

Abstract
We have generated a novel transgenic mouse model on a C57BL/6J genetic background that coexpresses KM670/671NL mutated amyloid precursor protein and L166P mutated presenilin 1 under the control of a neuron-specific Thy1 promoter element (APPPS1 mice). Cerebral amyloidosis starts at 6–8 weeks and the ratio of human amyloid (A)β42 to Aβ40 is 1.5 and 5 in pre-depositing and amyloid-depositing mice, respectively. Consistent with this ratio, extensive congophilic parenchymal amyloid but minimal amyloid angiopathy is observed. Amyloid-associated pathologies include dystrophic synaptic boutons, hyperphosphorylated tau-positive neuritic structures and robust gliosis, with neocortical microglia number increasing threefold from 1 to 8 months of age. Global neocortical neuron loss is not apparent up to 8 months of age, but local neuron loss in the dentate gyrus is observed. Because of the early onset of amyloid lesions, the defined genetic background of the model and the facile breeding characteristics, APPPS1 mice are well suited for studying therapeutic strategies and the pathomechanism of amyloidosis by cross-breeding to other genetically engineered mouse models.
Keywords: Alzheimer, amyloid, ageing, microglia transgenic mouse model
Introduction
A variety of transgenic mice have been generated to model cerebral amyloidosis, a hallmark of Alzheimer disease (AD) pathology. These models have been instrumental in studying the impact and mechanism of cerebral amyloidosis and to develop therapeutic strategies (Selkoe & Schenk, 2003). However, many of these models have limitations such as late onset of the pathology, mixed genetic backgrounds on which the mice have been generated, difficulties with breeding, gender differences in pathology and high variability in amyloid β (Aβ) levels. Therefore, we aimed to develop a transgenic mouse model that addresses most of these issues.
The most aggressive familial AD mutation identified so far is the Leu to Pro mutation at position 166 of presenilin 1 (PS1). This mutation in humans leads to disease onset as early as 24 years of age (Moehlmann et al, 2002; Bentahir et al, 2006). In transfected cells, the PS1-L166P mutation reveals the highest Aβ42 to Aβ40 ratio among all familial AD mutations studied so far (Moehlmann et al, 2002), which is consistent with the idea that a high Aβ42 to Aβ40 ratio is an important determinant for the onset of amyloid pathology (Herzig et al, 2004; Duering et al, 2005). Thus, we have generated transgenic mice coexpressing this mutated PS1 and KM670/671NL ‘Swedish' mutated amyloid precursor protein (APP), a familial AD mutation that has been previously used to generate transgenic mice (Hsiao et al, 1996; Sturchler-Pierrat et al, 1997). For both the PS1 and the APP constructs, the Thy1 minigene promoter was used to restrict expression to the postnatal brain and to achieve high levels of neuron-specific transgene expression. Finally, we have generated these transgenic mice on a C57BL/6J background to minimize genetic variability and because the C57BL/6J mice are the best-characterized mouse strain in the field of the neurobiology of ageing (Jucker & Ingram, 1997).
Results
Cerebral amyloidosis
Several transgenic lines were generated (Fig 1). Line 21 (APPPS1-21 mice) was selected for further analysis because it had high transgene expression levels, showed significant amyloid plaque formation (Fig 1), reproduced at a high rate heterozygotically (7.0±2.8 pups/litter (±s.d.); n=88 litters) and transgene segregation followed the expected mendelian inheritance (average males/litter: 3.6±1.6 of which 1.8±1.2 were transgenic; females: 3.4±2.0 of which 1.7±1.3 were transgenic). Transgene mapping revealed that both transgenes were integrated at the lower arm of chromosome 2 between 40 and 60 cM.
Figure 1.
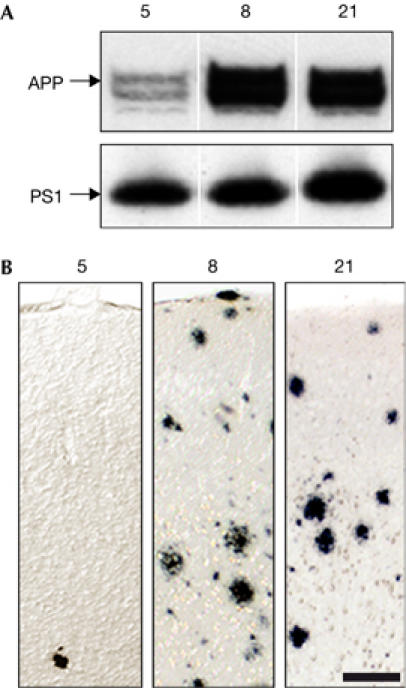
Amyloid precursor protein and presenilin 1 expression in APPPS1 transgenic lines. (A) Western blot showed similar human APP and PS1 expression in lines 8 and 21, whereas APP was lower in line 5. The upper APP band is glycosylated APP and the lower band is immature non-glycosylated APP. (B) Aβ-immunostaining showed only few amyloid deposits in the neocortex of 8-month-old male mice from line 5, whereas male mice from lines 8 and 21 showed abundant amyloidosis at this age, suggesting that high APP levels are more important than high PS1 levels for inducing cerebral amyloidosis. Scale bar, 50 μm. Aβ, amyloid β; APP, amyloid precursor protein; PS, presenilin 1.
Human APP expression in APPPS1-21 mice was about three times that of endogenous mouse APP (Fig 2) and was confined mainly to neurons in the neocortex, hippocampus and brain stem and to a lesser extent in the thalamus and striatum (supplementary Fig S1 online). Aβ levels showed a significant increase from 1 to 8 months of age, with Aβ42 exceeding Aβ40 several-fold (Table 1). The Aβ42 to Aβ40 ratio in pre-depositing 1-month-old mice was 1.6:1, whereas in depositing mice the ratio was about 5:1.
Figure 2.
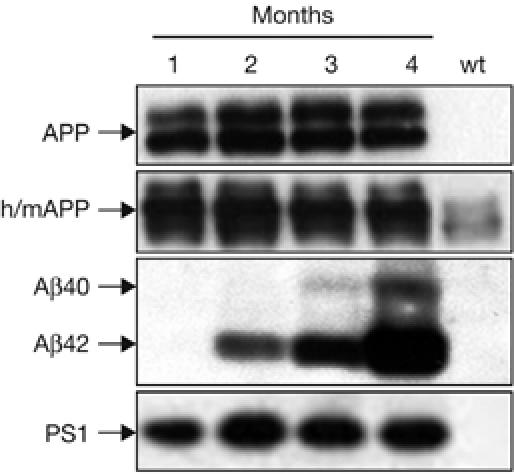
Amyloid precursor protein and amyloid β in APPPS1-21 mice. Human APP expression was constant between 2 and 4 months of age (antibody 6E10). Antibody 22C11 that recognizes both human and mouse (h/m) APP showed an approximate threefold overexpresssion of hAPP over endogenous mAPP. Human Aβ40 and Aβ42 (antibody 6E10) showed an exponential increase from 1 to 4 months of age, with Aβ42 levels exceeding Aβ40 several-fold. PS1 expression remained constant from 2 to 4 months of age. Female mice are shown. Aβ, amyloid β; APP, amyloid precursor protein; PS, presenilin 1; wt, wild type.
Table 1.
Human Aβx-40 and Aβx-42 were determined by enzyme-linked immunosorbent assay in hemibrains lacking cerebellum of 1- to 8-month-old female mice
| Age (months) | n | Aβ40 (pmol/g wet brain) | Aβ42 (pmol/g wet brain) | Ratio Aβ42 to Aβ40 |
|---|---|---|---|---|
| 1 |
8 |
0.9±0.1 |
1.5±0.4 |
1.6±0.2 |
| 2 |
6 |
35.1±4.9 |
169.9±26.9 |
5.1±0.7 |
| 4 |
8 |
633.0±152.5 |
2,456.9±380.3 |
4.4±0.4 |
| 8 | 5 | 3,277.4±263.5 | 14,863.8±659.3 | 4.6±0.4 |
| Values are the mean±s.e.m. | ||||
The first amyloid plaques in APPPS1-21 mice appeared in the neocortex at 6 weeks of age (Fig 3A). Because the Thy1 expression cassette is not fully active until postnatal day 14, amyloid deposition in APPPS1-21 mice is estimated to develop in a 4-week time period. These initial plaques were small, dense and nearly 100% congophilic (supplementary Fig S2 online). Amyloid deposits increased in size and number with ageing, and diffuse amyloid appeared at the plaque peripheries. At 8 months of age, three types of plaque were apparent: (i) small, congophilic dense core plaques as seen in younger mice, (ii) larger plaques with a dense core and a large halo of diffuse amyloid, and (iii) plaques with a dense core surrounded by a well-defined corona of diffuse amyloid, resembling the most abundant plaques in human AD (supplementary Fig S2 online).
Figure 3.

Age-related amyloid deposition in APPPS1-21 mice. (A) Aβ immunostaining in the frontal cortex of 1-, 2-, 4- and 8-month (mo)-old female transgenic mice. Note the early onset of mostly small and compact amyloid deposits at 2 months. (Another batch of mice was analysed at 6 weeks of age and amyloid deposits were already present at this age; results are not shown.) At 8 months of age, amyloid deposits cover virtually the entire neocortex, are larger in size and are surrounded by diffuse amyloid. (B) In the hippocampus, amyloid deposits develop later. Similar to the neocortex, initial deposits are small and compact. Scale bars, 200 μm. (C) Stereological analysis of amyloid load (%) showed a significant increase from 1 to 8 months of age for both neocortex (ctx) and hippocampus (hpx), P<0.001, with an earlier and greater amyloid deposition in the neocortex (five female mice/group). S.e.m. is shown. Aβ, amyloid β.
In the hippocampus, amyloid deposition occurred later and started in the dentate gyrus at 2–3 months of age and in CA1 at 4–5 months of age (Fig 3B). Amyloidosis in the striatum, thalamus, brain stem and other regions appeared between 3 and 5 months of age. At 8 months of age, virtually the entire forebrain was covered with amyloid. Consistent with our previous finding that a low ratio of Aβ42 to Aβ40 determines the amount of vascular amyloid (Herzig et al, 2004), no vascular amyloid was found in 2- to 4-month-old mice. In 8-month-old mice, vascular amyloid was rare and restricted to large pial vessels. At all ages, intraneuronal Aβ immunoreactivity was weak or absent, confirming previous observations that intraneuronal Aβ staining is not an indication for the onset and type of cerebral amyloidosis (Herzig et al, 2004).
The oldest mice available to this study were three female 19-month-old mice. These mice showed a neocortical and hippocampal amyloid load of 20.9±0.8% and 9.6±0.4% (±s.e.m.), respectively. Amyloid was observed in all brain regions including the cerebellum, which lacks detectable transgene expression (supplementary Fig S3 online).
Although this analysis was mainly restricted to female mice, pre-depositing and depositing 1- and 2-month-old male mice were also studied. Aβ40 and Aβ42 levels in male 1-month-old mice (n=6) were 1.7±0.1 and 1.9±0.2 pmol/g wet brain, respectively (±s.e.m.). In 2-month-old mice (n=8), the corresponding values were 34.8±5.9 and 191.0±31.1 pmol/g. Stereological assessment of amyloid showed no plaques in 1-month-old male mice and 0.92±0.11% and 0.05±0.02% amyloid load in 2-month-old male mice for neocortex and hippocampus, respectively (n=5/group). None of these results were statistically different from females (Table 1; Fig 3).
Amyloid-associated neuroinflammation and pathology
No signs of neuroinflammation were apparent in pre-depositing 1-month-old mice. However, when the first plaques appeared, distinct microgliosis was observed, together with clustering of hypertrophic Iba1-positive microglia around amyloid deposits (Fig 4A,B). Stereological analysis of microglia number in the neocortex of control mice showed about 1.4 × 106 microglia with a trend towards a decrease from 1 to 8 months of age. One-month-old APPPS1-21 mice were not different from non-transgenic control mice, whereas there was a significant threefold increase in microglia number in 8-month-old transgenic mice compared with control mice (Fig 4C). Astrocytosis that was assessed as an increase in glial fibrillary acidic protein-positive astrocytes occurred simultaneously to microgliosis (supplementary Fig S4 online).
Figure 4.
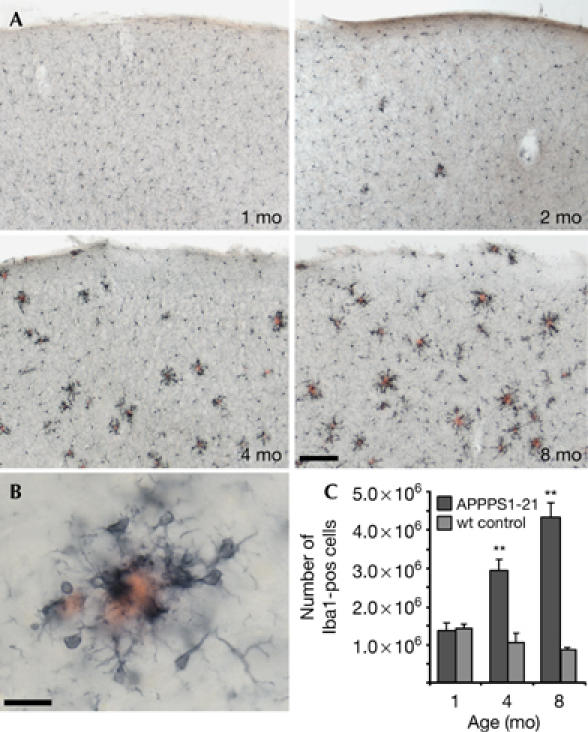
Amyloid-associated gliosis in APPPS1-21 transgenic mice. (A) Iba1-positive microglia and Congo red-stained amyloid in the neocortex of 1-, 2-, 4- and 8-month (mo)-old female transgenic mice. Activated and hypertrophic microglia appear concomitantly with the appearance of the first plaques and are tightly clustered around the amyloid. Scale bar, 100 μm. (B) High magnification reveals micoglia clustered around a plaque in an 8-month-old mouse. Scale bar, 20 μm. (C) Stereological quantification of total number of Iba1-positive cells in the neocortex of female APPPS1-21 mice (n=5/group) and non-transgenic control mice (n=3/group). Two-way analysis of variance showed significant effects of age (F2,18=7.6), transgene (F1,18=47.3) and age × transgene (F2,18=16.4; all P<0.01). Significant differences between transgenic and control were found at 4 and 8 months; **P<0.01. wt, wild type.
At 8 months of age, all congophilic amyloid plaques were surrounded by dystrophic synaptic boutons and hyperphosphorylated tau-positive neuritic structures (Fig 5A–C). No tangle formation was observed using the method of Gallyas. Although neuronal cell bodies were absent in the immediate vicinity of congophilic plaques, neurons in the neocortex appeared physically displaced rather than dying (Fig 5D). Global neuron loss in the neocortex was not apparent. By contrast, in regions with densely packed neuron layers, such as the dentate gyrus granule cell layer, neuron loss in the vicinity of amyloid plaques was more obvious (Fig 5E).
Figure 5.
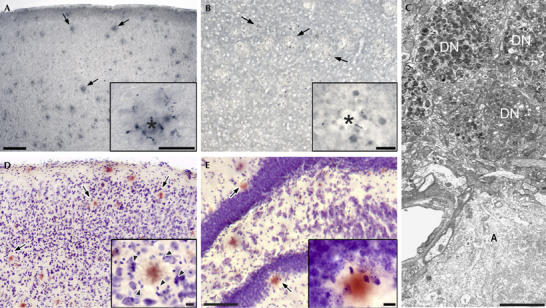
Amyloid-associated pathology in 8-month-old APPPS1-21 mice. (A) Immunostaining (AT8 antibody) shows hyperphosphorylated tau-positive neurites surrounding amyloid deposits (arrows) in the neocortex. The inset shows a plaque (asterisk) and abnormal neurites in higher magnification. (B) Synaptophysin-positive dystrophic terminals and boutons around amyloid plaques in layer III of the frontal cortex. (C) Electron micrograph of dystrophic neurites (DN) surrounding an amyloid plaque (A) in neocortical layer II. (D,E) Cresyl violet and congo red staining in the neocortex (D) reveals a ring of glial cells (arrowheads in the inset) surrounding the amyloid plaque. Neurons appear physically displaced but look normal in the periphery of the plaques. In the dentate gyrus (E), a thinning of the granule cell layer in the vicinity of amyloid plaques is apparent (arrows) although neurons with an unequivocally dying phenotype are rarely observed. Scale bars: 100 μm (A,B,D); 50 μm (E); 20 μm (insets); 5 μm (C).
Behavioural assessment
Open-field assessment of simple motor activity, exploratory behaviour and anxiety at 5 and 8 months of age did not indicate differences between transgenic and age-matched littermate control mice (results not shown). In the acquisition and reversal phase of a four-arm spatial maze task, no difference was observed at 5 months of age (Fig 6). However, when the same mice were tested again at 8 months of age, APPPS1-21 mice showed a significant impairment in reversal learning compared with the control mice (Fig 6).
Figure 6.

Cognitive deficits in APPPS1-21 mice. (A) At 5 months (mo) of age, no difference was observed in acquisition or reversal learning of a food-rewarded four-arm spatial maze task. (B) At 8 months of age, the same mice showed significant impaired reversal learning compared with their littermate wild-type control mice (two-way analysis of variance: F1,59=20.01, P=0.0001; post hoc Bonferroni tests: day 2, **P<0.01; day 4, *P<0.05; day 7, *P<0.05; n=12 for APPPS1-21, six females, six males; n=13 for control, six females, seven males). wt, wild type.
Discussion
Missense mutations in APP and PS1 lead to familial forms of AD by different mechanisms. Most PS1 mutations shift γ-secretase cleavage to increased Aβ42 production, which in turn accelerates cerebral amyloidosis in transgenic mice (Borchelt et al, 1997; Holcomb et al, 1998; Siman et al, 2000). An inverse correlation between the Aβ42 to Aβ40 ratio and the age of onset in familial AD has also been reported (Duering et al, 2005).
Mutations at position Leu 166 in PS1 lead to a severe course of AD pathology, with a very early onset in the third or fourth decade of life. So far, three mutations at position Leu 166 (L166A, L166P and L166H) have been described; the L166P mutation seems to be the most pathogenic (Ezquerra et al, 2000; Moehlmann et al, 2002; Pantieri et al, 2005). Expressing PS1-L166P in transfected cells resulted in the highest Aβ42 to Aβ40 ratio among several PS1 mutations (Moehlmann et al, 2002). However, in contrast to other PS1 mutations, the L166P mutation has been shown to markedly decrease Aβ40 production leaving Aβ42 levels unaffected (Bentahir et al, 2006). Interestingly, soluble Aβ42 levels in pre-depositing APPPS1-21 mice seem to be close to the levels reported in the most commonly used Tg2576 and APP23 mouse models, whereas Aβ40 levels in APPPS1-21 mice are four- to tenfold lower than that in the other models (supplementary Table 1 online). Hence, onset of amyloid deposition in APPPS1 mice occurs much earlier compared with the other transgenic models. This observation suggests that both increased Aβ42 levels and a reduction in Aβ40 can promote cerebral amyloidosis, a finding consistent with the emerging hypothesis that Aβ40 is a parenchymal ‘amyloid-inhibiting species' whereas Aβ42 is the ‘amyloid-promoting species' (McGowan et al, 2005; Kumar-Singh et al, 2006).
APPPS1-21 mice are a model of cerebral amyloidosis, and not directly of AD, as the mice do not model the tau pathology and robust neurodegeneration observed in AD. Neuron loss in the APPPS1-21 model seems to be similar to previous APP and APPPS1 transgenic mouse models, in which neuron death occurs only in close proximity to congophilic amyloid, giving rise to no significant global neuron loss in the neocortex and modest and mouse-model-specific neuronal loss in the hippocampal subregions (Bondolfi et al, 2002; Irizarry et al, 1997; Calhoun et al, 1998; Urbanc et al, 2002). Massive CA1 hippocampal neuronal loss due to high intracellular levels of Aβ42 has been reported in a recent APPPS1 knock-in transgenic mouse model (Casas et al, 2004), but it cannot be excluded that other factors could be responsible for the neuron loss observed in this model. Synaptic and neuritic pathology is clearly present in APPPS1-21 mice, similar to that observed in other congophilic amyloid-depositing mice (Masliah et al, 1996; Phinney et al, 1999). Because of the largely congophilic nature of the pathology, the microgliosis in APPPS1-21 mice might be stronger than in other amyloid-depositing mice, although rigorous stereological quantifications of microglia number has not been carried out previously (Frautschy et al, 1998; Matsuoka et al, 2001; Dudal et al, 2004; Stalder et al, 2005). The origin of the threefold increase in microglia number in APPPS1 mice has yet to be determined, but probably is a combination of both division of microglia and invasion of peripheral macrophages (Bondolfi et al, 2002; Stalder et al, 2005).
An impairment of behavioural flexibility and of switching from previously learned strategies to new strategies is a feature of AD (Rouleau et al, 1992). These cognitive functions and reversal learning have been shown to be dependent on the prefrontal cortex and hippocampal activity (Buzsaki et al, 1982). APPPS1-21 mice showed impairment in a reversal task at 8 months of age, whereas at an earlier time point, no deficit was apparent. Amyloid lesions in hippocampus develop with a delay of 1–3 months when compared with the neocortex: thus, the cognitive impairment observed in APPPS1-21 mice might coincide with the onset of significant amyloid in the hippocampus, although more detailed behavioural analysis is necessary to establish whether behavioural impairment precedes, coincides with or follows deposition of amyloid, as seen in other transgenic models of AD.
Several APP and APPPPS transgenic mice have been described in the literature. The present APPPS1-21 mouse combines several advantages of previous transgenic lines. (i) Mice have been generated on a pure C57BL/6J background that is known to reduce the variability of Aβ metabolism and deposition (Lehman et al, 2003). More recently, the mouse model has also been expanded into a BALB/cJ background. (ii) At least in young mice, no gender effects in Aβ level and amyloid deposition have been noted. (iii) Several APPPS1 lines have been generated that help to control for unwanted transgene insertion site effects. (iv) APPPS1-21 mice breed well, similar to wild-type C57BL/6J mice. (v) The APP and PS1 transgenes are coexpressed and the integration site has been located. (vi) The early onset of amyloid pathology allows a rapid readout and facilitates testing of therapeutic amyloid-targeting strategies. (vii) APPPS1-21 mice are a model of parenchymal amyloidosis and a welcome addition to the recently generated APPDutch mouse model that develops only vascular amyloid (Herzig et al, 2004). Because of these reasons, we anticipate that this new APPPS1 mouse model, now distributed to more than 30 laboratories worldwide, will be broadly valuable for addressing the various issues about the role of cerebral amyloidosis in AD and other proteopathies.
Methods
Generation of transgenic mice Thy1-APPKM670/671NL and Thy1-PS1L166P constructs were coinjected into male pronuclei of C57BL6J oocytes. Seven founders were identified, all of which were positive for both transgenes. The four male founders were further bred with C57BL/6J mice. APP and PS1 expression were assessed in three lines (lines 5, 8 and 21; Fig 1). See also supplementary information online.
Western blotting, morphological analysis and ELISA Western blotting, immunohistochemistry and ultrastructural analysis were carried out according to previously published protocols (Herzig et al, 2004). Aβ ELISA was performed using snap-frozen hemispheres. In mice, before amyloid deposition, Aβ was extracted by diethylamine, whereas formic acid was used to extract Aβ from amyloid-depositing mice. See also supplementary information online.
Stereology Amyloid load or microglia number was estimated on random sets of every twelfth systematically sampled 40-μm-thick NT12 or Iba1-immunostained sections throughout either neocortex or hippocampus within two-dimensional disectors (amyloid load) or using the optical fractionator technique with two-dimensional disectors (microglia), as described previously (Long et al, 1998; Bondolfi et al, 2002). See also supplementary information online.
Behavioural analysis An open-field test was used to assess motor activity, exploratory behaviour, anxiety and grooming episodes (Holscher et al, 2004). Spatial learning was assessed in a previously described radial four-arm maze task (Holscher et al, 2004). See also supplementary information online.
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
Supplementary Information
Acknowledgments
Breeding pairs of APPPS1 mice have been sent to academic and other non-profit organizations. We thank C. Schäfer, M. Herzig (Tübingen, Germany), H. Bluethmann (Basel, Switzerland), H. Steiner (Munich, Germany), C. Dorner-Ciossek (Biberach, Germany), M. Mazzella (Orangeburg, USA) and KOESLER (Rottenburg, Germany) for experimental help and advice. The generous antibody and complementary DNA donations from P. Paganetti, M. Staufenbiel, H. van der Putten (Basel, Switzerland) and K. Beyreuther (Heidelberg, Germany) are greatly appreciated. This work was supported by the German National Genome Network (NGFN2), European Union contract LSHM-CT-2003-503330 (APOPIS) in which the Swiss participants are funded by the Swiss State Secretariat for Education and Research and a fellowship to D.L. from the Boehringer Ingelheim Foundation. The authors have no competing financial interests.
References
- Bentahir M, Nyabi O, Verhamme J, Tolia A, Horre K, Wiltfang J, Esselmann H, De Strooper B (2006) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J Neurochem 96: 732–742 [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Calhoun M, Ermini F, Kuhn HG, Wiederhold KH, Walker L, Staufenbiel M, Jucker M (2002) Amyloid-associated neuron loss and gliogenesis in the neocortex of amyloid precursor protein transgenic mice. J Neurosci 22: 515–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS (1997) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: 939–945 [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Bors L, Nagy F, Eidelberg E (1982) Spatial mapping, working memory, and the fimbria–fornix system. J Comp Physiol Psychol 96: 26–34 [DOI] [PubMed] [Google Scholar]
- Calhoun ME, Wiederhold KH, Abramowski D, Phinney AL, Probst A, Sturchler-Pierrat C, Staufenbiel M, Sommer B, Jucker M (1998) Neuron loss in APP transgenic mice. Nature 395: 755–756 [DOI] [PubMed] [Google Scholar]
- Casas C et al. (2004) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am J Pathol 165: 1289–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F (2004) Inflammation occurs early during the Aβ deposition process in TgCRND8 mice. Neurobiol Aging 25: 861–871 [DOI] [PubMed] [Google Scholar]
- Duering M, Grimm MO, Grimm HS, Schroder J, Hartmann T (2005) Mean age of onset in familial Alzheimer's disease is determined by amyloid β 42. Neurobiol Aging 26: 785–788 [DOI] [PubMed] [Google Scholar]
- Ezquerra M, Carnero C, Blesa R, Oliva R (2000) A novel presenilin 1 mutation (Leu166Arg) associated with early-onset Alzheimer disease. Arch Neurol 57: 485–488 [DOI] [PubMed] [Google Scholar]
- Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM (1998) Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol 152: 307–317 [PMC free article] [PubMed] [Google Scholar]
- Herzig MC et al. (2004) Aβ is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci 7: 954–960 [DOI] [PubMed] [Google Scholar]
- Holcomb L et al. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97–100 [DOI] [PubMed] [Google Scholar]
- Holscher C, Schmid S, Pilz PK, Sansig G, van der Putten H, Plappert CF (2004) Lack of the metabotropic glutamate receptor subtype 7 selectively impairs short-term working memory but not long-term memory. Behav Brain Res 154: 473–481 [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99–102 [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997) APPSw transgenic mice develop age-related A β deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965–973 [DOI] [PubMed] [Google Scholar]
- Jucker M, Ingram DK (1997) Murine models of brain aging and age-related neurodegenerative diseases. Behav Brain Res 85: 1–26 [DOI] [PubMed] [Google Scholar]
- Kumar-Singh S et al. (2006) Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum Mutat 27: 686–695 [DOI] [PubMed] [Google Scholar]
- Lehman EJ et al. (2003) Genetic background regulates β-amyloid precursor protein processing and β-amyloid deposition in the mouse. Hum Mol Genet 12: 2949–2956 [DOI] [PubMed] [Google Scholar]
- Long JM, Kalehua AN, Muth NJ, Calhoun ME, Jucker M, Hengemihle JM, Ingram DK, Mouton PR (1998) Stereological analysis of astrocyte and microglia in aging mouse hippocampus. Neurobiol Aging 19: 497–503 [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y et al. (2001) Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer's disease. Am J Pathol 158: 1345–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan E et al. (2005) Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47: 191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moehlmann T et al. (2002) Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Aβ 42 production. Proc Natl Acad Sci USA 99: 8025–8030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantieri R, Pardini M, Cecconi M, Dagna-Bricarelli F, Vitali A, Piccini A, Russo R, Borghi R, Tabaton M (2005) A novel presenilin 1 L166H mutation in a pseudo-sporadic case of early-onset Alzheimer's disease. Neurol Sci 26: 349–350 [DOI] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M (1999) Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci 19: 8552–8559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouleau I, Salmon DP, Butters N, Kennedy C, McGuire K (1992) Quantitative and qualitative analyses of clock drawings in Alzheimer's and Huntington's disease. Brain Cogn 18: 70–87 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003) Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43: 545–584 [DOI] [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG (2000) Presenilin-1 P264L knock-in mutation: differential effects on Aβ production, amyloid deposition, and neuronal vulnerability. J Neurosci 20: 8717–8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalder AK et al. (2005) Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci 25: 11125–11132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C et al. (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287–13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbanc B, Cruz L, Le R, Sanders J, Ashe KH, Duff K, Stanley HE, Irizarry MC, Hyman BT (2002) Neurotoxic effects of thioflavin S-positive amyloid deposits in transgenic mice and Alzheimer's disease. Proc Natl Acad Sci USA 99: 13990–13995 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information