

Abstract
Menkes disease (MD) is an X-linked recessive disorder of copper metabolism. It is caused by mutations in the ATP7A gene encoding a copper-translocating P-type ATPase, which contains six N-terminal copper-binding sites (CBS1–CBS6). Most patients die in early childhood. We investigated the functional effect of a large frameshift deletion in ATP7A (including exons 3 and 4) identified in a patient with MD with unexpectedly mild symptoms and long survival. The mutated transcript, ATP7AΔex3+ex4, contains a premature termination codon after 46 codons. Although such transcripts are generally degraded by nonsense-mediated mRNA decay (NMD), it was established by real-time PCR quantification that the ATP7AΔex3+ex4 transcript was protected from degradation. A combination of in vitro translation, recombinant expression, and immunocytochemical analysis provided evidence that the ATP7AΔex3+ex4 transcript was protected from degradation because of reinitiation of protein translation. Our findings suggest that reinitiation takes place at two downstream internal codons. The putative N-terminally truncated proteins contain only CBS5 and CBS6. Cellular localization and copper-dependent trafficking of the major part of endogenous and recombinant ATP7AΔex3+ex4 proteins were similar to the wild-type ATP7A protein. Furthermore, the ATP7AΔex3+ex4 cDNA was able to rescue a yeast strain lacking the homologous gene, CCC2. In summary, we propose that reinitiation of the NMD-resistant ATP7AΔex3+ex4 transcript leads to the synthesis of N-terminally truncated and at-least-partially functional Menkes proteins missing CBS1–CBS4. This finding—that a mutation that would have been assumed to be null is not—highlights the need to examine the biochemical phenotype of patients to deduce the efficacy of copper therapy.
Menkes disease (MD [MIM 309400]) is an X-linked, multisystemic lethal disorder of impaired copper metabolism1 caused by mutations in ATP7A (MIM 300011). ATP7A contains 23 exons and encodes a P-type ATPase of 1,500 aa (EC 3.6.1.36). The ATP7A protein (GenBank accession number NP_000043) is a member of the CPx-type transmembrane ATPase family that performs ATP-driven translocation of metal cations across cellular membranes.2 The characteristic features of MD are due to reduced activity of copper-requiring enzymes. Classic MD is characterized by developmental delay, progressive mental retardation, vascular abnormalities, skeletal changes, loose joints, and early death (<3 years of age). However, MD shows clinical heterogeneity, as demonstrated by a variation in progression and severity of neurological symptoms and age of death. The mildest form, occipital horn syndrome (OHS), is primarily characterized by pronounced connective tissue symptoms (reviewed in Horn and Tümer3).
MD is closely related to Wilson disease (WD [MIM 277900]), an autosomal recessive disorder characterized by accumulation of copper in the liver, brain, and kidneys and associated with hepatic cirrhosis and neuronal degeneration.4,5 WD is caused by mutations in ATP7B (MIM 606882), which encodes another copper-transporting ATPase, ATP7B, that is highly homologous to ATP7A.6 ATP7A is expressed in most nonhepatic tissues,7 whereas ATP7B is expressed predominantly in liver.8
Unique for the Menkes and Wilson ATPases is the presence of six copper-binding sites (CBSs) in the N-terminal region of the proteins. Each CBS consists of the signature sequence of heavy metal–binding proteins, GMXCXXC, with which one copper atom is coordinated via the two cysteine residues. The ATPases receive copper ions via the small cytosolic chaperone Atox1.9–12 ATP7A and ATP7B are normally localized in the trans-Golgi network (TGN), where they transport copper to copper-requiring enzymes synthesized within the secretory compartments. In the presence of elevated copper concentrations, the ATP7A protein traffics to cytosolic vesicular compartments and to the plasma membrane, where it mediates copper efflux,13,14 whereas ATP7B in hepatocytes moves to pericanalicular vesicles.15
The role of the CBSs in copper transport has been investigated by in vitro experiments with isolated membrane vesicles from Chinese hamster ovary (CHO) cells, expressing ATP7A constructs with different mutations in the CBSs. It was shown that a protein with no intact CBSs retained at least 55% copper-transporting activity, indicating that the CBSs are not essential for copper-transporting activity of the Menkes protein.16 In contrast, however, it was found that CBS5 and CBS6 were necessary and sufficient for copper-induced trafficking.17
Transcripts with mutations that lead to premature termination codons (PTC) >50–55 nt upstream from the last exon, are normally degraded by a mechanism called nonsense-mediated mRNA decay (NMD).18 However, it has been shown that translation reinitiation abrogates NMD in mammalian cells.19
About 60 larger deletions have been reported in patients with MD.20–23 The majority have been identified in patients with classical symptoms. Here, we characterize the effect of a deletion including exons 3 and 4, which was identified in a patient with less-severe symptoms and long survival. We characterize the effect on the resulting mRNA transcript and protein product. Surprisingly, we found that the transcript, though it contains a PTC, was not recognized by the NMD machinery. Using a combination of in vitro translation, immunocytochemical analysis, and recombinant expression, we provide evidence that the mutated transcript, designated “ATP7AΔex3+ex4 transcript,” was protected from NMD because of reinitiation of protein translation at two downstream internal AUG codons, AUG461 and AUG475. The at-least-partial activity of the resulting N-terminally truncated proteins containing only CBS5 and CBS6 may contribute to the less-severe phenotype.
Material and Methods
Patient
A.B. was born in England in 1973 with a birth weight of 2.92 kg. At age 6 mo, he showed developmental delay, and, at age 2 years, he started having seizures. At age 4 years, he developed head control, and, at age 9 years, his motor and mental status was assessed to be like that of a 3-mo-old child. At age 17 years, A.B. had no speech, was hypotonic, and had brown, coarse hair. The copper content and ceruloplasmin values in serum were slightly reduced (7.3–8.9 μmol/liter copper, normal range 12.5–19 μmol/liter; 0.5–1.0 μmol/liter ceruloplasmin, normal range 1.3–2.9 μmol/liter).24 In general, serum copper and ceruloplasmin levels are ∼10% of normal in patients with MD.24 64Cu uptake and retention studies performed on fibroblasts from the patient showed that 72.2% of 64Cu taken up over 20 h was retained in the cells after an additional 24 h of efflux in normal medium (normal range 9.0%–35.8%).25 Copper absorption from the gut was virtually undetectable.24 A.B., who has never received any copper treatment, is 35 years old but is severely mentally delayed. A partial deletion of the ATP7A gene, including exons 3 and 4 (c.121-?_1338+?del), is demonstrated in the patient (described as “patient D11” elsewhere).21 A.B. has a maternal cousin with the same mutation and essentially the same less-severe symptoms.24 Skin samples were collected from the patients for diagnostic investigations. Similar samples from healthy persons were used as controls. The fibroblasts were cultured as described elsewhere.26
Characterization of ATP7A mRNA
Total RNA was isolated from ∼5×106 cells of cultured skin fibroblasts, with the use of the RNeasy Mini Kit (Qiagen). Single-stranded cDNA was synthesized by reverse transcription, with the use of the High-Capacity cDNA Archive Kit, in accordance with the manufacturer’s instructions (Applied Biosystems). cDNA was used for PCR amplification, with use of the primer pair ATP7Aex1U/ATP7Aex6L (table 1) flanking the genomic deletion and Platinum Pfx DNA polymerase, in accordance with the manufacturer’s instructions (Invitrogen). The obtained PCR product was subsequently sequenced on an ABI PRISM 310 Genetic Analyzer, with use of the primers ATP7Aex1 and ATP7Aex6L (table 1) and Big Dye Terminator v.3.1 Cycle Sequencing Kit, in accordance with the manufacturer’s instructions (Applied Biosystems).
Table 1. .
Primers Used in This Study
| Primer Name | Sequence (5′–3′ Direction) | Remarks |
| T7-134-(-111) | TAATACGACTCACTATAGGGAGAGCTACTGTGACTTCTCCGATTGT | In exon 1 |
| T7-M1 | TAATACGACTCACTATAGGGAGATGGATCCAAGTATGGGTGTGAAT | In exon 2 |
| T7-M461 | TAATACGACTCACTATAGGGAGATGCCGCTTTTGACTTCAACTAAT | In exon 5 |
| T7-kozak-M461 | TAATACGACTCACTATAGGGAGACCACCATGCCGCTTTTGACTTCAACTAAT | In exon 5 |
| T7-kozak-M475 | TAATACGACTCACTATAGGGAGACCACCATGACACCAGTTCAAGAC | In exon 5 |
| CATP7A642 | GATCTAAGTGACTTGCTGACCGATCCTTC | In exon 8 |
| ATP7Aex1U | CGCTAGCTACTGTGACTTCTCCGATTGT | In exon 1 |
| ATP7Aex1 | AACCATAGGATAGAGAAACC | In exon 1 |
| ATP7Aex6L | TGGGGGTTGTATAACAGCAGGAT | In exon 6 |
| ATP7Aex9L | AGACAATCCTGGAAGAATCTGGCGC | In exon 9 |
| ATP7Aex21 | AGGATTCGGATAAATTTTGTCTTTGCTCTA | In exon 21 |
| ATP7Aex22 | GCAAAACCAAACCAATGGGCATA | In exon 22 |
| ATP7A21/22probe | CATAGCTGCTGGAGTTTT | Probe (TaqMan) |
| Nhe-ATP7AUTR | CTGCACTAGCAAGCTAGCGGCCGCTCTAGAGAAACCAGGAATGTAA | In 5′ UTR; contains NheI site |
| Nhe-M461 | CTGCACTAGCAAGCTAGCACCACCATGCCGCTTTTGACTTCAAC | With NheI site and kozak |
| NheI-M475 | CTGCACTAGCAAGCTAGCACCACCATGACACCAGTTCAAGACAA | Contains NheI site |
| Nhe-M461-M475L | TGCACTAGCAAGCTAGCACCACCATGCCGCTTTTGACTTCAACTAATGAATTTTATACTAAAGGGCTGACAC | Contains NheI site |
| Yeast-M1 | CTGCACTAGCAAGCTAGCAAAAAAAAAAATGGATCCAAGTATGGGTGT | Contains NheI site |
| Yeast-M461 | CTGCACTAGCAAGCTAGCAAAAAAAAAAATGCCGCTTTTGACTTCAAC | Contains NheI site |
| Yeast-M475 | CTGCACTAGCAAGCTAGCAAAAAAAAAAATGACACCAGTTCAAGACAA | Contains NheI site |
| Yeast-M461-M475L | CTGCACTAGCAAGCTAGCAAAAAAAAAAATGCCGCTTTTGACTTCAACTAATGAATTTTATACTAAAGGGCTGACAC | Contains NheI site |
Quantitative RT-PCR by Real-Time PCR
For relative quantification of the mutated ATP7A transcript, real-time PCR amplification was performed on cDNA obtained from A.B. and from control mRNA samples. TaqMan FAM-probes and primers annealing to the junction between exons 21 and 22 in ATP7A (ATP7A 21/22 probe and primer pair ATP7Aex21/ATP7Aex22 obtained using Assays-by-Design) (table 1) and to the junction between exons 1 and 2 in ATP7A (TaqMan Gene Expression Assays [assay number Hs00921963_m1]) were purchased from Applied Biosystems. As endogenous control, a VIC-probe and primers to human GAPDH (Applied Biosystems [part number 4326317E]) were used. PCR amplification and detection was performed with an ABI7300 (Applied Biosystems), in accordance with the manufacturer’s instructions. The threshold cycle (CT) is defined as the fractional cycle number at which the fluorescence passes a fixed threshold. Standard curves of CT values compared with log cDNA concentrations were prepared by assaying threefold serial dilutions of control cDNA, from 66.67 ng/well to 0.27 ng/well, with the GADPH and the ATP7A assays, respectively.
In Vitro Expression
For in vitro expression, fragments encoding N-terminal parts of the mutated ATP7A protein were PCR amplified from cDNA with ATP7A-specific primers in high-salt buffer, with the use of Taq Plus Long, in accordance with the manufacturer’s instructions (Stratagene). All the forward primers (T7-134-[-111], T7-M1, T7-M461, T7-kozak-M461, and T7-kozak-M475) were designed to carry a T7 promoter and were used in combination with the reverse primer, cATP7A642 (table 1). Transcription and translation of the T7 promoter–containing PCR products were performed using the TnT T7-coupled reticolocyte lysate system (Promega) in the presence of 35S methionine (Amersham Pharmacia), in accordance with the manufacturer’s instructions. The synthesized proteins were separated by SDS-PAGE (10% gels) under reducing conditions, followed by fixing (fix-buffer: 10% v/v acetic acid and 50% v/v ethanol) and incubation for 30 min in Amplify Amersham NAMP 100 (Amersham Pharmacia). The gel was dried before exposure to Kodak X-Amat AR film, XAR-5, at −80°C for ⩾24 h.
Western Blot Analysis of Endogenously and Recombinantly Synthesized ATP7A Proteins
Fibroblasts were lysed in lysis buffer (50 mM Hepes [pH 7.6], 250 mM NaCl, 0.1% NP40, 5 mM EDTA, and 25 μl/ml Protease Inhibitor Cocktail III [CalBioChem]) on ice for 30 min. Cell debris was removed by centrifugation at 16,000 g for 5 min. Transiently transfected CHO-1K cells were scraped into 1 ml PBS containing 25 μl/ml Protease Inhibitor Cocktail III, were pelleted, and were resuspended in 50 μl PBS with protease inhibitors. Fibroblast and CHO lysates were heated to 70°C for 10 min and 95°C for 5 min, respectively, in the presence of LDS sample buffer (Pagegel Inc.) and dithiothreitol. Proteins were separated on 8% SDS gels (Pagegel Inc.) and were transferred to a precut nitrocellulose membrane (Pagegel Inc.). The membranes were blocked by incubation in blocking buffer (4% dry milk [Irma] in tris-buffered saline [TBS] with 0.1% Tween-20) for 3 h. Incubation with primary and secondary antibodies was performed overnight at 4°C and for 1–2 h at room temperature, respectively, in blocking buffer. The primary antibody, chicken anti-ATP7A (1:1,500) (ab13995 from Abcam, recognizing amino acids 1407–1500), was used for endogenous ATP7A proteins, whereas sc-789 rabbit anti-Myc (1:200) (Santa Cruz Biotechnology) was used for recombinant ATP7A proteins. The corresponding secondary horseradish peroxide–conjugated antibodies were rabbit anti-chicken (1:75,000) (Abcam ab6753) and swine anti-rabbit (1:1,000) (Dako). Blots were developed for 5 min with the use of SuperSignal West Pico Chemiluminescence kit (Pierce) and were exposed on Hyperfilm ECL (Amersham Biosciences) for 1–60 s.
ATP7A cDNA Constructs for Expression in Mammalian Cells
In this study, we used the plasmid encoding the human wild-type ATP7A cDNA sequence (GenBank accession number L06133.1 [nucleotides 113-4645, except for the polymorphisms 1152T→A, 1535C→T, 2151C→T, and 3677C→T]), described elsewhere.27 In this article, the construct is called “pCEP4ATP7Awt.” The ATP7A protein encoded by this construct contains a Myc tag fused inframe to the C-terminus. To generate an expression vector containing the ATP7AΔex3+ex4 cDNA sequence, we replaced a NheI-BclI fragment of pCEP4ATP7Awt (NheI located in the polylinker of pCEP4 and BclI located in exon 9 at c.2220 in L06133.1) with a corresponding fragment amplified from A.B. cDNA, with the primers Nhe-ATP7AUTR (located upstream from the normal AUG codon containing a NheI site) and ATP7Aex9L (table 1). The resulting plasmid is called “pCEP4ATP7AΔex3+ex4.” Likewise, the constructs pCEP4ATP7AΔ1−460, pCEP4ATP7AΔ1−474, and pCEP4ATP7AΔ1−460, M475L were prepared by PCR amplification, with the use of cDNA from A.B. and the forward primers Nhe-M461, Nhe-M475, or Nhe-M461-M475L, respectively, in combination with the antisense primer, ATP7Aex9L (table 1). All the cloned PCR products were amplified with Platinum Pfx DNA polymerase (Invitrogen), in accordance with the manufacturer’s instructions. To digest the plasmid with BclI, we propagated it in a methylation-deficient Escherichia coli strain, 2202 (araD139 Δ[ara-ABOIC-leu] 7679 gal E K Δ[lac] X74 relA rpsL spoT thi dam hsdR). After ligation, transformation was performed using XL1-Blue cells (recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac [F′ proAB laclqZΔM15 Tn10 (Tetr)]) (Stratagene). The cells were cultured at 30°C for 3 d, to obtain visible colonies. The entire ATP7A sequence was verified by automatic sequencing.
Transient Transfections
CHO-1K cells (ATCC number CCL-61) were cultured at 37°C with 5% CO2 in F12 growth medium (F12 medium [Gibco] supplemented with 10% v/v fetal calf serum [FCS], 100 U/ml penicillin, and 100 μg/ml streptomycin). The day before transfection, 2.5×105 cells were seeded in 9.6 cm2 dishes and were incubated for ∼24 h, to reach ∼60% confluence. For each transfection, 1.5 μg of large-scale plasmid DNA, together with 7 μl of lipofectamin (2 mg/ml) (Life Technologies), were used in accordance with the manufacturer’s instructions. Cells were incubated for 6 h, followed by the replacement of complexes with F12 growth medium, in which cells subsequently were cultured for at least 24 h before further studies.
Indirect Immunofluorescence Detection of ATP7A Proteins
Transiently transfected CHO-1K cells grown on glass coverslips were treated after 48 h, with either 50 μM bathocuproine disulfonic acid (BCS [Sigma]) or 200 μM CuCl2 in F12 medium for 2 h. Cells were fixed with −20°C cold methanol for 5 min at −20°C and were incubated in F12 with 10% v/v FCS for 30 min, to block unspecific signals. Doubled labeling was performed by incubation with antibodies for 1 h in blocking media. Primary antibodies used were rabbit anti-Myc (1:50) (Santa Cruz Biotechnologies) and mouse anti-GS28 (1:1,000) (BD Biosciences). Secondary antibodies were goat anti-rabbit, Alexa 488 (1:1,500), and donkey anti-mouse, Alexa 548 (1:1,500) (Molecular Probes). Coverslips were mounted with Dapi Antifade ES (Cytocell).
Fibroblasts obtained from A.B. and control persons were treated as described elsewhere,28 with a minor modification. Instead of 50 μM BCS, a concentration of 150 μM BCS was used. As positive controls, fibroblasts from unaffected persons were used, and, as negative controls, fibroblasts from a patient with MD with deletion of exons 3–23 (c.121-?-8333+?del) were used.
ATP7A cDNA Constructs for Expression in Yeast Cells
For expression of ATP7A constructs in yeast, the ATP7A sequences were cloned in the low-copy, URA3-encoding expression vector, pRS316GPD, under the control of the constitutively active glyceraldehydes-3-phosphate dehydrogenase (GPD) promoter. This vector was derived from pRS316,29 by insertion of the GDP promoter isolated from the multicopy expression vector, pG-1,30 with the use of BamHI and HindIII. The entire wild-type ATP7A cDNA sequence was isolated from pCEP4ATP7Awt and was inserted into pRS316GPD with the use of NotI. The resultant plasmid served as starting material for cloning of the different ATP7A cDNA variants modified to the yeast system. The fragment from SpeI (restriction site in the polylinker of pRS316) to BclI (restriction site in exon 9 in the ATP7A sequence) was swapped with different NheI- and BclI-digested PCR products. All forward primers (see below) containing a NheI restriction site and a poly A sequence (the translation-initiation consensus sequence in yeast is, presumably, 10 As31) were combined with the reverse primer, ATP7Aex9L (table 1). For preparation of pRS316GPDATP7Awt and pRS316GPDATP7AΔex3+ex4, control and A.B. cDNAs, respectively, were used as template with the forward primer yeast-M1 (table 1). For preparation of pRS316GPDATP7AΔ1−460, pRS316GPDATP7AΔ1−474, and pRS316GPDATP7AΔ1−460,M475L, the forward primers yeast-M461, yeast-M475, and yeast-M461-M475L, respectively, were used with control cDNA as template (table 1).
Yeast Transformation
We used the wild-type Saccharomyces cerevisiae strain 7 (MATα his3-200 leu2 trp1-101 ura3-52 ade5) and the ccc2 mutant strain 8 (MATα his3-200 leu2 trp1-101 ura3-52 ade5 Δccc2::LEU2) described elsewhere.32,33 Yeast transformations were performed using the Yeastmaker Yeast transformation system 2 kit (Clontech): strains 7 and 8 were cultured overnight at 30°C at 200 rpm in YPD-A medium (10 g/liter Bacto Yeast Extract [Difco], 20 g/liter Bacto peptone [Difco], 0.1 g/liter adenine [Sigma], and 2% glucose monohydrate [Merck]). Cells from 0.5 ml culture in log phase were pelleted, and 100 μg freshly denaturated (5 min at 100°C) Herring Testes Carrier DNA, with 1 μg transforming DNA (pRS316GPD-derived, with URA3 marker), was added. Half a milliliter of a solution containing 40% PEG 4000, 0.1 M LiAc, 10 mM Tris-HCl (pH 7.5) and 1 mM EDTA. In addition, 57 μl dimethyl sulfoxide was added, followed by incubation at room temperature for 15 min and heat shock at 42°C for an additional 15 min. The transformed cells were pelleted by centrifugation, were resuspended in 200 μl TE (10 mM Tris-HCl [pH 7.5] and 1 mM EDTA), were spread on synthetic-defined (SD) plates without uracil (27.4 g/liter, SD-ura-leu [Bio-101], 40 g/liter bacto agar [Difco], and 300 mg/liter l-leucin [Merck]), and were cultured at 30°C for 3 d.
Complementation Test
Uracil prototrophic colonies were tested for ATP7A protein function in iron-limited medium, with the use of a method modified from Forbes and Cox.34 Cells were cultured overnight at 30°C in liquid SD medium without uracil, reaching a late log phase. Cells were pelleted, were washed in sterile water, and were cultured overnight after a 1:10 dilution in liquid iron-limited medium (as described by Forbes and Cox,34 with the exception that tryptophan was included and uracil was excluded from the medium). Growth curves and complementation test on plates were prepared as described elsewhere.34 The plates were photographed after 3–4 d at 30°C and 37°C.
Results
Predicted Consequence of the Mutation in the Patient with Less-Severe MD
The patient, A.B., investigated in this study showed remarkably long survival and less-severe symptoms, in spite of what we would expect to be a severe mutation in the ATP7A gene. A.B. (patient D1121) has a partial deletion of the ATP7A gene, including exons 3 and 4 (c.121-?_1338+?del). This allele is designated in this article as “ATP7AΔex3+ex4.” To test the effect of the mutation on the resulting ATP7A mRNA transcript, we analyzed mRNA isolated from A.B. fibroblasts. RT-PCR amplification of the mRNA fragment, from exon 1 to exon 6, led to a 440-bp product (fig. 1, lane P), whereas the expected 1,650-bp product was obtained from control fibroblasts (fig. 1, lane C). Sequence analysis of the 440-bp PCR product revealed that exon 2 was followed directly by exon 5 (r.12_1338del). The deletion changes the reading frame and introduces a PTC in exon 5 (p.V41fsTX7) (fig. 2).
Figure 1. .
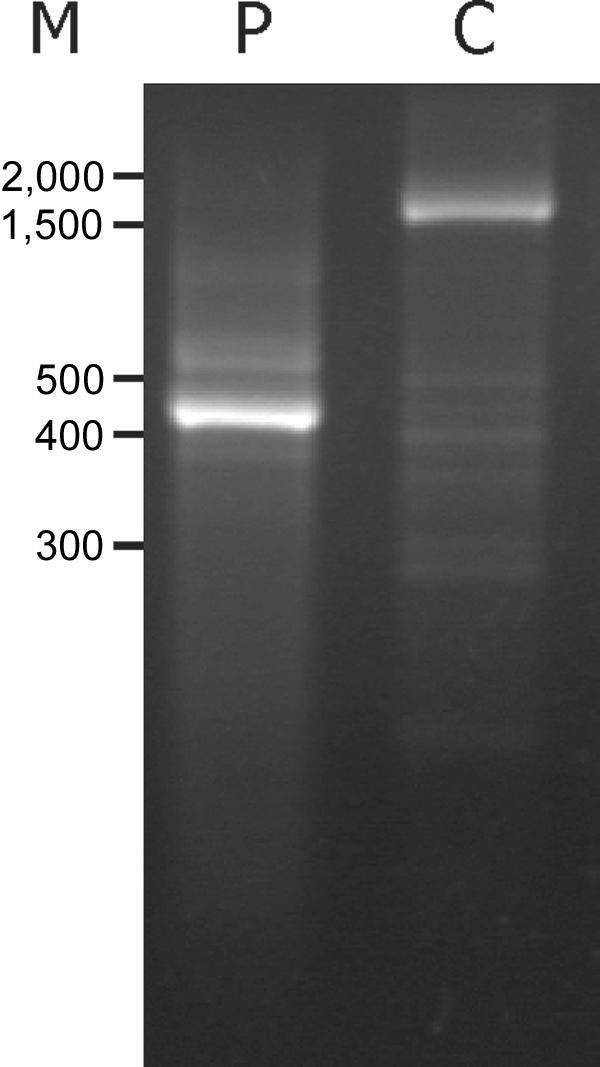
Characterization of patient ATP7A mRNA. PCR amplification of exons 1–6 was performed on cDNA obtained from RNA isolated from fibroblasts from patient A.B. (P) and from an unaffected control person (C). The PCR products were separated on a 1% agarose gel and were visualized by ethidium bromide staining. M, Sizes (in bp) of the marker DNA fragments.
Figure 2. .

Expected cDNA sequence of ATP7AΔex3+ex4. Lowercase letters indicate part of the 5′ UTR, and uppercase letters indicate exons 1, 2, and 5. The predicted normal amino acid sequence (one letter) is indicated below the DNA sequence. Italic letters above the DNA sequence indicate predicted amino acids as a consequence of frameshifting. The vertical lines indicate the exon 1/2 and exon 3/5 junctions. The normal translation-initiation ATG codon in exon 2 is underlined and highlighted. Alternative ATG codons in exon 2 are underlined, and the ATG461 and ATG475 codons in exon 5 are underlined and indicated by number. The position of the T7-containing forward primer T7c.-134-(-111) is indicated. The numbering is in accordance with the normal ATG1 start codon in exon 2.
Quantification of ATP7AΔex3+ex4 Transcript in Patient Cells
Transcripts with PTCs will normally be degraded by NMD.18 To investigate the stability of the ATP7AΔex3+ex4 transcript, the relative amount of this transcript was measured using real-time PCR. Two different ATP7A TaqMan assays were used—one detecting the junction between exons 21 and 22, located downstream from the deletion, and one detecting the junction between exons 1 and 2, upstream from the deletion. An assay detecting GAPDH was used as endogenous control, to normalize for differences in cDNA input in the samples. Patient and control cDNA samples (fourfold serial dilutions from 37.5 ng to 2.3 ng) were assayed with the GAPDH and the ATP7A assays. The results obtained from samples estimated to contain 9.4 ng of cDNA are given in table 2. For both ATP7A assays, we did not find any significant difference in the amount of ATP7A transcript in patient cells compared with that in control cells, when normalized to GAPDH. Results obtained using other cDNA concentrations did not differ from those reported in table 2 (data not shown). This result suggested that the mutation did not affect the stability of the transcript. One reason could be reinitation of protein translation, which has been found to abrogate NMD.19
Table 2. .
Expression of ATP7A Transcript in Patient Fibroblasts[Note]
| Sample | ATP7AN Valuea | ATP7AN Value Relative to Unaffected Controlsb |
| Probe against the exon 20/21 junction: | ||
| Control: | ||
| 1c | .91 | … |
| 2d | .83 | … |
| 3d | .89 | … |
| 4d | 1.20 | … |
| Average | .96 ± .16 | 1.00 ± .17 |
| Patient: | ||
| Experiment Ac | .99 | 1.03 |
| Experiment Bd | 1.39 | 1.46 |
| Experiment Ce | 1.00 | 1.04 |
| Average | 1.13 ± .23 | 1.18 ± .21 |
| Probe against the exon 1/2 junctionf: | ||
| Control: | ||
| 1 | 1.10 | … |
| 2 | 1.02 | … |
| 3 | 1.71 | … |
| Average | 1.28 ± .38 | 1.00 ± .29 |
| Patient (experiment D) | 1.31 | 1.03 |
Note.— Quantifications of transcripts in the patient cells were performed three independent times with the probe against the exon 21/22 junction (experiments A, B, and C) and one time with the probe against the exon 1/2 junction (experiment D), with essentially the same result. In experiment A, one unaffected control was included, and, in experiments B and D, three unaffected controls were included. In all experiments, the amounts of ATP7A and GAPDH RNA were calculated by linear regression of the lines generated by the standard curves, log cDNA concentration against CT. Averages of several experiments appear in bold type.
Normalized ATP7AN value is calculated by dividing the ATP7A RNA value by the GAPDH RNA value (SDs are shown).
ATP7AN value relative to unaffected control fibroblasts is calculated by dividing the normalized ATP7A value by the average ATP7AN value obtained from control fibroblasts (SDs are shown).
Experiment A: Y=-3.57X+24.41 for GAPDH; Y=-3.49X+32.96 for ATP7A.
Experiment B: Y=-3.49X+24.34 for GAPDH; Y=-3.10X+31.99 for ATP7A.
Experiment C: Y=-3.60X+25.87 for GAPDH; Y=-3.41X+31.29 for ATP7A.
Experiment D: Y=-3.49X+24.34 for GAPDH; Y=-3.10X+31.99 for ATP7A.
In Vitro Translation Indicates Initiation at Downstream AUG Codons
The ATP7AΔex3+ex4 protein is predicted to consist of only 46 amino acids (fig. 2) and is unlikely to have any copper-transporting function. To test whether the ATP7AΔex3+ex4 transcript encodes alternative protein products, we analyzed the part of the transcript encoding the N-terminal part of the protein, using RT-PCR combined with a coupled in vitro transcription/translation. The primer pair T7-M1/cATP7A642 (table 1) was used to amplify the coding sequence of cDNA from the ATG start codon 1, located in exon 2, to codon 642, located in exon 8. In vitro transcription/translation of the resulting PCR product, obtained from a control person, results in the formation of a 70-kDa protein product (fig. 3, lane 2), in concordance with the synthesis of 642 aa of the wild-type protein. The same test performed on A.B. cDNA leads to the formation of two protein products of ∼20.5 kDa and ∼23 kDa (fig. 3, lane 3), both of which are substantially larger than the expected 5-kDa product encoded by the 46 codons from the start codon to the PTC. This result indicates in vitro reinitiation of translation.
Figure 3. .

A coupled in vitro transcription/translation of the ATP7AΔex3+ex4 transcript indicates reinitiation of translation at the two internal codons AUG461 and AUG475. On A.B. (P) and control (C) cDNA, different primer pairs were used to PCR-amplify sequences encoding N-terminal fragments. The T7-containing PCR products were transcribed and translated in vitro, in the presence of 35S methionine, and were analyzed by SDS-PAGE. The used primer pairs were T7-M1/cATP7A642 (lanes 2 and 3), T7-M461/cATP7A642 (lanes 4 and 5), T7-kozak-M461/cATP7A642 (lanes 6 and 7), and T7-kozak-M475/cATP7A642 (lanes 8 and 9). Rainbow 14C-methylated proteins (Amersham) were used as molecular-weight markers (M).
To identify possible translation-reinitiation sites, we screened the mRNA sequence for AUG codons downstream from the PTC in the normal reading frame, at which translation-initiation would give rise to protein products of the observed sizes. Two codons, AUG461 and AUG475, corresponding to p.M461 and p.M475, respectively, were found in exon 5 (fig. 2). When a forward primer annealing to the sequence starting at the AUG461 codon was used, the amplified PCR product was devoid of exon 1, exon 2, and the part of exon 5 upstream from AUG461. In vitro transcription/translation of the resulting PCR product (T7-M461/cATP7A642) also leads to the formation of two protein products of ∼20.5 kDa and ∼23 kDa (fig. 3, lanes 4 and 5). Apparently identical products were obtained from both control and A.B. cDNA. The ∼20.5-kDa protein was the most abundant. When the translation start at the AUG461 codon was favored, with use of a forward primer containing a consensus eukaryotic translation-initiation signal (Kozak, ACCACC) upstream from the codon (T7-kozak-M461), the larger ∼23-kDa product became the most abundant (fig. 3, lanes 6 and 7). With use of a forward primer annealing to the sequence starting at the AUG475 codon (T7-kozak-M475), the possibility of translation start at AUG461 was abolished, and only the ∼20.5-kDa product was produced (fig. 3, lanes 8 and 9). The 20.5-kDa and 23-kDa products are in accordance with the expected size of proteins containing 168-aa residues (amino acids 475–642) and 189-aa residues (amino acids 461–642), respectively. These results suggest that protein translation in vitro is reinitiated at the two codons AUG461 and AUG475.
Two alternative AUG start codons in different reading frames are located in exon 2, upstream from the normal AUG start codon 1 (fig. 2). To test if the 5′ UTR region also including these alternative AUG codons had any effect on the results obtained by in vitro translation, we designed a T7-containing forward primer that anneals to the more-upstream–located sequence c.-134-(-111) in exon 1 (fig. 2). In vitro translation of the resulting PCR product leads again to the synthesis of the 20.5-kDa and 23-kDa products, with apparently the same efficiency (data not shown). Thus, the synthesis of these proteins was still possible in the presence of a part of the 5′ UTR region, mimicking to a higher degree the composition of the ATP7AΔex3+ex4 transcript present in A.B. On the other hand, the presence of the 5′ UTR region seems to not enhance the efficiency of the synthesis of the 20.5-kDa and 23-kDa products.
Analysis of ATP7A Protein in Cell Lysate Obtained from Cultured Patient Fibroblasts
To test for the presence of alternative ATP7AΔex3+ex4-encoded proteins in A.B. cells, we performed western blotting with total cell lysate obtained from cultured fibroblasts. We used an anti-ATP7A antibody raised against the 15 most C-terminal amino acids. An ATP7Awt protein of ∼170 kDa was detected in unaffected control fibroblasts but not in the negative control fibroblasts (fig. 4, lanes C and N, respectively). No ATP7A signal was obtained in fibroblasts from A.B. (fig. 4, lane P).
Figure 4. .
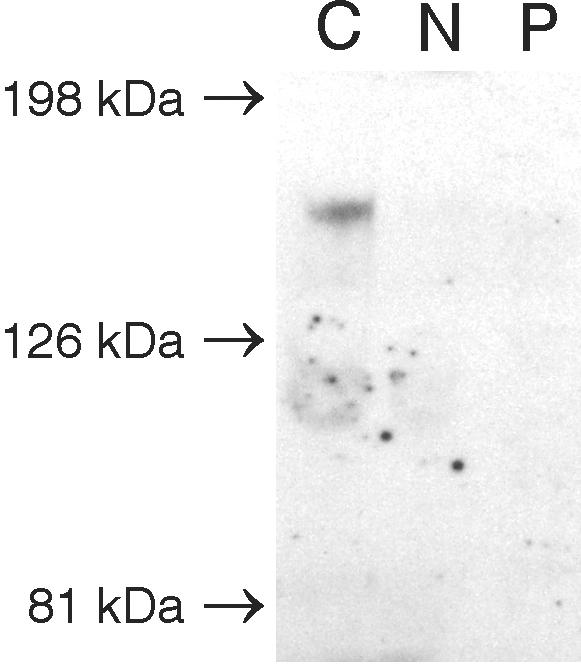
No ATP7AΔex3+ex4 protein could be identified by western blotting analysis in cell extract from A.B. fibroblasts. Proteins in total cell lysates from fibroblasts were separated by SDS-PAGE, followed by immunoblotting with antibody to the ATP7A protein. The results of lysates obtained from positive control cells (C), negative control cells (c.121-?-8333+?del) (N), and A.B. cells (P) are shown. Kaleidoscope Prestained standard was used as a molecular marker (Biorad).
Cellular Localization of the Protein Variants in Cultured Patient Fibroblasts
Although it was not possible to identify any ATP7A proteins in cell extract obtained from the patient by western blotting, we wanted to test if ATP7A proteins could be identified by immunofluorescence (IF). We used the same primary anti-ATP7A antibody, combined with not only a secondary but also a tertiary Alexa 488–conjugated antibody, to enhance the signal. With the use of this system, it was possible to identify the ATP7AΔex3+ex4 protein in A.B. fibroblasts (figs. 5 and 6). At physiological conditions, the copper content in cells is very low, and, to reflect this condition, cells were treated with BCS, which specifically complexes Cu(I) ions. A closely to completely overlapping signal from the ATP7Awt protein and the TGN-localized GS28 SNARE protein35 was observed in almost all the positive control cells after treatment with 150 μM BCS (figs. 5 and 6A). In the A.B. cells, the ATP7A signal was faint and slightly more diffused. However, the ATP7AΔex3+ex4 protein was visible in about half of the cells, and, in the major part of these, the signal was located in TGN, in similarity with the signal present in positive control fibroblasts (figs. 5, 6A, and 6B). However, in contrast to what we observed for the positive control cells, a reduced concentration of BCS (50 μM) was not sufficient to obtain concentrated Golgi localization of the ATP7A signal in the A.B. cells (data not shown). No specific ATP7A signal could be detected in the negative control cells (fig. 5). A nonspecific signal was observed as spheres in the nucleus and as spots in the cytoplasm, but the nonspecific signal was easily distinguishable from the much stronger signal observed in the control and A.B. cells.
Figure 5. .

The endogenously and recombinantly synthesized ATP7AΔex3+ex4 protein shows a copper-dependent trafficking. Fibroblasts and transiently transfected CHO cells were cultured in the presence of CuCl2 or BCS, as indicated, to test the effect of the deletion of exons 3 and 4 on the copper-dependent trafficking. Fibroblasts from A.B. (P), positive control fibroblasts from unaffected persons (C), and negative control fibroblasts from a patient with MD with deletion of exons 3–23 (c.121-?-8333+?del) (N) were analyzed. The analyzed CHO cells were transfected with pCEP4ATP7AΔex3+ex4 and with different constructs optimized to express the truncated ATP7A proteins initiated at p.M461 (pCEP4ATP7AΔ1−460 and pCEP4ATP7AΔ1−460,M475L) and at p.M475 (pCEP4ATP7AΔ1−474). CHO cells transfected with pCEP4ATP7Awt and empty vector (EV) pCEP4 were used as positive and negative controls, respectively. Fixed cells were subjected to double-IF staining for the ATP7A protein (green) and the TGN marker GS28 (red). The combined images are shown (yellow). Images were viewed using a 100× oil objective.
Figure 6. .

Quantification of the effect of CBS1–CBS4 on cellular localization by IF microscopy. A, Percentage of positive cells with ATP7A signal concentrated in TGN, compared with positive cells with more diffuse staining. The cells were treated with CuCl2 or BCS, as indicated. The total number of positive cells is defined as 100%. A.B. fibroblasts (P), positive control fibroblasts (C), and negative control fibroblasts (N), in addition to CHO cells transfected with pCEP4ATP7Awt, pCEP4ATP7AΔex3+ex4, pCEP4ATP7AΔ1−460, pCEP4ATP7AΔ1−474, pCEP4ATP7AΔ1−460,M475L, and EV pCEP4 were investigated. More than 100 cells of each type were analyzed. B, Percentages of cells with a positive ATP7A signal, a less-positive signal, and no signal were determined. A.B. fibroblasts (P) and positive control fibroblasts (C) treated with BCS were investigated. More than 100 cells of each type were analyzed.
Furthermore, we analyzed whether the mutation affects the copper-dependent trafficking normally observed for ATP7Awt.13,14 In cells cultured in media supplemented with 200 μM CuCl2, a diffuse ATP7A staining was found in both the positive control cells and the A.B. cells. A part of the ATP7A staining was still overlapping with the Golgi-located GS28, but a fraction was also scattered in the cytoplasma or was located perinuclear (fig. 5). No cells with persistent, concentrated, TGN-located ATP7A signal were observed (fig. 6A).
These results suggest that mutated ATP7AΔex3+ex4 proteins are synthesized in A.B. fibroblasts and that the C-terminal part of the proteins is identical to the ATP7Awt protein, since an antibody against the 15 most C-terminal amino acids recognized it. In accordance with the intensity of the signal (fig. 6B), the amount of ATP7AΔex3+ex4 was reduced in comparison to the amount of ATP7Awt synthesized in the positive control cells, which might explain the lack of signal with the use of western blotting. In addition, the ATP7AΔex3+ex4 proteins might be sensitive to the lysis treatment performed during western blotting. The mutation probably affects the cellular copper concentration but not, or only slightly, the copper-dependent trafficking of the ATP7AΔex3+ex4 proteins. An increase in intracellular copper concentration is expected, secondary to the impaired copper efflux measured in the A.B. fibroblasts.
Expression of ATP7A Variants in Cultured CHO Cells
To test whether it was possible to synthesize in vivo the protein products predicted by the in vitro study, we generated a recombinant construct encoding the ATP7AΔex3+ex4 cDNA sequence (fig. 7) and introduced it into the plasmid pCEP4ATP7Awt, in place of the ATP7Awt sequence. Plasmids expressing the wild-type and mutant alleles were transiently transfected into CHO cells. Proteins encoded by these constructs contain an inframe, C-terminal Myc tag and are recognized by an antibody against this epitope. The proteins synthesized were examined by western blotting (fig. 8). From the pCEP4ATP7Awt plasmid, a 170-kDa protein is synthesized, and a protein of 120 kDa is synthesized from pCEP4ATP7AΔex3+ex4. The 170-kDa protein correlates with the expected size of the 1,500-aa-long wild-type protein. The 120-kDa protein correlates with a truncated protein containing only two CBSs. This indicates that protein translation not only in vitro but also in vivo is reinitiated at an internal AUG codon. Since proteins encoded by pCEP4ATP7Awt as well as by pCEP4ATP7AΔex3+ex4 were recognized by the antibody against Myc, this result also suggests that the internal reinitiation AUG codon is in the same reading frame as the normal AUG codon. This result suggests that the ATP7AΔex3+ex4 proteins lack the first four CBSs, whereas the remaining C-terminal fragment of the proteins is identical to the ATP7Awt protein. It is, however, not possible to decide whether one or two protein products are synthesized from pCEP4ATP7AΔex3+ex4.
Figure 7. .

Schematic diagram of the ATP7A constructs. Exons 1–6 of ATP7A are depicted as separate small boxes. Exons 7–23 are depicted as a big black box. The predicted N-terminal amino acids are indicated. All the constructs encode an inframe C-terminal Myc tag.
Figure 8. .

Western blotting shows that a protein of ∼120 kDa is synthesized from pCEP4ATP7AΔex3+ex4, which is consistent with reinitation at ATG461 and ATG475. Cell lysates from CHO cells transiently transfected with pCEP4ATP7AΔex3+ex4, pCEP4ATP7AΔ1−460, pCEP4ATP7AΔ1−474, and pCEP4ATP7AΔ1−460,M475L were subjected to SDS-PAGE and immunoblotting, with the use of antibody to the Myc epitope tag. CHO cells transfected with pCEP4ATP7Awt and EV pCEP4 were used as positive and negative controls, respectively. Aliquots of 3–15 μl total cell lysate (∼6–30 μg total protein) were loaded to obtain approximately equal signal intensity in all lanes.
The molecular weight of the pCEP4ATP7AΔex3+ex4-encoded proteins suggests that in vivo reinitiation also takes place at the internal codons AUG461 and AUG475. To test this hypothesis further, we generated two constructs optimized to express the truncated ATP7A proteins initiated at p.M461 (r.1_1380del) and p.M475 (r.1_1422del). These constructs, designated “pCEP4ATP7AΔ1−460” and “pCEP4ATP7AΔ1−474” were devoid of exon 1, exon 2, and the part of exon 5 upstream from the expected restart AUG codons; furthermore, they were cloned with a Kozak consensus sequence upstream from the selected AUG codon (fig. 7). To be able to examine a protein product initiated at p.M461 alone, without the presence of a product initiated at p.M475, we generated a third construct, pCEP4ATP7AΔ1−460,M475L, that encodes a protein initiated at p.M461 combined with the mutation p.M475L (r1_1422del1423A→C). The proteins synthesized from these three constructs migrate in the same way as the protein synthesized from pCEP4ATP7AΔex3+ex4 (fig. 8), which supports the hypothesis that reinitiation also takes place at the AUG461 and AUG475 codons in vivo.
To investigate the cellular localization of the recombinant, truncated ATP7A proteins, we analyzed CHO cells transiently transfected with the different plasmids. By double IF studies, we found that the major part of the ATP7Awt protein encoded by pCEP4ATP7Awt colocalizes with GS28 in the presence of 50 μM BCS (figs. 5 and 6A). The localization of the protein variants encoded by pCEP4ATP7AΔex3+ex4 was slightly more diffused, in agreement with what we observed for the endogenous ATP7AΔex3+ex4 protein synthesized in A.B. fibroblasts (fig. 6A). However, a large fraction of the recombinantly synthesized ATP7AΔex3+ex4 protein also seems to be correctly located in the TGN (fig. 6A). Similarly, a large fraction of pCEP4ATP7AΔ1−460-, pCEP4ATP7AΔ1−474-, and pCEP4ATP7AΔ1−460,M475L-encoded protein products colocalized with GS28 (fig. 6A). To investigate if the protein variants were able to traffic in response to copper, we incubated transfected CHO cells with 200 μM CuCl2. As a result of copper treatment, the intensities of ATP7Awt and of the different ATP7A variants in TGN decreased, and a staining throughout the cytoplasma appeared in the major part of the cells (figs. 5 and 6A). Indistinguishable staining patterns were seen for all tested protein variants.
Since the cellular localization and copper-dependent trafficking of the recombinantly and the endogenously synthesized ATP7AΔex3+ex4 proteins are similar, these results support the hypothesis that the endogenously synthesized ATP7AΔex3+ex4 variants are identical to proteins encoded by pCEP4ATP7AΔex3+ex4, which might be identical to the proteins encoded by pCEP4ATP7AΔ1−460 and pCEP4ATP7AΔ1−474. Furthermore, lack of the four N-terminal CBS1–CBS4 did not appear to substantially affect the localization or trafficking of the ATP7A protein, in agreement with results published elsewhere.36
Complementation of a Δccc2 Yeast Strain by Expression of Constructs Encoding ATP7A Variants
To investigate the biological function of the mutated ATP7A proteins, we cloned the wild-type and different mutated cDNA forms into the low-copy yeast expression vector, pRS316GPD, for introduction into the yeast strain Δccc2 lacking the CCC2 gene, the yeast homologue to ATP7A. In S. cerevisiae, Ccc2p is responsible for the incorporation of copper into the copper-requiring ferroxidase Fet3p, a ceruloplasmin homologue necessary for high-affinity iron uptake.32 As a consequence, a Δccc2 yeast strain is deficient in high-affinity iron uptake, a phenotype that is detected by reduced growth under iron-limited conditions (fig. 9A). The wild-type yeast strain showed no growth phenotype on iron-limited medium (fig. 9A). All the constructs—pRS316GPDATP7Awt, pRS316GPDATP7AΔex3+ex4, pRS316GPDATP7AΔ1−460, pRS316GPDATP7AΔ1−474, and pRS316GPDATP7AΔ1−460,M475L—encoding recombinant ATP7A proteins were able to complement the Δccc2 iron-deficient phenotype at 30°C; thus, they allowed cells to grow under iron-limited conditions (fig. 9A), indicating that they were at least partially functional. These data suggest that reinitiation of the ATP7AΔex3+ex4 cDNA sequence containing the 46-codon ORF upstream from the expected restart AUG codons is possible not only in mammals but also in yeast. Interestingly, at 37°C, the construct pRS316GPDATP7AΔex3+ex4 was no longer able to complement, whereas complementation was still observed with the remaining constructs (fig. 9D). At 37°C, the truncated protein is likely less stable, and the reinitiation efficiency might be too low to produce a sufficient amount for complementation. Addition of copper (fig. 9B and 9E) or iron (fig. 9C and 9F) rescued all transformed cells at both 30°C and 37°C. To more closely define the complementation efficiency of the mutant constructs, we analyzed the doubling time at 30°C in liquid iron-limited medium. The doubling time was calculated on the basis of OD600 measurements of exponentially growing cultures. The growth rate of the Δccc2 yeast cells transformed with the different mutants was not significantly different from the pRS316GPDATP7Awt-transformed cells (Student t test P<.05) (fig. 10). The almost normal growth rate at 30°C supports the notion that the ATP7AΔex3+ex4 protein is at least partially functional, consistent with the less-severe phenotype of A.B.
Figure 9. .

N-terminally truncated Menkes proteins complement a ccc2 yeast strain by restoring high-affinity iron uptake. The Δccc2 strain 8 was transformed with EV (pRS316GPD), pRS316GPDATP7Awt, pRS316GPDATP7AΔex3+ex4, pRS316GPDATP7AΔ1−460, pRS316GPDATP7AΔ1−474, and pRS316GPDATP7AΔ1−460,M475L. The wild-type strain 7 was transformed with EV. The transformed colonies were cultured as described in the “Material and Methods” section. At optical density OD600=0.1, an aliquot of 5 μl was streaked on plates with iron-limited medium (A and D). Rescue of the Δccc2 yeast can also be accomplished by addition of either a high concentration of copper (B and E) or iron (C and F) to the iron-limited medium.32,55 Plates were photographed after 4 d at 30°C or 37°C, as indicated. Schematic illustration of the plates is shown.
Figure 10. .

Complementation efficiency of N-terminal–truncated Menkes proteins synthesized in Δccc2 yeast. The growth in iron-limited medium of wild-type strain 7 harboring pRS316GPD (EV) (n=5) and of Δccc2 strain 8 harboring pRS316GPDATP7Awt (n=4), pRS316GPDATP7AΔex3+ex4 (n=3), pRS316GPDATP7AΔ1−460 (n=3), pRS316GPDATP7AΔ1−474 (n=3), and pRS316GPDATP7AΔ1−460,M475L (n=3) was determined by OD600 measurements. OD600 (log scale) was plotted in comparison with time, and, in accordance with the equation describing exponential growth (Y=a*bx), the doubling time was calculated as log2/logb. SDs are shown.
Discussion
Here, we report, for the first time, moderation of the clinical phenotype in a patient with MD by partially functional truncated proteins synthesized as a result of translation reinitiation. The patient had a less-severe phenotype, despite a large frameshift gene deletion. Surprisingly, we found that, although it contains a PTC, the transcript was protected from NMD. The predicted product of the ATP7AΔex3+ex4 transcript consists of only 46 aa, which is extremely unlikely to mediate any copper translocation. Therefore, we used an in vitro translation system to test the hypothesis that the ATP7AΔex3+ex4 transcript encodes alternative proteins. We found that the transcript directed the synthesis of N-terminally truncated proteins by reinitiation at the downstream codons AUG461 and AUG475. Transfection studies confirmed the synthesis of proteins of a size consistent with the predicted reinitiation codons. Using an antibody against the C-terminus, we detected endogenously synthesized ATP7A in patient fibroblasts, which supports the formation of an N-terminally truncated protein.
Translation Initiation from Internal AUG Codons
Translation in eukaryotes is normally initiated at the first AUG codon closest to the 5′ end, as long as it resides in the optimal context: GCCRCCAUGG. The purine R in position −3 (G or A, where A is preferred) and the G in position +4 are the most important (reviewed in Kozak37). However, studies have shown that, when a PTC shortly follows the first AUG codon, the ribosomes move forward to reinitiate at downstream internal AUG codons.38 By investigating plasmid constructs, reinitiation was found to be optimal after translation of an ORF between 13 and 55 codons.38,39 Since the first ORF in ATP7AΔex3+ex4 is 46 codons in length, the conditions for reinitiation are met in this study, and we propose that translation initiates at the normal AUG1 codon, producing a 46-aa-long peptide, followed by reinitiation at AUG461 and AUG475.
It has been demonstrated elsewhere40 that reinitiation is not limited to the closest AUG beyond the 5′-proximal ORF. If the ribosomal subunits, which only gradually become competent, are not yet ready when they reach the first AUG codon downstream from the PTC, they continue scanning, initiating at an AUG codon(s) further downstream.40 This phenomenon might explain why we found reinitiation at both the AUG461 codon and the AUG475 codon. Alternatively, it might be the result of leaky scanning, since the AUG461 resides in the weak context UCAUCGGAAAUGC (fig. 2).
Very few studies have previously demonstrated reinitiation of translation in patients suffering from inherited disorders. Thus, in patients suffering from peroxisome-biogenesis disorder, methylmalonic acidemia, or androgen resistance, reinitiation has been demonstrated after translation of ORFs of 8, 17, or 59 codons, respectively.41–43 Interestingly, in a patient suffering from peroxisome-biogenesis disorder, because of two different mutations in the PEX12 alleles, reinitiation of one of two mutated transcripts was proposed to modulate the phenotype.41 To our knowledge, this is the only previous demonstration of translation reinitiation proposed to modulate the disease phenotype of a patient. Since peroxisome-biogenesis disorder due to PEX12 mutations is autosomally inherited, interpretation is more complicated than for the present X-linked MD.
Abrogation of NMD
Transcripts with PTC are generally low in abundance, because of decay.18,44 However, some exceptions from this rule have been described. Transcripts containing PTC positioned in close proximity to the initial codon in triosephosphate isomerase constructs and in β-globin and BRCA1 mRNA have been found to be, at least partially, NMD resistant.19,45,46 For triosephosphate isomerase constructs, this resistance was caused by reinitation of translation,19 whereas it does not seem to be the case for β-globin mRNA.46
The normal amount of ATP7A transcript in cultured skin fibroblasts from the patient indicates that the ATP7AΔex3+ex4 transcript containing a PTC at codon 47 is not recognized by the NMD machinery. We found that the level of ATP7A transcripts containing a PTC after ORFs of 305 and 425 codons was <15% of the level found in controls (data not shown [L. B. Møller, unpublished results]). Testing the 5′ ends of these mutated cDNAs by the in vitro transcription/translation system did not reveal any reinitiation (data not shown). Thus, PTC-containing ATP7A transcripts seem to behave as predicted from the literature18,19,44 and are not always protected against NMD. In conclusion, our results suggest that the ATP7AΔex3+ex4 transcript containing an early PTC is protected from degradation as a consequence of reinitiation.
Partially Functional Activity of the N-Terminally Truncated Proteins Containing CBS5 and CBS6
We observed cellular localization and copper-dependent trafficking of endogenous and recombinant ATP7AΔex3+ex4 proteins similar to that of the ATP7Awt protein, with one major exception. A higher concentration of BCS was necessary to obtain Golgi localization of the endogenous ATP7AΔex3+ex4 proteins, compared with ATP7Awt. The higher cellular copper content observed in the A.B. fibroblasts might explain this difference in sensitivity to the copper chelator BCS.
This observation is consistent with previously published results, showing that CBS1–CBS4 in neither ATP7A nor ATP7B is necessary for copper-dependent trafficking.36,47 However, since CHO cells expressing the ATP7AΔex3+ex4 construct also had a lower percentage of TGN-concentrated ATP7A signal compared with the ATP7Awt-expressing cells, we cannot exclude the possibility that the deletion of the four CBSs makes the ATP7A protein more sensitive to copper.
The Δccc2 strain was complemented by constructs expressing the ATP7AΔex3+ex4 proteins, indicating that the proteins are able to load Fet3p with copper to regain high-affinity iron uptake. In a previous study, a similar construct encoding a Menkes protein without the first four CBSs was found to complement almost as well.17 The minor difference in complementation efficiency can be explained by the difference in the constructs; the construct of Mercer’s group encodes a protein lacking amino acids 8–485, whereas the proteins tested in this study lack the amino acids 1–460 or 1–474. For the Wilson protein, it has been demonstrated that the sixth CBS alone is sufficient for complementation.48 It was found that a Wilson cDNA construct with all the cysteines in CBS1–CBS5 mutated, as well as a construct where CBS1–CBS5 were deleted, were able to complement the Δccc2 strain.48
Consistent with the functional properties of the ATP7AΔex3+ex4 proteins, serum levels of copper and ceruloplasmin in A.B. were reduced to only ∼50% of normal. In patients with typical MD, levels are reduced to ∼10%.24 The hair had no pili torti, as is normally seen in MD patients. A.B. has had a long survival, similar to patients with OHS, whereas patients with classical MD typically die before they are 3 years old. That the overall activity of the ATP7AΔex3+ex4 proteins was not sufficient to obtain homeostatic copper balance was, however, clear. Although A.B., in contrast to patients with typical MD, was able to achieve minor improvements, he has severe neurological symptoms: mental delay, epileptic seizures, and no speech.24 A reduced amount of endogenously synthesized ATP7AΔex3+ex4 proteins could explain the lack of full complementation. From previous studies, it has been shown that, even when optimal conditions for reinitiation are present, the degree of reinitiation is not 100%.39 Similarly, we found a reduced in vitro reinitiation level, using the ATP7AΔex3+ex4 cDNA sequence as template, compared with the normal initiation level obtained using ATP7Awt cDNA as template. The lack of detection of the endogenous ATP7AΔex3+ex4 proteins by western blotting and the faint IF signal in the major part of the cells support this suggestion.
Another contributing factor could be that the region containing CBS1–CBS4, indeed, has an important function in maintaining normal copper homeostasis in humans—a function that was not assessed here. For both ATP7A and ATP7B, an interaction between the copper chaperone, Atox1, and CBS1–CBS4 has been demonstrated.9,10,12 CBS2 and CBS4 have been identified as important sites in ATP7B for this interaction.11,12 It was proposed that binding of copper to CBS2 works as a switch, allowing subsequent loading of other sites.11
The diffuse localization pattern observed for the endogenous ATP7AΔex3+ex4 proteins, especially in the presence of low concentration of BCS, is similar to the ATP7A localization pattern in Atox1−/− cells.49 Consistent with the proposed role for Atox1 in copper delivery to the secretory pathway, a marked increase in intracellular copper content secondary to impaired copper efflux was observed in Atox1−/− cells. We speculate that copper efflux, in both cases, is hampered because of a lack of interaction between ATP7A and Atox1. Several disease-causing mutations in ATP7B have been shown to interrupt the interaction between the ATP7B protein and Atox1,50 stressing the importance of this interaction for copper homeostasis.
The Biochemical Phenotype of a Patient with MD and Efficacy of Copper Therapy
The treatment of MD involves copper replacement therapy, where parenteral injections of copper histidine bypass the intestinal blockage of dietary copper absorption, raising the circulating copper levels. Moreover, copper crossing the blood-brain barrier is required to prevent the classical neurological symptoms in patients with MD. It has been shown elsewhere that the efficacy of copper therapy in patients with MD depends on the presence of residual ATP7A protein function.51 Patients with MD with truncated and/or mislocated, but partially functional, ATP7A proteins have been copper treated with success.52–54 These results and the present case highlight the importance of investigating the biochemical phenotype of a patient to provide an estimation of copper-therapy success, even though the genotype predicts a null mutation.
Conclusion
Reinitiation of NMD-resistant transcripts, combined with a PTC located close to the initiation codon, might be suspected in patients with less-severe symptoms of any type of genetic disease. In the present case, a partial activity of a truncated Menkes protein probably contributed to a less-severe phenotype. The correlation between NMD and reinitiation has previously been studied entirely with the use of minigene transfection, where mutations were introduced by site-directed mutagenesis. To our knowledge, this is the first direct demonstration of abrogation to NMD combined with translation reinitiation of an endogenous transcript.
Acknowledgments
We thank Michael J. Francis, for the generous gift of the plasmid pCEP4 containing the wild-type Menkes cDNA, and Vicky L. Kirby, for the multicopy expression vector pG-1. We also thank Andrew Dancis and Theresa Dunn, for S. cerevisiae strains 7 and 8, and Carsten Petersen, for E. coli strain 2202. This study was supported by The Danish Medical Research Council, The Novo Nordisk Foundation, the Foundation of 1870, and a DANSYNC grant from the Danish National Research Council.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for ATP7A [accession number L06133.1] and ATP7A [accession number NP_000043])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for MD, ATP7A, WD, and ATP7B)
References
- 1.Danks DM, Campbell PE, Walker-Smith J, Stevens BJ, Gillespie JM, Blomfield J, Turner B (1972) Menkes’ kinky-hair syndrome. Lancet 1:1100–1102 10.1016/S0140-6736(72)91433-X [DOI] [PubMed] [Google Scholar]
- 2.Møller JV, Juul B, Le Maire M (1996) Structural organization, ion transport, and energy transduction of P-type ATPases. Biochim Biophys Acta 1286:1–51 [DOI] [PubMed] [Google Scholar]
- 3.Horn N, Tümer Z (2002) Menkes disease and the occipital horn syndrome. In: Royce P, Steinmann B (eds) Connective tissue and its heritable disorders, 2nd ed. Wiley-Liss, New York, pp 651–685 [Google Scholar]
- 4.Cox DW (1996) Molecular advances in Wilson disease. Prog Liver Dis 14:245–264 [PubMed] [Google Scholar]
- 5.Cuthbert JA (1998) Wilson’s disease: update of a systemic disorder with protean manifestations. Gastroenterol Clin North Am 27:655–681 10.1016/S0889-8553(05)70025-X [DOI] [PubMed] [Google Scholar]
- 6.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW (1993) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 5:327–337 10.1038/ng1293-327 [DOI] [PubMed] [Google Scholar]
- 7.Vulpe C, Levinson B, Whitney S, Packma S, Gitschier J (1993) Isolation of a candidate gene for Menkes disease and evidence that it encodes a copper-transporting ATPase. Nat Genet 3:7–13 10.1038/ng0193-7 [DOI] [PubMed] [Google Scholar]
- 8.Hung IH, Suzuki M, Yamaguchi Y, Yuan DS, Klausner RD, Gitlin JD (1997) Biochemical characterization of the Wilson disease protein and functional expression in the yeast Saccharomyces cerevisiae. J Biol Chem 272:21461–21466 10.1074/jbc.272.34.21461 [DOI] [PubMed] [Google Scholar]
- 9.Larin D, Mekios C, Das K, Ross B, Yang AS, Gilliam TC (1999) Characterization of the interaction between the Wilson and Menkes disease proteins and the cytoplasmic copper chaperone, HAH1p. J Biol Chem 274:28497–28504 10.1074/jbc.274.40.28497 [DOI] [PubMed] [Google Scholar]
- 10.Strausak D, Howie MK, Firth SD, Schlicksupp A, Pipkorn R, Multhaup G, Mercer JF (2003) Kinetic analysis of the interaction of the copper chaperone Atox1 with the metal binding sites of the Menkes protein. J Biol Chem 278:20821–20827 10.1074/jbc.M212437200 [DOI] [PubMed] [Google Scholar]
- 11.Walker JM, Huster D, Ralle M, Morgan CT, Blackburn NJ, Lutsenko S (2004) The N-terminal metal-binding site 2 of the Wilson’s disease protein plays a key role in the transfer of copper from Atox1. J Biol Chem 279:15376–15384 10.1074/jbc.M400053200 [DOI] [PubMed] [Google Scholar]
- 12.van Dongen EM, Klomp LW, Merkx M (2004) Copper-dependent protein-protein interactions studied by yeast two-hybrid analysis. Biochem Biophys Res Commun 323:789–795 10.1016/j.bbrc.2004.08.160 [DOI] [PubMed] [Google Scholar]
- 13.Petris MJ, Mercer JF, Culvenor JG, Lockhart P, Gleeson PA, Camakaris J (1996) Ligand-regulated transport of the Menkes copper P-type ATPase efflux pump from the Golgi apparatus to the plasma membrane: a novel mechanism of regulated trafficking. EMBO J 15:6084–6095 [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi Y, Heiny ME, Suzuki M, Gitlin JD (1996) Biochemical characterization and intracellular localization of the Menkes disease protein. Proc Natl Acad Sci USA 93:14030–14035 10.1073/pnas.93.24.14030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaefer M, Roelofsen H, Wolters H, Hofmann WJ, Muller M, Kuipers F, Stremmel W, Vonk RJ (1999) Localization of the Wilson’s disease protein in human liver. Gastroenterology 117:1380–1385 10.1016/S0016-5085(99)70288-X [DOI] [PubMed] [Google Scholar]
- 16.Voskoboinik I, Strausak D, Greenough M, Brooks H, Petris M, Smith S, Mercer JF, Camakaris J (1999) Functional analysis of the N-terminal CXXC metal-binding motifs in the human menkes copper-transporting P-type ATPase expressed in cultured mammalian cells. J Biol Chem 274:22008–22012 10.1074/jbc.274.31.22008 [DOI] [PubMed] [Google Scholar]
- 17.Mercer JF, Barnes N, Stevenson J, Strausak D, Llanos RM (2003) Copper-induced trafficking of the Cu-ATPases: a key mechanism for copper homeostasis. Biometals 16:175–184 10.1023/A:1020719016675 [DOI] [PubMed] [Google Scholar]
- 18.Maquat LE (1995) When cells stop making sense: effects of nonsense codons on RNA metabolism in vertebrate cells. RNA 1:453–465 [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J, Maquat LE (1997) Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J 16:826–833 10.1093/emboj/16.4.826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tümer Z, Tønnesen T, Horn N (1994) Detection of genetic defects in Menkes disease by direct mutation analysis and its implications in carrier diagnosis. J Inherit Metab Dis 17:267–270 10.1007/BF00711804 [DOI] [PubMed] [Google Scholar]
- 21.Tümer Z, Møller LB, Horn N (2003) Screening of 383 unrelated patients affected with Menkes disease and finding of 57 gross deletions in ATP7A. Hum Mutat 22:457–464 10.1002/humu.10287 [DOI] [PubMed] [Google Scholar]
- 22.Poulsen L, Horn N, Heilstrup H, Lund C, Tümer Z, Møller LB (2002) X-linked recessive Menkes disease: identification of partial gene deletions in affected males. Clin Genet 62:449–457 10.1034/j.1399-0004.2002.620605.x [DOI] [PubMed] [Google Scholar]
- 23.Poulsen L, Horn N, Møller LB (2002) X-linked recessive Menkes disease: carrier detection in the case of a partial gene deletion. Clin Genet 62:440–448 10.1034/j.1399-0004.2002.620604.x [DOI] [PubMed] [Google Scholar]
- 24.Haas RH, Robinson A, Evans K, Lascelles PT, Dubowitz V (1981) An X-linked disease of the nervous system with disordered copper metabolism and features differing from Menkes disease. Neurology 31:852–859 [DOI] [PubMed] [Google Scholar]
- 25.Tønnesen T, Garrett C, Gerdes AM (1991) High 64Cu uptake and retention values in two clinically atypical Menkes patients. J Med Genet 28:615–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Møller LB, Tümer Z, Lund C, Petersen C, Cole T, Hanusch R, Seidel J, Jensen LR, Horn N (2000) Similar splice-site mutations of the ATP7A gene lead to different phenotypes: classical Menkes disease or occipital horn syndrome. Am J Hum Genet 66:1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Francis MJ, Jones EE, Levy ER, Ponnambalam S, Chelly J, Monaco AP (1998) A Golgi localization signal identified in the Menkes recombinant protein. Hum Mol Genet 7:1245–1252 10.1093/hmg/7.8.1245 [DOI] [PubMed] [Google Scholar]
- 28.Møller LB, Bukrinsky JT, Mølgaard A, Paulsen M, Lund C, Tümer Z, Larsen S, Horn N (2005) Identification and analysis of 21 novel disease-causing amino acid substitutions in the conserved part of ATP7A. Hum Mutat 26:84–93 10.1002/humu.20190 [DOI] [PubMed] [Google Scholar]
- 29.Sikorski RS, Hieter P (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schena M, Picard D, Yamamoto KR (1991) Vectors for constitutive and inducible gene expression in yeast. Methods Enzymol 194:389–398 [DOI] [PubMed] [Google Scholar]
- 31.Vega Laso MR, Zhu D, Sagliocco F, Brown AJ, Tuite MF, McCarthy JE (1993) Inhibition of translational initiation in the yeast Saccharomyces cerevisiae as a function of the stability and position of hairpin structures in the mRNA leader. J Biol Chem 268:6453–6462 [PubMed] [Google Scholar]
- 32.Yuan DS, Stearman R, Dancis A, Dunn T, Beeler T, Klausner RD (1995) The Menkes/Wilson disease gene homologue in yeast provides copper to a ceruloplasmin-like oxidase required for iron uptake. Proc Natl Acad Sci USA 92:2632–2636 10.1073/pnas.92.7.2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu D, Beeler TJ, Dunn TM (1995) Sequence, mapping and disruption of CCC2, a gene that cross-complements the Ca(2+)-sensitive phenotype of csg1 mutants and encodes a P-type ATPase belonging to the Cu(2+)-ATPase subfamily. Yeast 11:283–292 10.1002/yea.320110310 [DOI] [PubMed] [Google Scholar]
- 34.Forbes JR, Cox DW (1998) Functional characterization of missense mutations in ATP7B: Wilson disease mutation or normal variant? Am J Hum Genet 63:1663–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Subramaniam VN, Peter F, Philp R, Wong SH, Hong W (1996) GS28, a 28-kilodalton Golgi SNARE that participates in ER-Golgi transport. Science 272:1161–1163 [DOI] [PubMed] [Google Scholar]
- 36.Strausak D, La Fontaine S, Hill J, Firth SD, Lockhart PJ, Mercer JF (1999) The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J Biol Chem 274:11170–11177 10.1074/jbc.274.16.11170 [DOI] [PubMed] [Google Scholar]
- 37.Kozak M (2002) Pushing the limits of the scanning mechanism for initiation of translation. Gene 299:1–34 10.1016/S0378-1119(02)01056-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozak M (2001) Constraints on reinitiation of translation in mammals. Nucleic Acids Res 29:5226–5232 10.1093/nar/29.24.5226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luukkonen BG, Tan W, Schwartz S (1995) Efficiency of reinitiation of translation on human immunodeficiency virus type 1 mRNAs is determined by the length of the upstream open reading frame and by intercistronic distance. J Virol 69:4086–4094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozak M (1987) Effects of intercistronic length on the efficiency of reinitiation by eucaryotic ribosomes. Mol Cell Biol 7:3438–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang CC, Gould SJ (1998) Phenotype-genotype relationships in complementation group 3 of the peroxisome-biogenesis disorders. Am J Hum Genet 63:1294–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ledley FD (1990) Perspectives on methylmalonic acidemia resulting from molecular cloning of methylmalonyl CoA mutase. Bioessays 12:335–340 10.1002/bies.950120706 [DOI] [PubMed] [Google Scholar]
- 43.Zoppi S, Wilson CM, Harbison MD, Griffin JE, Wilson JD, McPhaul MJ, Marcelli M (1993) Complete testicular feminization caused by an amino-terminal truncation of the androgen receptor with downstream initiation. J Clin Invest 91:1105–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wormington M (2003) Zero tolerance for nonsense: nonsense-mediated mRNA decay uses multiple degradation pathways. Mol Cell 12:536–538 10.1016/S1097-2765(03)00362-9 [DOI] [PubMed] [Google Scholar]
- 45.Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S (2002) The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet 11:2805–2814 10.1093/hmg/11.23.2805 [DOI] [PubMed] [Google Scholar]
- 46.Inacio A, Silva AL, Pinto J, Ji X, Morgado A, Almeida F, Faustino P, Lavinha J, Liebhaber SA, Romao L (2004) Nonsense mutations in close proximity to the initiation codon fail to trigger full nonsense-mediated mRNA decay. J Biol Chem 279:32170–32180 10.1074/jbc.M405024200 [DOI] [PubMed] [Google Scholar]
- 47.Cater MA, Forbes J, La Fontaine S, Cox D, Mercer JF (2004) Intracellular trafficking of the human Wilson protein: the role of the six N-terminal metal-binding sites. Biochem J 380:805–813 10.1042/BJ20031804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forbes JR, Hsi G, Cox DW (1999) Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J Biol Chem 274:12408–12413 10.1074/jbc.274.18.12408 [DOI] [PubMed] [Google Scholar]
- 49.Hamza I, Prohaska J, Gitlin JD (2003) Essential role for Atox1 in the copper-mediated intracellular trafficking of the Menkes ATPase. Proc Natl Acad Sci USA 100:1215–1220 10.1073/pnas.0336230100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hamza I, Schaefer M, Klomp LW, Gitlin JD (1999) Interaction of the copper chaperone HAH1 with the Wilson disease protein is essential for copper homeostasis. Proc Natl Acad Sci USA 96:13363–13368 10.1073/pnas.96.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaler SG (1996) Menkes disease mutations and response to early copper histidine treatment. Nature Genet 13:21–22 10.1038/ng0596-21 [DOI] [PubMed] [Google Scholar]
- 52.Ambrosini L, Mercer JFB (1999) Defective copper-induced trafficking and localization of the Menkes protein in patients with mild and copper-treated classical Menkes disease. Hum Mol Genet 8:1547–1555 10.1093/hmg/8.8.1547 [DOI] [PubMed] [Google Scholar]
- 53.Kaler SG, Das S, Levinson B, Goldstein DS, Holmes CS, Patronas NJ, Packman S, Gahl WA (1996) Successful early copper therapy in Menkes disease associated with a mutant transcript containing a small in-frame deletion. Biochem Mol Med 57:37–46 10.1006/bmme.1996.0007 [DOI] [PubMed] [Google Scholar]
- 54.Kim BE, Smith K, Petris MJ (2003) A copper treatable Menkes disease mutation associated with defective trafficking of a functional Menkes copper ATPase. J Med Genet 40:290–295 10.1136/jmg.40.4.290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stearman R, Yuan DS, Yamaguchi-Iwai Y, Klausner RD, Dancis A (1996) A permease-oxidase complex involved in high-affinity iron uptake in yeast. Science 271:1552–1557 [DOI] [PubMed] [Google Scholar]