

Abstract
Paramecium bursaria chlorella virus, a large DNA virus that replicates in unicellular Chlorella-like algae, encodes an RNA triphosphatase which is involved in the synthesis of the RNA cap structure found at the 5′ end of the viral mRNAs. The Chlorella virus RNA triphosphatase is the smallest member of the metal-dependent RNA triphosphatases that include enzymes from fungi, DNA viruses, protozoans and microsporidian parasites. In the present study, we investigated the ability of various vanadate oxoanions to inhibit the phosphohydrolase activity of the enzyme. Fluorescence spectroscopy and CD studies were used to directly monitor the binding of decavanadate to the enzyme. Moreover, competition assays show that decavanadate is a potent non-competitive inhibitor of the phosphohydrolase activity, and mutagenesis studies indicate that the binding of decavanadate does not involve amino acids located in the active site of the enzyme. In order to provide additional insight into the relationship between the enzyme structure and decavanadate binding, we correlated the effect of decavanadate binding on protein structure using both CD and guanidinium chloride-induced denaturation as structural indicators. Our data indicated that no significant modification of the overall protein architecture was occurring upon decavanadate binding. However, both fluorescence spectroscopy and CD experiments clearly revealed that the binding of decavanadate to the enzyme significantly decreased the structural stability of the enzyme. Taken together, these studies provide crucial insights into the inhibition of metal-dependent RNA triphosphatases by decavanadate.
Keywords: chlorella virus, circular dichroism, denaturation studies, enzymology, fluorescence spectroscopy
Abbreviations: ANS, 1-anilino-8-naphthalenesulfonate; DTT, dithiothreitol; GdnCl, guanidinium chloride
INTRODUCTION
Hydrolysis of the γ-phosphate of nascent mRNAs by RNA triphosphatases constitutes the first step in the synthesis of the cap structure found at the 5′ end of eukaryotic mRNAs [1]. The cap structure is critical for translation, stability and transport of mRNAs from the nucleus to the cytoplasm [2]. Not surprisingly, many viruses have developed elegant strategies allowing the synthesis of a cap structure at the 5′ end of their viral mRNAs [3]. Because of their importance in mRNA metabolism, the viral enzymes involved in the synthesis of this structure appear as appealing targets for the development of antivirals.
Two families of RNA triphosphatases have been identified through the purification and characterization of mammalian, yeast, protozoan and viral enzymes. The first family consists of RNA triphosphatases from metazoans and plants that possess the ability to catalyse the hydrolysis of the γ-phosphate of mRNAs [4–6]. These cation-independent enzymes belong to the cysteine phosphatase family and catalyse a two-step reaction that involves a covalent cysteinyl-phosphoenzyme intermediate [6]. The second family of RNA triphosphatases, encompassing enzymes from fungi, viruses, protozoan and microsporidian parasites, is characterized by three collinear motifs which include amino acids that are essential for activity [7–11]. Catalysis by these enzymes involves the attack of a water molecule in close proximity to the γ-phosphate with no formation of a covalent intermediate [7]. The members of this family can also hydrolyse nucleoside triphosphates to nucleoside diphosphates [12,13].
Analysis of the crystal structure of both the Saccharomyces cerevisiae [7] and mouse RNA triphosphatases [4], in conjunction with the recent crystallization of the baculovirus RNA triphosphatase [14], has brought new insights into the enzymatic reactions catalysed by these proteins. For instance, the structure of the S. cerevisiae enzyme revealed a distinctive active site that is characterized by an eight-stranded β-barrel that defines a topologically closed tunnel [7]. Based upon the crystal structure, a catalytic mechanism has been proposed in which the essential acidic amino acids located on the base of the tunnel would co-ordinate the essential divalent cation that is adjacent to the γ-phosphate [7]. The essential bivalent cation would allow the activation of the γ-phosphate for attack by water, and the stabilization of a pentaco-ordinate phosphorane transition state [7]. Moreover, amino acids located on the walls and roof of the tunnel appear to contribute to the co-ordination of the γ-phosphate, and to the stabilization of the negative charge on the γ-phosphate that arises during the formation of the transition state [7]. On the other hand, analysis of both the mouse and baculovirus RNA triphosphatase crystals revealed a completely different mechanism of action [4,14]. Despite sharing the HCXXXXXR(S/T) P-loop signature motif and the core α/β fold with the other members of the cysteine phosphatase family, the mechanism of action of these RNA triphosphatases is different from the one utilized by the other members of the cysteine phosphatase family [15–18]. The main difference occurs in the absence of a carboxylate general acid catalyst in the RNA triphosphatase enzymes. Moreover, the crystal structures of the mouse and baculovirus enzymes revealed the importance of backbone amides and side chains located in the P-loop motif for the binding of the hydrolysable phosphate, and in the stabilization of the transition state [4,14]. Indeed, analysis of the baculovirus enzyme identified a direct catalytic role of an asparagine residue located in the P-loop motif (HCXXXXNRT) that would be involved in orienting a water molecule for its attack on the phosphoenzyme [14].
Vanadium is a transition element that occurs in a number of oxometalate forms known as vanadates [19]. Orthovanadate (VO43−) is a phosphate analogue that has been effectively used as a mechanistic probe of enzymes that catalyse phosphoryl-transfer reactions [19–22]. Orthovanadate has been shown to inhibit the enzymatic activity of protein phosphatases by acting as a transition state mimic [23]. It has been reported previously that orthovanadate can inhibit the metal-independent mouse RNA triphosphatase in a concentration-dependent fashion [5]. Orthovanadate binds to metal-independent phosphatases as a transition state analogue, adopting a trigonal bypiramidal structure, thereby inhibiting the phosphohydrolase reaction [23]. In contrast, the phosphohydrolase activity of metal-dependent RNA triphosphatases such as the S. cerevisiae RNA triphosphatase is not inhibited by orthovanadate [24].
Additional vanadate oxoanions such as metavanadate (VO3−) and decavanadate (V10O286−) have been shown previously to inhibit a number of different phosphohydrolase reactions by affecting the architecture of proteins and by reducing the binding of the nucleic acid substrates [19–21]. Could these vanadate oxoanions inhibit the phosphohydrolase activity of metal-dependent RNA triphosphatases? In the present study, we investigated the interaction between different vanadate oxoanions and the metal-dependent RNA triphosphatase of the paramecium bursaria chlorella virus, a large DNA virus that replicates in unicellular Chlorella-like algae [25]. The chlorella virus RNA triphosphatase is the smallest member of the metal-dependent RNA triphosphatases and is therefore well suited for mechanistic studies [26]. Our results show that decavanadate inhibits the phosphohydrolase activity of the chlorella virus RNA triphosphatase. Using a combination of fluorescence spectroscopy, CD and denaturation assays, we investigated the mechanism by which decavanadate inhibits the RNA triphosphatase activity of chlorella virus.
EXPERIMENTAL
chlorella virus RNA triphosphatase expression and purification
A plasmid for the expression of a full-length chlorella virus RNA triphosphatase protein was generated by inserting the A449R gene between the NdeI and BamHI cloning sites of the pET-28a expression plasmid (Novagen). In this context, the enzyme is fused in-frame with a N-terminal peptide containing six tandem histidine residues, and expression of the His-tagged protein is driven by a T7 RNA polymerase promoter. The resulting recombinant plasmid was transformed into Escherichia coli BL21(DE3), and a 1000 ml culture was grown at 37 °C in Luria–Bertani medium containing 30 μg/ml kanamycin until the attenuance (D600) reached 0.5. The culture was adjusted to 2% ethanol and 0.4 mM isopropyl β-D-thiogalactoside (IPTG), and the incubation continued at 18 °C for 20 h. The cells were harvested by centrifugation, and the pellet stored at −80 °C. All subsequent procedures were performed at 4 °C. Thawed bacterial pellets were resuspended in 50 ml of lysis buffer A [50 mM Tris/HCl (pH 7.5), 150 mM NaCl and 10% sucrose] and cell lysis achieved by the addition of lysozyme and Triton X-100 to final concentrations of 50 μg/ml and 0.1% respectively. The lysates were sonicated to reduce viscosity, and any insoluble material removed by centrifugation at 13000 rev./min for 45 min. The soluble extract was applied to a 5 ml column of Ni-nitrilotriacetic acid–agarose (Qiagen) that had been equilibrated with buffer A containing 0.1% Triton X-100. The column was washed with the same buffer, and then eluted stepwise with buffer B [50 mM Tris/HCl (pH 8.0), 0.1 M NaCl and 10% glycerol] containing 50, 100, 200, 500 and 1000 mM imidazole. The polypeptide composition of the column fractions was monitored by SDS/PAGE. The recombinant protein was retained on the column and recovered in the 500 mM imidazole eluate. Following dialysis against buffer C [50 mM Tris/HCl (pH 8.0), 50 mM NaCl, 2 mM DTT (dithiothreitol) and 10% glycerol], the enzyme was stored at −80 °C. The protein concentration was determined using the Bio-Rad dye binding method using BSA as a standard.
Phosphohydrolase assays
ATPase reactions were performed in a buffer containing 50 mM Tris/HCl (pH 7.0), 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP and enzyme as indicated. The reactions were incubated at 37 °C for 15 min, and stopped by the addition of formic acid (1.25 M). RNA triphosphatase reactions were performed in a buffer containing 50 mM Tris/HCl (pH 7.0), 5 mM DTT, 1 mM MgCl2, 1 pmol of 5′-[γ-32P]RNA and enzyme as indicated. The reaction mixtures were incubated at 37 °C for 15 min, and stopped by the addition of formic acid (1.25 M). Aliquots of the phosphohydrolase reactions were applied to a polyethyleneimine–cellulose TLC plate. Aliquots of the ATPase reactions were developed with 1 M formic acid and 0.5 M LiCl, whereas aliquots of the RNA triphosphatase reactions were developed with 0.75 M potassium phosphate (pH 4.3). The release of [32P]Pi was evaluated by scanning the TLC plate with a PhosphorImager (Molecular Dynamics). The average for two single independent experiments is presented.
Preparation of RNA substrates
A radiolabelled RNA substrate of 84 nucleotides was synthesized with the MAXIscript® kit (Ambion) using T7 RNA polymerase. The RNA transcript was synthesized in the presence of [γ-32P]GTP from the pBS-KSII+ plasmid (Stratagene) that had been linearized with HindIII. The RNA substrate was purified by denaturing 10% PAGE and visualized by UV shadowing. The corresponding band was excised, and then eluted from the gel by an overnight incubation in 0.1% SDS/0.5 M ammonium acetate. The RNA was precipitated with ethanol and quantified by spectrophotometry.
Fluorescence measurements
Fluorescence was measured using an Hitachi F-2500 fluorescence spectrophotometer. Excitation was performed at a wavelength of 290 nm. Background emission was eliminated by subtracting the signal from either buffer alone or buffer containing the appropriate quantity of substrate.
The extent to which vanadate oxoanions bind to the protein was determined by monitoring the fluorescence emission of a fixed concentration of proteins and titrating with a given ligand. The binding can be described by eqn (1):
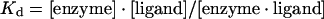 |
(1) |
where Kd is the apparent dissociation constant, [enzyme] is the concentration of the protein, [enzyme·ligand] is the concentration of complexed protein and [ligand] is the concentration of unbound ligand.
The proportion of ligand-bound protein as described by eqn (1) is related to measured fluorescence emission intensity by eqn (2):
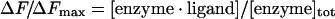 |
(2) |
where ΔF is the magnitude of the difference between the observed fluorescence intensity at a given concentration of ligand and the fluorescence intensity in the absence of ligand, Fmax is the difference at infinite [ligand], and [enzyme]tot is the total protein concentration.
If the total ligand concentration, [ligand]tot, is in large molar excess relative to [enzyme]tot, then it can be assumed that [ligand] is approximately equal to [ligand]tot. Eqns (1) and (2) can then be combined to give eqn (3):
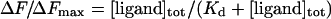 |
(3) |
The Kd values were determined from a non-linear least-squares regression analysis of titration data by using eqn (3). The average for two single independent experiments is presented.
CD spectroscopy measurements
CD measurements were performed with a Jasco J-810 spectropolarimeter. The samples were analysed in quartz cells with pathlengths of 1 mm. Far-UV and near-UV wavelength scans were recorded from 200 to 250 nm and from 250 to 340 nm respectively. All the CD spectra were corrected by subtraction of the background for the spectrum obtained with either buffer alone or buffer containing the ligand. The average of three wavelength scans is presented. The ellipticity results were expressed as mean residue ellipticity, [θ], in degrees·cm2·dmol−1.
Equilibrium unfolding experiments
A 2 μM solution of purified protein was adjusted to the desired final concentration of GdnCl (guanidinium chloride) and incubated for 30 min at 25 °C. The intrinsic fluorescence of the protein was then monitored as a function of the GdnCl concentration. The parameters ΔGu (Gibbs free energy of unfolding), ΔG°u (Gibbs free energy of unfolding in the absence of denaturant), m (co-operativity of unfolding), and Cm (midpoint concentration of denaturant required to unfold 50% of the protein) were obtained as previously outlined [27] using eqn (4):
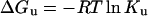 |
(4) |
and eqn (5):
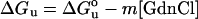 |
(5) |
Alternatively, thermal transitions were monitored by following the change in CD ellipticity of the protein (15 μM) at 222 nm. The samples were heated from 20 °C to 95 °C, at a heating rate of 1 °C/min. The ellipticity results were expressed as mean residue ellipticity, [θ], in degrees·cm2·dmol−1.
ANS binding measurements
Binding of ANS (1-anilino-8-naphthalenesulfonate) was evaluated by measuring the fluorescence enhancement of ANS (50 μM) upon excitation at a wavelength of 380 nm. The emission spectra were integrated from 400 to 600 nm. The average of two single independent experiments is presented.
RESULTS
Expression and purification of the chlorella virus RNA triphosphatase
In order to investigate the ability of various vanadate oxoanions to inhibit the enzymatic activity of viral RNA triphosphatases, the chlorella virus RNA triphosphatase was expressed in E. coli as described in the Experimental section. The enzyme was sequentially purified from soluble bacterial extracts by affinity chromatography. SDS/PAGE analysis showed that the 24.6 kDa protein was the predominant polypeptide in the purified fraction (Figure 1A). The concentration of chlorella virus RNA triphosphatase in this fraction was estimated to be 460 μg/ml. All subsequent experiments were performed using this purified phosphocellulose fraction.
Figure 1. Expression and purification of the chlorella virus RNA triphosphatase.

(A) Aliquots (3 μg) of various protein purification steps were analysed by electrophoresis through 12.5% PAGE containing 0.1% SDS and visualized by staining with Coomassie Blue. The positions and sizes (in kDa) of the molecular-mass markers are indicated on the left. The protein was eluted from the Ni-nitrilotriacetic acid–agarose with buffer [50 mM Tris/HCl (pH 8.0), 0.1 M NaCl, and 10% glycerol] containing 50 (lane 1), 100 (lane 2), 200 (lane 3) and 500 mM (lane 4) imidazole. (B) ATPase activity of the enzyme. ATPase reactions were performed in a buffer containing 50 mM Tris/HCl (pH 7.0), 5 mM DTT, 2 mM MnCl2, 1 mM [γ-32P]ATP and 240 nM of the enzyme. The reactions were incubated at 37 °C for 15 min and aliquots of the phosphohydrolase reactions were analysed by TLC. A double-reciprocal plot is shown in the inset.
Inhibition of the phosphohydrolase activity by orthovanadate, metavanadate and decavanadate
In solution vanadium occurs in a number of oxometalate forms known as vanadates, which have been used as mechanistic probes for a number of different enzymes that catalyse phosphoryl-transfer reactions [19–22]. The ability of orthovanadate (VO43−), metavanadate (VO3−) and decavanadate (V10O286−) to inhibit the phosphohydrolase activity of the chlorella virus RNA triphosphatase was initially investigated by measuring the release of radiolabelled inorganic phosphate from 250 μM [γ-32P]ATP during a 15 min reaction at 30 °C in the presence of 2 mM manganese. In the absence of vanadate oxoanions, the extent of inorganic phosphate release was proportional to input protein (Figure 1B), and an apparent Km value of 20 μM was determined by a double-reciprocal plot for the ATP substrate. Addition of decavanadate to the reaction had a pronounced effect on the phosphohydrolase reaction. Half of the reaction was inhibited at a decavanadate concentration of 100 μM (Figure 2A). Moreover, the presence of 1 mM decavanadate resulted in >90% inhibition of the nucleotide phosphohydrolase activity. The fact that decavanadate elicits a 50% inhibition when present at a concentration 20-fold less than input manganese strongly argues against the possibility that the inhibition results from the simple chelation of the essential manganese ions. Metavanadate was also shown to inhibit the ATPase activity, albeit to a lesser extent (50% inhibition at 150 μM). Interestingly, the addition of orthovanadate, a phosphate analogue that has been shown previously to significantly inhibit the metal-independent activity of the mouse RNA triphosphatase [5], had no significant influence on the nucleotide phosphohydrolase reaction catalysed by the chlorella virus RNA triphosphatase (results not shown). High concentrations of orthovanadate only resulted in limited inhibition of the reaction (50% inhibition at 4 mM). Note that both decavanadate and metavanadate were also shown to be potent inhibitors of the RNA triphosphatase reaction, resulting in a 50% inhibition at concentrations of 600 μM and 900 μM respectively (Figure 2B). In contrast, orthovanadate was not a very potent inhibitor of the RNA triphosphatase activity [50% inhibition at 3.5 mM (results not shown)].
Figure 2. Effect of vanadate oxoanions on the chlorella virus RNA triphosphatase phosphohydrolase activity.

(A) ATPase assays were performed in the presence of increasing concentrations of decavanadate (●) or metavanadate (○). (B) RNA triphosphatase assays were performed in the presence of decavanadate (●) or metavanadate (○). RNA triphosphatase reactions were performed in a buffer containing 50 mM Tris/HCl (pH 7.0), 5 mM DTT, 1 mM MgCl2, 1 pmol of 5′[γ-32P]RNA and 40 nM of the chlorella virus RNA triphosphatase. The reactions were incubated at 37 °C for 30 min and aliquots of the RNA triphosphatase reactions were analysed by TLC.
Binding of vanadate to the enzyme
Vanadate oxoanions have been shown to inhibit phosphoryl transfer reactions by a number of different mechanisms [19–22]. Given the fact that limited studies have focused on the effect of polymeric oxovanadates (such as decavanadate) on phosphohydrolase activity, we initially decided to use fluorescence emission spectroscopy to monitor the direct binding of the decavanadate oxoanion to the chlorella virus RNA triphosphatase. The fluorescence emission spectrum of the purified enzyme in standard buffer at 22 °C is shown in Figure 3(A). In order to obtain the maximal emission peak at the low concentrations of protein required to accurately determine Kd values, excitation was carried out at 290 nm, which is specific for tryptophan [28]. The chlorella virus RNA triphosphatase harbours three tryptophan residues which provided a significant fluorescence signal upon excitation at 290 nm. The emission maximum of the enzyme (333 nm) is blue-shifted relative to that of free L-tryptophan, which under the same conditions is observed to be at 350 nm. The λmax of tryptophan is highly sensitive to the polarity of the microenvironment in which its indole side chain is located. Blue-shifts of protein emission spectra have been ascribed to shielding of the tryptophan residues from the aqueous phase [29]. This shielding is the result of the protein's three-dimensional structure. Accordingly, denaturation of the protein with 8 M urea results in a red-shift of λmax towards 350 nm (Figure 3A).
Figure 3. Binding of decavanadate to the chlorella virus RNA triphosphatase.

(A) Background corrected fluorescence emission spectra of the enzyme. 1, purified enzyme in 50 mM Tris/HCl and 50 mM potassium acetate (pH 7.5); 2, purified protein after a 2 h exposure to an 8 M solution of urea at 25 °C. Fluorescence spectra were recorded at an excitation wavelength of 290 nm. (B) Increasing amounts of decavanadate were added to a 480 nM solution of the enzyme in binding buffer [50 mM Tris/HCl and 50 mM potassium acetate, (pH 7.5)] and the emission spectrum was scanned from 310 to 440 nm. (C) A saturation isotherm was generated by plotting the change in fluorescence intensity at 333 nm as a function of added decavanadate.
Increasing amounts of decavanadate were then added to the purified chlorella virus RNA triphosphatase and the fluorescence intensity was monitored. We observed that the binding of decavanadate to the enzyme resulted in a modification of the intensity of the intrinsic fluorescence of the protein. As a consequence, we were able to evaluate the Kd values for this vanadate oxoanion by titrating the binding of increasing amounts of decavanadate to a fixed concentration of the enzyme. The addition of increasing amounts of decavanadate produced a decrease in the fluorescence intensity (Figure 3B). The corresponding saturation isotherm generated by plotting the change in fluorescence intensity as a function of added decavanadate is shown in Figure 3(C). Quenching saturated at micromolar vanadate concentrations, and a Kd value of 70 μM was determined for decavanadate from a fit of Equation 3 to the generated saturation isotherm. Note that the Kd values for metavanadate (60 μM) and orthovanadate (810 μM) were also determined.
Effect of decavanadate on RNA binding
Vanadate oxoanions have previously been shown to inhibit the nucleic acid binding activity of a number of enzymes involved in phosphoryl transfer [21]. We were thus interested in determining whether the interaction of decavanadate with the chlorella virus RNA triphosphatase could influence the RNA binding activity of the enzyme. Therefore, fluorescence spectroscopy was used to monitor the RNA binding activity of the enzyme. Addition of increasing concentrations of an RNA substrate of 30 nucleotides to the protein resulted in a significant decrease in emission fluorescence intensity (Figure 4A). Quenching saturated around 100 μM, and higher concentrations of RNA did not cause a further decrease in emission fluorescence intensity, suggesting that the reaction had come to an equilibrium. An apparent Kd of 42 μM could be estimated for the RNA substrate (Figure 4B). The effect of decavanadate on the RNA binding reaction was then investigated by performing the binding reactions in the presence of 1 mM decavanadate, a concentration that produced a 90% inhibition of the ATPase activity. Our titration assays indicated that the presence of decavanadate had no significant effect of the RNA binding activity of the chlorella virus RNA triphosphatase (Kd of 40 μM). Higher concentrations of decavanadate had no effect on the RNA binding activity of the enzyme (results not shown).
Figure 4. Effect of decavanadate on the RNA binding activity.

(A) Increasing amounts of RNA were added to a 480 nM solution of the enzyme in binding buffer [50 mM Tris/HCl and 50 mM KOAc, (pH 7.5)] and the emission spectrum was scanned from 310 to 440 nm. (B) A saturation isotherm was generated by plotting the change in fluorescence intensity at 333 nm as a function of added RNA. The RNA binding assays were performed in the absence (■) or presence of 1 mM decavanadate (○).
Effect of decavanadate on the structure of the enzyme
Vanadate oxoanions can inhibit the phosphoryl transfer reaction of certain ATPases by modifying the architecture of the proteins [21]. Analysis of the far-UV region (200–250 nm) of CD spectra can provide useful information on the secondary structural features of a protein while the CD spectra in the near-UV region (250–300 nm) reflect the environments of the aromatic amino acid side chains, giving information about the tertiary structure of a protein. In an effort to determine if the binding of decavanadate to the chlorella virus RNA triphosphatase results in the modification of the enzymatic structure, CD spectra were recorded both in the presence and the absence of decavanadate. Examination of the far-UV region (200–250 nm) of the CD spectra of the protein–decavanadate complex revealed that the helical content of the enzyme is only slightly increased in the presence of decavanadate (Figure 5A). Analysis of the near-UV region of the CD spectra (250–300 nm) of the chlorella virus RNA triphosphatase in both the absence and presence of decavanadate was also performed. As shown in Figure 5(A), no significant modification of the CD signal was noted upon incubation with decavanadate. Overall, the CD spectra suggested that no significant structural modifications of the overall protein architecture were occurring upon binding of decavanadate.
Figure 5. Structural consequences of decavanadate binding.

(A) Far-UV and near-UV CD spectra of the chlorella virus RNA triphosphatase. CD spectra were recorded for the enzyme both in the absence (1) and presence (2) of 2 mM decavanadate. In each case the enzyme concentration was 15 μM and the spectra were recorded from 200 to 400 nm. The average of three wavelength scans is presented. (B) Binding of ANS to the chlorella virus RNA triphosphatase during urea denaturation. The chlorella virus RNA triphosphatase protein was incubated in the absence (■) or presence (○) of 2 mM decavanadate and unfolded with various concentrations of urea at 22 °C for 30 min. Fluorescence emission was monitored after ANS addition (50 μM) at an excitation wavelength of 380 nm. The integrated fluorescence area between 400 and 600 nm was evaluated.
In order to gain additional insights into the potential structural modifications that could occur upon decavanadate binding, we investigated the binding of a structural fluorescent reporter to the enzyme. The exposure of hydrophobic areas of the chlorella virus RNA triphosphatase was evaluated by measuring the binding of ANS to the protein. ANS binds with high affinity to hydrophobic patches on proteins, resulting in an enhancement of ANS intrinsic fluorescence [30]. Our data revealed that the unliganded enzyme binds very weakly to ANS, probably reflecting limited hydrophobic regions at the surface of the protein (Figure 5B). No significant modification of the ANS fluorescence was observed when the protein was incubated in the presence of saturating concentrations of decavanadate. This indicates that the binding of decavanadate to the enzyme does not involve significant hydrophobic exposure on the surface of the protein. Binding of the ANS reporter was also performed in the presence of increasing urea concentrations. As expected, increasing the urea concentration resulted in a decrease in ANS emission intensity, as the protein became unfolded by the denaturant. However, no significant differences were observed when the denaturation assays were performed in the presence of the protein–decavanadate complex. Overall, these results indicate that the binding of the decavanadate oxoanion to the chlorella virus RNA triphosphatase does not increase hydrophobic exposure on the surface of the enzyme.
Thermodynamic stability
The binding of vanadate oxoanions to phosphohydrolases has been shown previously to alter the structural integrity of proteins [21]. To investigate whether the binding of decavanadate to the chlorella virus RNA triphosphatase could modify the stability of the enzyme, denaturation assays were performed with GdnCl, a strong protein denaturant. Modification of the intrinsic protein fluorescence of the enzyme was monitored as a function of the GdnCl concentration. The addition of increasing concentrations of GdnCl to the protein caused the quenching of the intrinsic fluorescence intensity and a red-shift in the emission maximum, reflecting the transfer of tryptophan residues to a more polar environment (results not shown). The change of the integrated fluorescence intensity as a function of the GdnCl concentration is shown in Figure 6(A). The chlorella virus RNA triphosphatase structure reacts very sensitively to the slightest concentration changes in the lower concentration range between 1.0 and 2.0 M where the strongest effects on emission changes are observed. No radical changes could be seen at GdnCl concentrations higher than 2.5 M. The protein displays a smooth transition curve, indicating a co-operative unfolding event. The thermodynamic unfolding parameters were determined and are presented in Table 1. The denaturation experiments were then performed in the presence of decavanadate to evaluate whether the binding of this substrate could modify the stability of the protein. Incubation of the enzyme with saturating amounts of decavanadate resulted in a significant modification of the enzyme stability. Lower concentrations of GdnCl were required to unfold the protein with decavanadate. The transitions extended from approx. 0.4–1.0 M. Analysis of the thermodynamic parameters revealed that the protein–decavanadate complex is thermodynamically less stable than the unliganded enzyme (Table 1). While half of the unliganded chlorella virus RNA triphosphatase was denatured at 1.15 M GdnCl, half denaturation of the protein bound to decavanadate occurred at 0.55 M. The effects of decavanadate on the structural stability are also highlighted by the significant differences in the values of the respective Gibbs free energies of unfolding (Table 1). Our data indicate that the Gibbs free energy of unfolding of the enzyme bound to decavanadate is decreased by 2.83 kJ·mol−1. Overall, these results indicate that the formation of the enzyme–decavanadate complex destabilizes the protein architecture, rendering it more susceptible to chemical denaturation. Note that the destabilizing effect of decavanadate was also observed when the protein was bound to the ATP substrate (Table 1).
Figure 6. Unfolding equilibrium of the chlorella virus RNA triphosphatase.

(A) Transition curves for GdnCl-induced unfolding of the enzyme were determined. The protein was pre-incubated in the absence (■) or presence of 1 mM decavanadate (○), and denatured with increasing concentrations of GdnCl. Equilibrium unfolding transitions were monitored by integration of the fluorescence intensity. (B) Thermal denaturation of the chlorella virus RNA triphosphatase enzyme. Thermal denaturation was recorded for the unliganded protein (1), or the protein incubated in the presence of 2 mM decavanadate (2). CD spectra were recorded at a constant wavelength of 222 nm from 20 to 90 °C at a protein concentration of 15 μM.
Table 1. Thermodynamic unfolding parameters measured by equilibrium GdnCl denaturation.
The parameters ΔG°u (Gibbs free energy of unfolding in the absence of denaturant), m (cooperativity of unfolding), and Cm (midpoint concentration of denaturant required to unfold 50% of the protein) were determined by GdnCl denaturation and from the integration of the fluorescence intensity. The differences in Cm and ΔG°u values in comparison to the free enzyme are also shown (ΔCm and ΔΔG°u respectively). cvRTPase, chlorella virus RNA triphosphatase.
| Protein | Cm (M) | ΔCm (M) | m (kJ·mol−1·M−1) | ΔG°u (kJ·mol−1) | ΔΔG°u (kJ·mol−1) |
|---|---|---|---|---|---|
| cvRTPase | 1.15 | 0.00 | 3.89 | 7.11 | 0.00 |
| cvRTPase· | 0.55 | −0.60 | 3.84 | 4.28 | −2.83 |
| decavanadate | |||||
| cvRTPase·ATP | 0.80 | −0.35 | 6.66 | 6.42 | −0.69 |
| cvRTPase·ATP· | 0.50 | −0.65 | 1.41 | 3.46 | −3.65 |
| decavanadate |
The effects of decavanadate on the structural stability of the enzyme were also assessed by CD spectroscopy. Thermal denaturation assays were performed both in the presence and absence of the vanadate oxoanion, and unfolding of the enzyme was evaluated by monitoring the changes in the α-helix content of the protein (222 nm). Thermal denaturation of the unliganded chlorella virus RNA triphosphatase initially revealed a midpoint of thermal transition (Tm) of 56.8 °C (Figure 6B). Addition of saturating concentrations of decavanadate (2 mM) resulted in a shift of the Tm value to 51.2 °C (Table 2). Evaluation of the thermodynamic parameters of unfolding revealed that the binding of the oxometalate ion to the chlorella virus RNA triphosphatase reduces the structural stability of the enzyme (Table 2). Comparison of the denaturation enthalpies clearly revealed that the protein with bound decavanadate is less stable than the unliganded enzyme. The differences amount to 47.3 kJ·mol−1 for the chlorella virus RNA triphosphatase with bound decavanadate. Note that all of the denaturation assays, performed either with increasing GdnCl concentrations or by thermal denaturation, revealed monophasic unfolding curves, suggestive of a two-state unfolding model. No intermediate form could be detected during the unfolding process.
Table 2. Thermodynamic unfolding parameters measured by thermal denaturation.
The thermodynamic parameters of unfolding ΔHVH (van't Hoff enthalpy of denaturation) and ΔS (entropy of denaturation) were determined by thermal denaturation of the unliganded cvRTPase (chlorella virus RNA triphosphatase) and the protein bound to decavanadate. Temperature-induced denaturations were monitored by CD spectroscopy at 222 nm.
| Protein | Tm (°C) | ΔHVH (kJ·mol−1) | ΔS (J·mol−1·K−1) |
|---|---|---|---|
| cvRTPase | 56.8 | −192.0 | −582 |
| cvRTPase·decavanadate | 51.2 | −144.7 | −446 |
Mode of decavanadate inhibition and mechanistic implications
Inhibition of the phosphohydrolase reaction by decavanadate presents several interesting scenarios: (i) does decavanadate bind to the active site of the enzyme or, (ii) does it bind at a different site on the protein, thereby hindering the phosphohydrolase reaction? In order to answer these questions, ATPase and RNA triphosphatase assays were performed in the presence of various decavanadate concentrations (0.1, 0.2 and 0.5 mM). Consistent with a non-competitive inhibition, the addition of decavanadate resulted in a decrease in the apparent Vmax value, whereas no significant changes were made to the Km value for the ATP substrate (Figures 7A and 7B). These data indicate that decavanadate and the ATP substrate are not competing for the same site on the protein. Similarly, our competition assays indicate that RNA and decavanadate are not competing for the same active site (Figure 7C). The addition of increasing concentrations of decavanadate had no significant effect on the Km value for the RNA substrate (45 μM).
Figure 7. Non-competitive inhibiton of the phosphohydrolase activity by decavanadate.

(A) ATPase assays were performed in the absence (■) or presence of 0.1 mM (○), 0.2 mM (△), or 0.5 mM decavanadate (□). (B) Lineweaver–Burk representations of the ATPase assays performed in the absence (■) or presence of 0.1 mM (□) or 0.5 mM decavanadate (○). (C) Lineweaver–Burk representations of the RNA triphosphatase assays performed in the absence (■) or presence of 0.1 mM (□), 0.2 mM (●), or 0.5 mM decavanadate (○). (D) Consequences of ATP binding on the structure of the chlorella virus RNA triphosphatase. CD spectra were recorded for the enzyme both in the absence (1) and presence (2) of 1 mM ATP. In each case the enzyme concentration was 15 μM and the spectra were recorded from 200 to 400 nm. The average of three wavelength scans is presented.
Additionally, fluorescence spectroscopy assays were performed to confirm that decavanadate was not binding to the same site as ATP in the catalytic centre of the chlorella virus RNA triphosphatase. Remarkably, the binding of the nucleotide substrate (up to 10 mM) to the chlorella virus RNA triphosphatase protein could not be detected even by fluorescence spectroscopy, indicating that no significant modification of the solvent accessibility of the tryptophan residues occurs following the binding of ATP. This is in contrast with the results that were obtained when decavanadate was used as a ligand, and clearly indicates that decavanadate and ATP are not binding to the same site. Since the binding of ATP is not detected by fluorescence spectroscopy, this raises the possibility that the active site of the chlorella virus RNA triphosphatase is preformed, and that the binding of the ATP substrate does not involve significant conformational changes. Therefore, CD analyses were performed to monitor potential structural modifications that could occur upon substrate (ATP) binding. Analysis of the near-UV region (250–300 nm) of the CD spectra indicated that binding of ATP does not significantly alter the tertiary structure of the enzyme. However, analysis of the far-UV region (200–250 nm) of the CD spectra suggested that the binding of ATP results in an increased helical content (Figure 7D).
Mutational analysis
Mutational and crystallographic studies of the closely related RNA triphosphatase from S. cerevisiae has previously revealed the importance of specific essential amino acids that are required for catalytic activity [7,13]. Several of these amino acids participate directly in catalysis via co-ordination of the γ-phosphate of the ATP (RNA) substrate [7,13]. Moreover, previous mutagenesis studies performed on the chlorella virus RNA triphosphatase indicate that the corresponding residues of the viral enzyme are also essential for the catalytic activity of the enzyme [31]. In order to investigate further the decavanadate binding activity of the chlorella virus RNA triphosphatase, we synthesized a series of three enzymatic mutants that have been shown previously to be essential for the activity of the enzyme [31]. The Arg76, Lys129 and Arg131 amino acids that are thought to be involved in the co-ordination of the γ-phosphate were therefore substituted for alanine and the mutant polypeptides were expressed and purified in parallel with the wild-type enzyme (Figure 8A). The effects of single alanine mutations on the decavanadate binding activity were investigated by fluorescence spectroscopy. Our results indicate that the mutants can still efficiently bind decavanadate, indicating that the binding of decavanadate does not involve amino acids located in the active site of the enzyme (Figure 8B). Not surprisingly, the introduction of these mutations in the chlorella virus RNA triphosphatase had a more significant impact on the RNA binding activity of the protein (Figure 8C). Substitution of both the Arg76 and Arg131 by alanine resulted in a significant decrease in the ability of the enzyme to bind RNA (13% and 40% of the wild-type activity respectively). However, substitution of the Lys129 by alanine had a less pronounced impact on the RNA binding activity (83% of the wild-type activity).
Figure 8. Characterization of the RNA triphosphatase alanine mutants.

(A) The purified proteins were analysed by electrophoresis on 12.5% PAGE gels containing 0.1% SDS and visualized by staining with Coomassie Blue. Aliquots (3 μg) of the 200 mM fraction of the wild-type protein (lane 1), R76A (lane 2), K129A (lane 3) and R131A (lane 4) mutants were analysed. The positions and sizes (in kDa) of the molecular-mass markers are indicated on the left. (B) Fluorescence spectroscopy assays were performed by incubating the wild-type protein (□), R76A (○), K129A (△) and R131A (◇) mutants (480 nM) with increasing amounts of decavanadate. Excitation was performed at 290 nm, and emission was monitored from 310 to 440 nm. The saturation isotherms were generated by plotting the change in the fluorescence intensity at 333 nm as a function of added decavanadate. (C) Fluorescence spectroscopy assays were performed by incubating the wild-type protein (□), R76A (○), K129A (△) and R131A (◇) mutants (480 nM) with increasing amounts of RNA. Excitation was performed at 290 nm, and emission was monitored from 310 to 440 nm. The saturation isotherms were generated by plotting the change in the fluorescence intensity at 333 nm as a function of added RNA.
DISCUSSION
The metal-dependent RNA triphosphatase activity of proteins from fungi, viruses, protozoan and microsporidian parasites has been extensively characterized [7–13]. Essential amino acids involved in the binding of both the substrate and the metal ion cofactors have been identified, and mutational studies have confirmed their importance in catalysis [7–13]. However, the intimate details of the phosphohydrolase reaction catalysed by these enzymes remain to be addressed. In the past few years, vanadate oxoanions have been effectively used both as mechanistic probes and inhibitors of enzymes that catalyse phosphoryl-transfer reactions [19–22]. In the present study, we investigated the ability of vanadate oxoanions to inhibit the phosphohydrolase activity of the chlorella virus RNA triphosphatase, the smallest identified member of the metal-dependent RNA triphosphatases.
The present study highlights the inhibitory effect of decavanadate on the RNA triphosphatase activity of the chlorella virus RNA triphosphatase. Under our experimental conditions, decavanadate appeared as the most inhibitory vanadate oxoanion, although metavanadate also produced an inhibitory effect. Limited information is available on the mechanism of inhibition of phosphohydrolases by decavanadate. The oxoanion has previously been shown to inhibit the ATPase activity of the Pseudomonas aeruginosa MutS protein [21]. Binding studies clearly demonstrated that decavanadate was a potent inhibitor of the DNA binding activity of the MutS protein [21]. Moreover, structural assays indicated that the conformation of the P. aeruginosa MutS protein was affected by the presence of decavanadate [21]. In contrast, addition of decavanadate to the chlorella virus RNA triphosphatase had no effect on the RNA binding activity of the enzyme as judged from fluorescence spectroscopy assays in the current study. Furthermore, we have looked for conformational changes upon decavanadate binding using spectroscopic approaches. However, no significant modification of the overall protein architecture was observed through CD analyses. Nevertheless, denaturation experiments clearly indicated that the binding of decavanadate to the chlorella virus RNA triphosphatase had a pronounced effect on the structural stability of the enzyme. Both fluorescence spectroscopy and CD assays indicated that the formation of the enzyme–decavanadate complex destabilized the architecture of the enzyme.
Growing evidence suggests that the enzymatic reaction catalysed by enzymes that belong to the metal-dependent RNA triphosphatase family requires a very precise alignment between1 the residues in the active site of the protein, the substrate and the essential metal-ion cofactor [32]. For instance, the fact that the RNA triphosphatase activity is supported by magnesium, but not by manganese or cobalt, while the ATPase activity is activated by manganese and cobalt, highlights the delicate differences that exist between the two phosphohydrolase activities. The metal ions probably modify the co-ordination geometry, or induce minor local conformational perturbations in the active-site residues that ultimately influence substrate specificity. In the present study, differences were also noted in the concentrations of decavanadate that were required to inhibit half of the ATPase (100 μM) and RNA triphosphatase activities (600 μM), underscoring the subtle differences in the requirements for the ATPase and RNA triphosphatase activities. Our inhibition studies suggest that the modification of the structural stability of the enzyme that is induced upon decavanadate binding has a more pronounced effect on the ATPase activity.
In an attempt to gain additional insights into the inhibition of metal-dependent RNA triphosphatases by decavanadate, inhibition studies were also performed on related enzymes. Interestingly, our biochemical assays provided no evidence for the inhibition of the metal-dependent S. cerevisiae RNA triphosphatase by decavanadate (results not shown). Although no crystal structures are yet available for the chlorella virus RNA triphosphatase, sequence alignments and mutagenesis studies suggest that the enzyme lacks several structural elements that are found in the yeast enzyme [26,31]. More specifically, the chlorella virus RNA triphosphatase appears to be lacking key elements that surround the active site of the yeast enzyme [7,26,31]. These elements include domains involved in protein–protein interactions and an α-helix found in the active site of the S. cerevisiae RNA triphosphatase [7]. It is tempting to speculate that the absence of these key elements in the chlorella virus RNA triphosphatase plays a pivotal role in the decavanadate-induced structural instability. This hypothesis is further supported by the fact that our fluorescence spectroscopy assays indicated that decavanadate can bind to the S. cerevisiae RNA triphosphatase (results not shown), albeit with a lower affinity (Kd=340 μM). Decavanadate, however, displayed no inhibitory effect on the phosphohydrolase activity of the yeast enzyme. Moreover, subtle mechanistic differences between the chlorella virus RNA triphosphatase and the corresponding enzyme from S. cerevisiae have previously been noted through structure-activity studies [31]. The critical differences mainly concerned residues indirectly involved in catalysis through their interactions with other amino acids [31]. Structural differences between the chlorella virus RNA triphosphatase and the metal-dependent RNA triphosphatase of other large DNA viruses (e.g. vaccinia virus) have also been observed through mutagenesis studies [31]. It is therefore not surprising that decavanadate displayed no inhibitory activity against the vaccinia virus RNA triphosphatase (results not shown).
Because of the crucial role of the cap structure for mRNA metabolism and stability, the development of potent inhibitors targeting the enzymes involved in the synthesis of the cap structure of pathogens appears as an attractive challenge. For instance, an elegant study recently demonstrated the ability of non-nucleoside inhibitors to inhibit the mRNA guanylylation of the human respiratory syncitial virus viral transcripts [33]. These guanylyltransferase inhibitors were shown to exhibit antiviral activities both in vitro and in a mouse model of infection [33]. The broad spectrum antiviral ribavirin triphosphate, a nucleoside analogue, has also been shown to directly interact with the guanylyltransferase of vaccinia virus thereby inhibiting the guanylylation of RNA transcripts [34]. Ribavirin can actually be used as a substrate by the vaccinia virus enzyme and transferred to an acceptor RNA molecule [34]. Finally, tripolyphosphate, a phosphate derivative, is a potent inhibitor of the chlorella virus RNA triphosphatase [31]. In fact, the chlorella virus enzyme harbours a tripolyphosphatase activity [31]. Because of the high overall similarity between the members of the metal-dependent RNA triphosphatase family, it has been suggested that a mechanism-based inhibitor might display broad spectrum activity against other members of the family. This is observed for tripolyphosphate which displays activity against the RNA triphosphatases of chlorella virus [31], Schizosaccharomyces pombe [34], and S. cerevisiae [35]. However, decavanadate displayed no inhibitory effect on the phosphohydrolase activity of the yeast enzyme suggesting that the subtle structural and mechanistic differences which exist between the S. cerevisiae and chlorella virus RNA triphosphatases are important determinants for the action of decavanadate.
In order to get additional insights into the phosphohydrolase reaction catalysed by the chlorella virus RNA triphosphatase, we have looked for potential conformational changes that could result from the binding of ATP. Our spectroscopic data indicate that the binding of the ATP substrate does not lead to significant structural modifications of the protein architecture. This suggests a model in which the chlorella virus RNA triphosphatase possesses a preformed active site where major domain rearrangements are not needed to form a catalytically active site. In fact, the binding of the ATP substrate to the enzyme could not even be detected by fluorescence spectroscopy, indicating that no significant modification of the solvent accessibility of the tryptophan residues occurs following binding of the substrate. This is in agreement with the model that has been previously proposed for the S. cerevisiae RNA triphosphatase [32] in which the β strands that constitute the active site of the yeast enzyme are already positioned prior to the binding of the substrate (ATP or RNA).
The high intrinsic fluorescence signal of the chlorella virus RNA triphosphatase allowed us to monitor the binding of the decavanadate oxoanion with a high degree of sensitivity. The decrease in fluorescence intensity observed upon saturation of the enzyme with a ligand can be produced by contact of the quenching agent with the indole side chain of a tryptophan and/or by alterations in the microenvironments of tryptophan residues distal to the ligand binding site. The fact that no conformational changes are occurring upon decavanadate binding strongly suggests that the oxanion binds in close proximity to a tryptophan residue. The absence of a crystal structure of the chlorella virus RNA triphosphatase makes it difficult to precisely evaluate the location of the three tryptophan residues that are found in the enzyme. Although the protein lacks several structural elements that are found in the S. cerevisiae RNA triphosphatase, sequence alignments and homology modelling studies suggest that two of the three tryptophan residues (Trp87 and Trp139) are located on the walls of the tunnel-like active centre [7,26,31]. Nonetheless, our competition assays clearly indicate that decavanadate and the phosphohydrolase substrates (ATP or RNA) are not competing for the same site on the enzyme. Similarly, our mutational studies also confirmed that the binding of decavanadate does not involve key residues located in the active site of the protein. The future determination of the crystal structure of the chlorella virus RNA triphosphatase will undoubtedly reveal key structural features of the enzyme, and provide additional insights on the phosphohydrolase reaction catalysed by the enzyme.
Acknowledgments
We thank Dr James Van Etten for generously providing the chlorella virus DNA (Department of Plant Pathology, University of Nebraska, Lincoln, NE, U.S.A.). This work was supported by a grant from the CIHR (Canadian Institutes for Health Research). M.B. is a New Investigator Scholar from the CIHR.
References
- 1.Shuman S. Structure, mechanism, and evolution of the mRNA capping apparatus. Prog. Nucleic Acids Res. Mol. Biol. 2000;66:1–40. doi: 10.1016/s0079-6603(00)66025-7. [DOI] [PubMed] [Google Scholar]
- 2.Furuichi Y., Shatkin A. J. Viral and cellular mRNA capping: past and prospects. Adv. Virol. Res. 2000;55:135–184. doi: 10.1016/S0065-3527(00)55003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisaillon M., Lemay G. Viral and cellular enzymes involved in synthesis of mRNA cap structure. Virology. 1997;236:1–7. doi: 10.1006/viro.1997.8698. [DOI] [PubMed] [Google Scholar]
- 4.Changela A., Ho C. K., Martins A., Shuman S., Mondragon A. Structure and mechanism of the RNA triphosphatase component of mammalian mRNA capping enzyme. EMBO J. 2001;20:2575–2586. doi: 10.1093/emboj/20.10.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wen Y., Yue Z., Shatkin A. J. Mammalian capping enzyme binds RNA and uses protein tyrosine phosphatase mechanism. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12226–12231. doi: 10.1073/pnas.95.21.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denu J. M., Dixon J. E. Protein tyrosine phosphatases: mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol. 1998;2:633–641. doi: 10.1016/s1367-5931(98)80095-1. [DOI] [PubMed] [Google Scholar]
- 7.Lima C. D., Wang L. K., Shuman S. Structure and mechanism of yeast RNA triphosphatase: an essential component of the mRNA capping apparatus. Cell. 1999;99:533–543. doi: 10.1016/s0092-8674(00)81541-x. [DOI] [PubMed] [Google Scholar]
- 8.Yu L., Martins A., Deng L., Shuman S. Structure-function analysis of the triphosphatase component of vaccinia virus mRNA capping enzyme. J. Virol. 1997;71:9837–9843. doi: 10.1128/jvi.71.12.9837-9843.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross C. H., Shuman S. RNA 5′-triphosphatase, nucleoside triphosphatase, and guanylyltransferase activities of baculovirus LEF-4 protein. J. Virol. 1998;72:10020–10028. doi: 10.1128/jvi.72.12.10020-10028.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ho C. K., Schwer B., Shuman S. Genetic, physical, and functional interactions between the triphosphatase and guanylyltransferase components of the yeast mRNA capping apparatus. Mol. Cell. Biol. 1998;18:5189–5198. doi: 10.1128/mcb.18.9.5189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamada-Okabe T., Mio T., Matsui M., Kashima Y., Arisawa M., Yamada-Okabe H. Isolation and characterization of the Candida albicans gene for mRNA 5′-triphosphatase: association of mRNA 5′-triphosphatase and mRNA 5′-guanylyltransferase activities is essential for the function of mRNA 5′-capping enzyme in vivo. FEBS Lett. 1998;453:49–54. doi: 10.1016/s0014-5793(98)01037-0. [DOI] [PubMed] [Google Scholar]
- 12.Bisaillon M., Shuman S. Structure-function analysis of the active site tunnel of yeast RNA triphosphatase. J. Biol. Chem. 2001;276:17261–17266. doi: 10.1074/jbc.M100980200. [DOI] [PubMed] [Google Scholar]
- 13.Ho C. K., Pei Y., Shuman S. Yeast and viral RNA 5′ triphosphatases comprise a new nucleoside triphosphatase family. J. Biol. Chem. 1998;273:34151–34156. doi: 10.1074/jbc.273.51.34151. [DOI] [PubMed] [Google Scholar]
- 14.Changela A., Martins A., Shuman S., Mondragon A. Crystal structure of baculovirus RNA triphosphatase complexed with phosphate. J. Biol. Chem. 2005;280:17848–17856. doi: 10.1074/jbc.M500885200. [DOI] [PubMed] [Google Scholar]
- 15.Barford D., Flint A. J., Tonks N. K. Crystal structure of human protein tyrosine phosphatase 1B. Science. 1994;263:1397–1404. [PubMed] [Google Scholar]
- 16.Denu J. M., Dixon J. E. A catalytic mechanism for the dual-specific phosphatases. Proc. Natl. Acad. Sci. U.S.A. 1995;92:5910–5914. doi: 10.1073/pnas.92.13.5910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fauman E. B., Cogswell J. P., Lovejoy B., Rocque W. J., Holmes W., Montana V. G., Piwnica-Worms H., Rink M. J., Saper M. A. Crystal structure of the catalytic domain of the human cell cycle control phosphatase, Cdc25A. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 18.Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., Shoelson S. E. Crystal structure of the tyrosine phosphatase SHP-2. Cell. 1998;92:441–450. doi: 10.1016/s0092-8674(00)80938-1. [DOI] [PubMed] [Google Scholar]
- 19.Messmore J. M., Raines R. T. Decavanadate inhibits catalysis by ribonuclease A. Arch. Biochem. Biophys. 2000;381:25–30. doi: 10.1006/abbi.2000.1951. [DOI] [PubMed] [Google Scholar]
- 20.Huyer G., Liu S., Kelly J., Moffat J., Payette P., Kennedy B., Tsapraillis G., Gresser M., Ramachandran C. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 1997;272:843–851. doi: 10.1074/jbc.272.2.843. [DOI] [PubMed] [Google Scholar]
- 21.Pezza R. J., Villarreal M. A., Montich G. G., Argarana C. E. Vanadate inhibits the ATPase activity and DNA binding capability of bacterial MutS. A structural model for the vanadate–MutS interaction at the Walker A motif. Nucleic Acids Res. 2002;30:4700–4708. doi: 10.1093/nar/gkf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan C., Roberts M. F. Vanadate is a potent competitive inhibitor of phospholipase C from Bacillus cereus. Biochim. Biophys. Acta. 1996;1298:58–68. doi: 10.1016/s0167-4838(96)00114-8. [DOI] [PubMed] [Google Scholar]
- 23.Gresser M. J., Tracey A. S. In: Vanadium in Biological Systems. Chasteen N. D., editor. Netherlands: Kluwer Academic publishers; 1990. pp. 63–79. [Google Scholar]
- 24.Rodriguez C. R., Tagaki T., Cho E. J., Buratowski S. A Saccharomyces cerevisiae RNA 5′-triphosphatase related to mRNA capping enzyme. Nucleic Acids Res. 1999;27:2181–2188. doi: 10.1093/nar/27.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Etten J. L., Meints R. H. Giant viruses infecting algae. Annu. Rev. Microbiol. 1999;53:447–494. doi: 10.1146/annurev.micro.53.1.447. [DOI] [PubMed] [Google Scholar]
- 26.Ho C. K., Gong C., Shuman S. RNA triphosphatase component of the mRNA capping apparatus of Paramecium bursaria Chlorella virus 1. J. Virol. 2001;75:1744–1750. doi: 10.1128/JVI.75.4.1744-1750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pace C. N. Evaluating contribution of hydrogen bonding and hydrophobic bonding to protein folding. Methods Enzymol. 1995;259:538–554. doi: 10.1016/0076-6879(95)59060-9. [DOI] [PubMed] [Google Scholar]
- 28.Painter G. R., Wright L. L., Hopkins S., Furman P. A. Initial binding of 2′-deoxynucleoside 5′-triphosphates to human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem. 1991;266:19362–19368. [PubMed] [Google Scholar]
- 29.Eftink M. R., Ghiron C. A. Fluorescence quenching studies with proteins. Anal. Biochem. 1981;114:199–227. doi: 10.1016/0003-2697(81)90474-7. [DOI] [PubMed] [Google Scholar]
- 30.Labowicz J. R. Principles of Fluorescence Spectroscopy. 2nd Ed. New York: Kluwer/Plenum; 1999. pp. 237–266. [Google Scholar]
- 31.Gong C., Shuman S. Chlorella virus RNA triphosphatase. Mutational analysis and mechanism of inhibition by tripolyphosphate. J. Biol. Chem. 2002;277:15317–15324. doi: 10.1074/jbc.M200532200. [DOI] [PubMed] [Google Scholar]
- 32.Bisaillon M., Bougie I. Investigating the role of metal ions in the catalytic mechanism of the yeast RNA triphosphatase. J. Biol. Chem. 2003;278:33963–33971. doi: 10.1074/jbc.M303007200. [DOI] [PubMed] [Google Scholar]
- 33.Liuzzi M., Mason S. W., Cartier M., Lawetz C., McCollum R. S., Dansereau N., Bolger G., Lapeyre N., Gaudette Y., et al. Inhibitors of respiratory syncytial virus replication target cotranscriptional mRNA guanylylation by viral RNA-dependent RNA polymerase. J. Virol. 2005;79:13105–13115. doi: 10.1128/JVI.79.20.13105-13115.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bougie I., Bisaillon M. The broad spectrum antiviral nucleoside ribavirin as a substrate for a viral RNA capping enzyme. J. Biol. Chem. 2004;279:22124–22130. doi: 10.1074/jbc.M400908200. [DOI] [PubMed] [Google Scholar]
- 35.Pei Y., Schwer B., Hausmann S., Shuman S. Characterization of Schizosaccharomyces pombe RNA triphosphatase. Nucleic Acids Res. 2001;29:387–396. doi: 10.1093/nar/29.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]