]
- 13.Overturf K., Al-Dhalimy M., Ou C. N., Finegold M., Grompe M. Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am. J. Pathol. 1997;151:1273–1280. [PMC free article] [PubMed] [Google Scholar]
- 14.Michalopoulos G. K., DeFrances M. C. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 15.Mead J. E., Braun L., Martin D. A., Fausto N. Induction of replicative competence (“priming”) in normal liver. Cancer Res. 1990;50:7023–7030. [PubMed] [Google Scholar]
- 16.Locker J., Tian J., Carver R., Concas D., Cossu C., Ledda-Columbano G. M., Columbano A. A common set of immediate-early response genes in liver regeneration and hyperplasia. Hepatology. 2003;38:314–325. doi: 10.1053/jhep.2003.50299. [DOI] [PubMed] [Google Scholar]
- 17.Jansen-Durr P., Meichle A., Steiner P., Pagano M., Finke K., Botz J., Wessbecher J., Draetta G., Eilers M. Differential modulation of cyclin gene expression by MYC. Proc. Natl. Acad. Sci. U.S.A. 1993;90:3685–3689. doi: 10.1073/pnas.90.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradham C. A., Hatano E., Brenner D. A. Dominant-negative TAK1 induces c-Myc and G0 exit in liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G1279–G1289. doi: 10.1152/ajpgi.2001.281.5.G1279. [DOI] [PubMed] [Google Scholar]
- 18a.European Economic Community. Current Protocols for Protection of Animals Used for Experimental and Other Scientific Purposes, Council Directive 86/609/EEC. 1986.
- 19.Kountouras J., Billing B. H., Scheuer P. J. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br. J. Exp. Pathol. 1984;65:305–311. [PMC free article] [PubMed] [Google Scholar]
- 20.Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 21.Sandoval J., Rodríguez J. L., Tur G., Serviddio G., Pereda J., Boukaba A., Sastre J., Torres L., Franco L., López-Rodas G. RNAPol-ChIP: a novel application of chromatin immunoprecipitation to the analysis of real-time gene transcription. Nucleic Acids Res. 2004;32:e88. doi: 10.1093/nar/gnh091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson N. L., Mead J. E., Braun L., Goyette M., Shank P. R., Fausto N. Sequential protooncogene expression during rat liver regeneration. Cancer Res. 1986;46:3111–3117. [PubMed] [Google Scholar]
- 23.Iavarone A., Garg P., Lasorella A., Hsu J., Israel M. A. The helix–loop–helix protein Id-2 enhances cell proliferation and binds to the retinoblastoma protein. Genes Dev. 1994;8:1270–1284. doi: 10.1101/gad.8.11.1270. [DOI] [PubMed] [Google Scholar]
- 24.Lasorella A., Iavarone A., Israel M. A. Id2 specifically alters regulation of the cell cycle by tumor suppressor proteins. Mol. Cell. Biol. 1996;16:2570–2578. doi: 10.1128/mcb.16.6.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasorella A., Noseda M., Beyna M., Yokota Y., Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature (London) 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 26.Chen C. R., Kang Y., Siegel P. M., Massagué J. E2F4/5 and p107 as Smad cofactors linking the TGFβ receptor to c-myc repression. Cell. 2002;110:19–32. doi: 10.1016/s0092-8674(02)00801-2. [DOI] [PubMed] [Google Scholar]
- 27.Brehm A., Miska E. A., McCance D. J., Reid J. L., Bannister A. J., Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature (London) 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 28.Luo R. X., Postigo A. A., Dean D. C. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 29.Magnaghi-Jaulin L., Groisman R., Naguibneva I., Robin P., Lorain S., Le Villain J. P., Troalen F., Trouche D., Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature (London) 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 30.Iavarone A., Massagué J. E2F and histone deacetylase mediate transforming growth factor β repression of cdc25A during keratinocyte cell cycle arrest. Mol. Cell. Biol. 1999;19:916–922. doi: 10.1128/mcb.19.1.916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tsuda H., Hirohashi S., Shimosato Y., Terada M., Hasegawa H. Clonal origin of atypical adenomatous hyperplasia of the liver and clonal identity with hepatocellular carcinoma. Gastroenterology. 1988;95:1664–1666. doi: 10.1016/s0016-5085(88)80093-3. [DOI] [PubMed] [Google Scholar]
- 32.Yasui H., Hino O., Ohtake K., Machinami R., Kitagawa T. Clonal growth of hepatitis B virus-integrated hepatocytes in cirrhotic liver nodules. Cancer Res. 1992;52:6810–6814. [PubMed] [Google Scholar]
- 33.Findor J., He X. S., Sord J., Terg R., Gershwin M. E. Primary biliary cirrhosis and hepatocellular carcinoma. Autoimmun. Rev. 2002;1:220–225. doi: 10.1016/s1568-9972(02)00050-2. [DOI] [PubMed] [Google Scholar]
- 34.Webber E. M., Godowski P. J., Fausto N. In vivo response of hepatocytes to growth factors requires an initial priming stimulus. Hepatology. 1994;19:489–497. [PubMed] [Google Scholar]
- 35.Makino R., Hayashi K., Sugimura T. c-Myc transcript is induced in rat liver at a very early stage of regeneration or by cycloheximide treatment. Nature (London) 1984;310:697–698. doi: 10.1038/310697a0. [DOI] [PubMed] [Google Scholar]
- 36.Togo S., Makino H., Kobayashi T., Morita T., Shimizu T., Kubota T., Ichikawa Y., Ishikawa T., Okazaki Y., Hayashizaki Y., Shimada H. Mechanism of liver regeneration after partial hepatectomy using mouse cDNA microarray. J. Hepatol. 2004;40:464–471. doi: 10.1016/j.jhep.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Siegel P. M., Shu W., Massagué J. Mad upregulation and Id2 repression accompany transforming growth factor (TGF)-mediated epithelial cell growth suppression. J. Biol. Chem. 2003;278:35444–35450. doi: 10.1074/jbc.M301413200. [DOI] [PubMed] [Google Scholar]
- 38.Haggerty T. J., Zeller K. L., Osthus R. C., Wonsey D. R., Dang C. V. A strategy for identifying transcription factor binding sites reveals two classes of genomic c-Myc target sites. Proc. Natl. Acad. Sci. U.S.A. 2003;100:5313–5318. doi: 10.1073/pnas.0931346100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fernandez P. C., Frank S. R., Wang L., Schroeder M., Liu S., Greene J., Cocito A., Amati B. Genomic targets of the human c-Myc protein. Genes Dev. 2003;17:1115–1129. doi: 10.1101/gad.1067003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wartiovaara K., Barnabe-Heider F., Miller F. D., Kaplan D. R. N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J. Neurosci. 2002;22:815–824. doi: 10.1523/JNEUROSCI.22-03-00815.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zebedee Z., Hara E. Id proteins in cell cycle control and cellular senescence. Oncogene. 2001;20:8317–8325. doi: 10.1038/sj.onc.1205092. [DOI] [PubMed] [Google Scholar]
- 42.Hara E., Yamaguchi T., Nojima H., Ide T., Campisi J., Okayama H., Oda K. Id-related genes encoding helix–loop–helix proteins are required for G1 progression and are repressed in senescent human fibroblasts. J. Biol. Chem. 1994;269:2139–2145. [PubMed] [Google Scholar]
- 43.Rusell G. R., Lasorella A., Dettin L. E., Iavarone A. Id2 drives differentiation and suppresses tumor formation in the intestinal epithelium. Cancer Res. 2004;64:7220–7225. doi: 10.1158/0008-5472.CAN-04-2095. [DOI] [PubMed] [Google Scholar]
- 44.Kirillova I., Chaisson M., Fausto N. Tumor necrosis factor induces DNA replication in hepatic cells through nuclear factor κB activation. Cell Growth Differ. 1999;10:819–828. [PubMed] [Google Scholar]
- 45.Klappacher G. W., Lunyak V. V., Sykes D. B., Sawka-Verhelle D., Sage J., Brard G., Ngo S. D., Gangadharan D., Jacks T., Kamps M. P., et al. An induced Ets repressor complex regulates growth arrest during terminal macrophage differentiation. Cell. 2002;109:169–180. doi: 10.1016/s0092-8674(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 46.Mori S., Nishikawa S. I., Yokota Y. Lactation defect in mice lacking the helix–loop–helix inhibitor Id2. EMBO J. 2000;19:5772–5781. doi: 10.1093/emboj/19.21.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim J. W., Wang X. W. Gene expression profiling of preneoplastic liver disease and liver cancer: a new era for improved early detection and treatment of these deadly diseases? Carcinogenesis. 2003;24:363–369. doi: 10.1093/carcin/24.3.363. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X. K., Huang D. P., Qiu D. K., Chiu J. F. The expression of c-myc and c-N-ras in human cirrhotic livers, hepatocellular carcinomas and liver tissue surrounding the tumors. Oncogene. 1990;5:909–914. [PubMed] [Google Scholar]
- 49.Pascale R. M., Simile M. M., De Miglio M. R., Muroni M. R., Calvisi D. F., Asara G., Casabona D., Frau M., Seddaiu M. A., Feo F. Cell cycle deregulation in liver lesions of rats with and without genetic predisposition to hepatocarcinogenesis. Hepatology. 2002;35:1341–1350. doi: 10.1053/jhep.2002.33682. [DOI] [PubMed] [Google Scholar]
- 50.Block T. M., Mehta A. S., Fimmel C. J., Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093–5107. doi: 10.1038/sj.onc.1206557. [DOI] [PubMed] [Google Scholar]