

Abstract
The general transcription factor TFIIB has a central role in the assembly of the preinitiation complex at the promoter, providing a platform for the entry of RNA polymerase II/TFIIF. We used an RNA interference (RNAi)-based system in which TFIIB expression is ablated in vivo and replaced with a TFIIB derivative that contains a silent mutation and is refractory to the RNAi. Using this approach, we found that transcriptionally defective TFIIB amino-terminal mutants showed distinct effects on the basis of their ability to compete with wild-type TFIIB in vivo. Moreover, analysis of the TFIIB mutant derivatives by chromatin immunoprecipitation showed that promoter occupancy by TFIIB is dependent on the association with RNA polymerase II. Together, our results support a mode of preinitiation complex assembly in which TFIIB/RNA polymerase II recruitment to the promoter occurs in vivo.
Keywords: transcription, RNA polymerase II, TFIIB, promoter
Introduction
The recruitment of RNA polymerase II (Pol II) to the promoter of a gene requires general transcription factors (GTFs; reviewed by Butler & Kadonaga, 2002; Hahn, 2004). TFIID binds to the TATA box (when present) within the promoter, which is then stabilized by transcription factor IIB (TFIIB). TFIIB can also make sequence-specific contact with the core promoter DNA (Lagrange et al, 1998; Qureshi & Jackson, 1998; Evans et al, 2001; Deng & Roberts, 2005). The assembly of TFIIB at the promoter is a prerequisite for the recruitment of Pol II, which enters the preinitiation complex (PIC) being formed along with TFIIF. TFIIE, TFIIH and other factors can then enter the complex. Several previous studies have suggested the existence of a preformed Pol II holoenzyme complex containing GTFs in addition to other components (reviewed by Lee & Young, 2000; Gill, 2001).
TFIIB has a crucial role in the PIC, showing absolute requirement at all promoters for the recruitment of Pol II. TFIIB contains a zinc ribbon at the amino terminus and a carboxy-terminal core domain composed of α-helices that form two direct repeats (Bagby et al, 1995; Nikolov et al, 1995; Zhu et al, 1996). In between the zinc ribbon and core domain lies the most conserved region of TFIIB, termed the B-finger (Chen & Hahn, 2003, 2004; Bushnell et al, 2004). Recently, intact TFIIB has been crystallized as a complex with Pol II (Bushnell et al, 2004). In this structure, the B-finger lies within the Pol II catalytic centre, which is consistent with previous observations that mutations in this domain can cause alterations in the start site of transcription (Pinto et al, 1994; Faitar et al, 2001; Fairley et al, 2002).
So far, most analyses of mammalian TFIIB function have used in vitro transcription systems. In this study, we have used an RNA interference (RNAi)-based system to replace endogenous TFIIB in living cells. We uncover distinct mechanistic effects of TFIIB mutations that demonstrate the requirement for TFIIB to associate with RNA Pol II to form a stable PIC in vivo.
Results and Discussion
Replacement of TFIIB in living cells using RNAi
We developed an RNAi-based approach to analyse TFIIB mutants in mammalian cells (see supplementary Fig S1 online). The vector pSUPER was used to drive expression of a short interfering RNA (siRNA) that targets the endogenous TFIIB transcript. Two silent point mutations were introduced into the human TFIIB complementary DNA (under the control of a cytomegalovirus (CMV) promoter) in the region targeted by the siRNA (smTFIIB). By simultaneous co-transfection of these plasmids, endogenous TFIIB expression can be ablated and replaced by a TFIIB derivative that is not targeted by the siRNA. In addition, the transfected smTFIIB encodes a T7 tag fused to the C terminus; this results in a slower migration in SDS–polyacrylamide gel electrophoresis (SDS–PAGE), which can easily be distinguished from endogenous TFIIB.
We generated plasmids driving the expression of smTFIIB that also harbour either the C34A or the R66E mutation. These TFIIB mutations, which perturb the zinc ribbon and B-finger, respectively, have previously been shown to have transcriptional defects (Buratowski & Zhou, 1993; Hawkes et al, 2000). pcDNA-smTFIIBT7, or derivatives containing either the C34A or the R66E mutation, were transfected as above with pSUPER RNAi and a reporter containing the AdML promoter linked to chloramphenicol acetyltransferase (CAT) and controlled by nine GAL4 DNA-binding sites. Where indicated, a plasmid driving the expression of GAL4(1–94)-VP16 was included. After the transfection procedure, CAT activity was measured (Fig 1A). Below the graph is an immunoblot of each transfection sample with anti-TFIIB antibodies. The results show that when the expression of endogenous TFIIB is ablated by RNAi, neither TFIIB R66E nor TFIIB C34A is able to restore transcription to a level observed with wild-type smTFIIBT7. The immunoblot with anti-TFIIB antibodies demonstrates the replacement of endogenous TFIIB with the smTFIIB derivatives. We also performed the same experiment, but using empty pSUPER vector (Fig 1B). As reported previously, TFIIB R66E is a potent inhibitor of transcription in living cells in the presence of endogenous TFIIB (Hawkes et al, 2000). By contrast, TFIIB C34A does not inhibit transcription under the same conditions. Together, the results in Fig 1 show distinct mechanistic defects caused by the C34A and R66E mutations in TFIIB. Although an undefined amount of endogenous TFIIB will remain in cells treated with RNAi, our results indicate that TFIIB has become sufficiently limiting to discriminate between the effects of the co-transfected mutant TFIIB derivatives. Thus, TFIIB R66E fails to support transcription when it is the sole source of TFIIB and also inhibits transcription in the presence of endogenous TFIIB. By contrast, TFIIB C34A fails to support transcription when it is the only source of TFIIB, but does not inhibit transcription in the presence of endogenous TFIIB.
Figure 1.
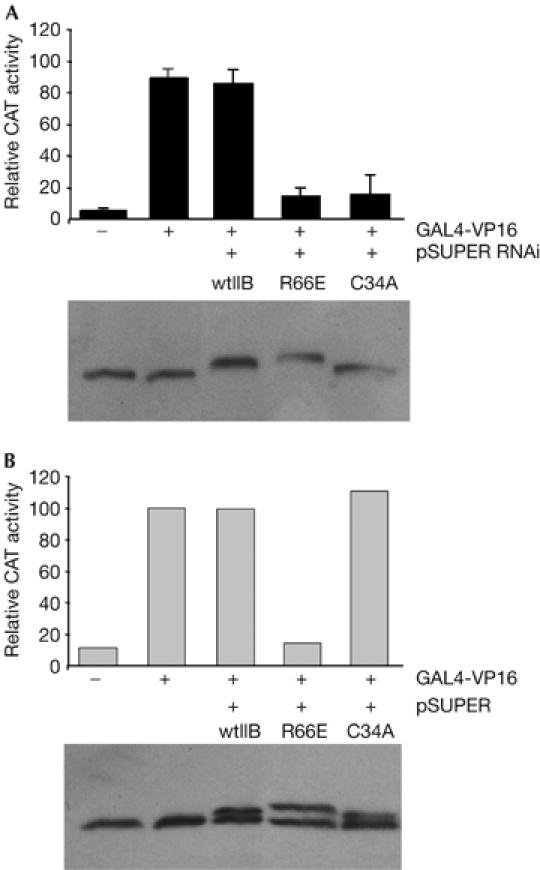
Analysis of TFIIB function in vivo by replacement of endogenous TFIIB. (A) Cells were transfected with pSUPER RNAi and the indicated smTFIIB derivatives. CAT activity was measured and is presented graphically: each bar is the mean of three independent experiments with standard deviation. Below is an immunoblot of the transfected cells performed with anti-TFIIB antibodies. (B) As in (A), except that empty pSUPER vector was used instead of pSUPER RNAi. CAT, chloramphenicol acetyltransferase; RNAi, RNA interference; TFIIB, general transcription factor IIB; wtIIB, wild-type TFIIB.
Distinct effects of TFIIB N-terminal mutations
We considered next whether the differential abilities of TFIIB C34A and TFIIB R66E to compete with endogenous TFIIB were promoter specific. Transfection experiments were performed in which wild-type TFIIB, TFIIB C34A or TFIIB R66E were ectopically expressed along with GAL4-VP16 and either the AdE4 promoter or the human immunodeficiency virus long terminal repeat (HIV LTR). In each case transcripts were detected by primer extension (Fig 2A). The results show that at both the AdE4 and HIV LTR promoters, TFIIB R66E is a potent inhibitor of transcription, whereas TFIIB C34A has no effect. We also found that the HSV thymidine kinase and human IGF II core promoters show similar effects with the TFIIB derivatives (data not shown). Thus, the inert and inhibitory effects of TFIIB C34A and TFIIB R66E, respectively, do not seem to be promoter specific.
Figure 2.
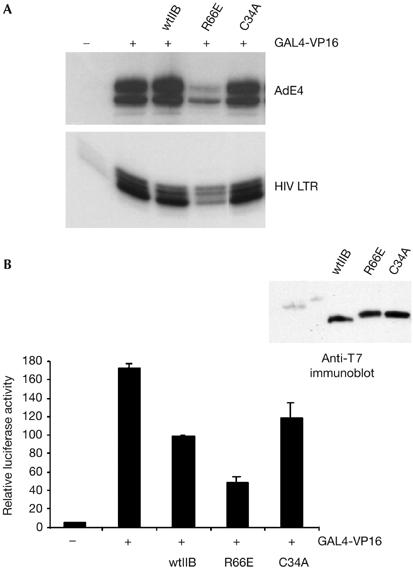
Distinct effect of transcriptionally defective TFIIB mutants in vivo. (A) Plasmids driving the expression of wild-type TFIIB (wtIIB) or the indicated mutants (2 μg) were introduced into embryonic kidney 293 cells with 2 μg of the reporter G9E4CAT (top) or HIV LTR CAT (bottom) and a plasmid driving the expression of GAL4(1–94)-VP16 (0.2 μg). After 48 h, RNA was prepared and CAT transcripts were detected by primer extension and autoradiography. (B) A 293-derivative cell line that contains G9ML-Luciferase stably integrated was transfected as in (A), but without the reporter. After 48 h, cells were lysed and luciferase activity was measured and is presented graphically. A blot (anti-T7) showing expression of the TFIIB derivatives is shown at top right. CAT, chloramphenicol acetyltransferase; HIV LTR, human immunodeficiency virus long terminal repeat; TFIIB, general transcription factor IIB.
We determined next whether the different effects that we observed with TFIIB R66E and TFIIB C34A could be recapitulated when we monitored expression of a stably integrated promoter rather than an episomal transfected reporter. A 293 cell line containing an integrated G9AdML-Luciferase cassette was transfected with a plasmid driving the expression of GAL4(1–94)-VP16 along with wild-type TFIIB, TFIIB R66E or TFIIB C34A. After 48 h, luciferase activity was measured (Fig 2B). We note that expression of wild-type TFIIB caused a reduction in the activity of the endogenous reporter. However, as we observed with the episomal reporter, overexpression of TFIIB R66E was an inhibitor of transcription but overexpression of TFIIB C34A was without significant effect when compared with wild-type TFIIB. Thus, the distinct effects of TFIIB C34A and TFIIB R66E are generally observed at different promoters in either an episomal or an integrated context.
Mechanism of inhibition by TFIIB B-finger mutation
We sought next to determine the mechanistic basis for the effects that we observed with the TFIIB derivatives C34A and R66E. We first assessed, by band shift, their ability to assemble with TATA-binding protein (TBP) at the promoter and also to recruit Pol II/TFIIF (Fig 3A). Both TFIIB C34A and TFIIB R66E were able to form a TFIIB–TBP–AdML promoter complex that was indistinguishable from that observed with wild-type TFIIB. However, TFIIB C34A showed a clear defect in the formation of a complex containing Pol II/TFIIF, which is consistent with previous reports using other zinc ribbon mutations in TFIIB (Buratowski & Zhou, 1993; Tubon et al, 2004). In addition, as we have reported previously, TFIIB R66E does not show a defect in the recruitment of Pol II/TFIIF to the promoter (Glossop et al, 2004). Thus, both TFIIB C34A and TFIIB R66E can form a complex with TBP at the promoter. However, TFIIB R66E, but not TFIIB C34A, is able to extend PIC formation to incorporate Pol II and TFIIF.
Figure 3.
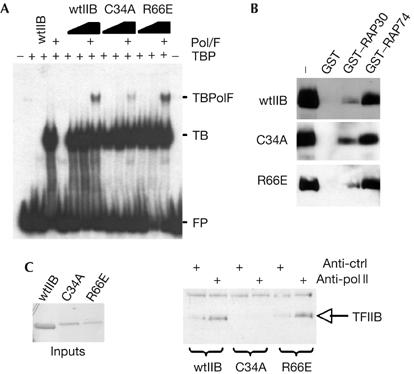
Analysis of preinitiation complex formation by TFIIB derivatives. (A) Electrophoretic mobility shift assay to analyse the formation of a Pol II/TFIIF–TFIIB–TBP–AdML promoter complex. Where indicated, the reactions contained 4 ng TBP, 50 ng Pol II, 10 ng TFIIF and 25 or 50 ng TFIIB (or derivatives). Complexes were resolved by non-denaturing electrophoresis and detected by autoradiography. FP is free probe, TB is TFIIB–TBP promoter complex and TBpolF is Pol II–TFIIF–TFIIB–TBP promoter complex. (B) GST, GST–RAP30 or GST–RAP74 (each at 1 μg) was used as an affinity matrix in binding assays with recombinant wild-type TFIIB (wtIIB; 500 ng) or the indicated TFIIB derivatives. Stably bound proteins were resolved by SDS–PAGE, immunoblotted with anti-TFIIB antibodies and detected by enhanced chemiluminescence. (C) Highly purified Pol II (50 ng) was incubated with wild-type TFIIB (wtIIB) or the indicated TFIIB mutant derivative (100 ng of each protein; input shown as a Coomassie-stained gel at left). Immunoprecipitation was performed with either anti-Pol II or anti-promyelocytic leukaemia protein (anti-ctrl) antibodies and the products were resolved by SDS–PAGE, immunoblotted with anti-T7 antibodies and then detected by enhanced chemiluminescence. GST, glutathione S-transferase; Pol II, polymerase II; SDS–PAGE, SDS–polyacrylamide gel electrophoresis; TBP, TATA-binding protein; TFIIB, general transcription factor IIB.
TFIIB can interact with Pol II and also with both the TFIIF subunits (RAP30 and RAP74; Ha et al, 1993; Fang & Burton, 1996). Therefore, we were interested to determine which of these interactions was perturbed in TFIIB C34A. We performed binding assays using glutathione S-transferase (GST), GST–RAP30 or GST–RAP74 as an affinity matrix. Wild-type TFIIB, TFIIB C34A and TFIIB R66E showed similar binding profiles to each of the TFIIF subunits (Fig 3B). Immunoprecipitation was performed to monitor the ability of wild-type TFIIB, TFIIB C34A and TFIIB R66E to interact with Pol II. Consistent with our previous studies, both wild-type TFIIB and TFIIB R66E co-immunoprecipitated with Pol II (Hawkes et al, 2000). By contrast, TFIIB C34A did not co-immunoprecipitate with Pol II. Thus, TFIIB C34A failed to recruit Pol II/TFIIF to the promoter owing to a defect in interaction with Pol II.
Our interaction data suggest that both TFIIB C34A and TFIIB R66E can form a complex with TBP at the promoter and that TFIIB R66E, but not TFIIB C34A, can extend PIC assembly further to incorporate Pol II/TFIIF. Thus, the inhibitory effect of TFIIB R66E may be due to its ability to interact with Pol II. To test this possibility, we generated a TFIIB derivative that contained both C34A and R66E mutations. Plasmids driving the expression of wild-type TFIIB, TFIIB R66E, TFIIB C34A or the compound mutant TFIIB C34A:R66E were transfected in 293 cells with the G9MLCAT reporter and GAL4(1–94)-VP16 expression plasmid. CAT transcripts were analysed by primer extension and are presented in Fig 4. The results show that TFIIB R66E inhibits transcription as before, but the compound mutant TFIIB C34A:R66E does not inhibit transcription. Thus, the C34A mutation renders TFIIB R66E incapable of competing with endogenous TFIIB to inhibit transcription, which suggests that inhibition of transcription requires an intact TFIIB–Pol II interaction. Analysis of TFIIB zinc ribbon mutant G30A further demonstrated that a TFIIB derivative that is defective in Pol II recruitment cannot compete with wild-type TFIIB to inhibit transcription (see supplementary Fig S2 online).
Figure 4.
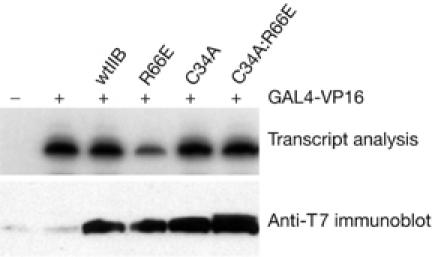
Analysis of the mode of inhibition by TFIIB R66E in vivo. Plasmids driving the expression of wild-type TFIIB (wtIIB) or the indicated mutants (2 μg) were introduced into embryonic kidney 293 cells with 2 μg of the reporter G9MLCAT and a plasmid driving the expression of GAL4(1–94)-VP16 (0.2 μg). After 48 h, RNA was prepared and CAT transcripts were detected by primer extension. An autoradiograph detecting the transcripts is shown. CAT, chloramphenicol acetyltransferase; TFIIB, general transcription factor IIB.
To examine further the basis for the differential effects of the TFIIB derivatives C34A and R66E, we tested their ability to assemble at a promoter in living cells. 293 cells were transfected with a plasmid driving the expression of wild-type TFIIB, TFIIB C34A or TFIIB R66E. After 48 h, chromatin immunoprecipitation was performed with T7 antibodies to specifically concentrate chromatin associated with each TFIIB derivative. Real-time PCR was then performed to monitor precipitation of the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) promoter (Fig 5A). Both wild-type TFIIB and the TFIIB mutant derivative R66E assembled at the GAPDH promoter; by contrast, TFIIB C34A did not. We observed similar results when we examined assembly of these same TFIIB derivatives at the β-tubulin promoter (supplementary Fig S3 online). These data suggest that TFIIB R66E inhibits transcription by virtue of its ability to compete with wild-type TFIIB to assemble at the promoter. Furthermore, the results show a direct correlation between TFIIB–Pol II contact and not TFIIB–TBP contact in the recruitment of TFIIB to a promoter in vivo. It is not clear why TFIIB R66E should fail to form a functional PIC, but it is likely that TFIIB R66E is specifically defective in a crucial step in initiation. Interestingly, previous structural analysis locates TFIIB residue R66 close to the active site of Pol II in a PIC (Bushnell et al, 2004). Thus, a direct effect on Pol II may underlie the defect of TFIIB R66E.
Figure 5.
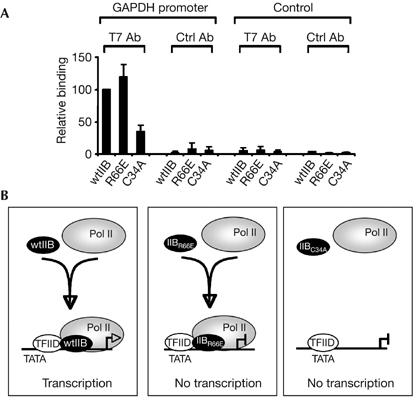
Assembly of TFIIB amino-terminal mutants at the promoter in vivo. (A) 293 cells were transfected with 2 μg of a plasmid driving the expression of wild-type TFIIB or the indicated derivative. After 24 h, chromatin immunoprecipitation was performed with anti-T7 antibodies to specifically isolate chromatin associated with the transfected TFIIB derivatives. Real-time quantitative PCR was performed with primers to amplify the GAPDH promoter region (left) or a control non-promoter region (right). Results shown are the average of three independent experiments, each normalized to input. (B) A model of TFIIB–Pol II association with the promoter is shown (discussed in the text). Assembly of wild-type TFIIB (wtIIB) and the TFIIB mutants (IIBR66E and IIBC34A) is depicted. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; Pol II, polymerase II; TFIIB, general transcription factor IIB.
Together, our results indicate that TFIIB cannot assemble at the promoter simply by virtue of its ability to interact with TBP and promoter DNA. Instead, the assembly of TFIIB at the promoter is determined by its interaction with Pol II (see model in Fig 5B). Studies in yeast also suggest that the inhibitory effect of some TFIIB mutants correlates with an intact TFIIB N terminus and also the core domain that associates with TBP (Gonzalez-Couto et al, 1997). Our present data agree with this idea and further suggest that stable assembly with TFIID at a promoter in vivo requires the association of TFIIB with Pol II. Previous studies in yeast demonstrated that recruitment of TBP to the promoter requires intact components of the holoenzyme (Kuras & Struhl, 1999; Li et al, 1999). Similarly, yeast TFIIB does not assemble at the promoter if Pol II or mediator is defective (Ranish et al, 1999). Thus, the interdependence of the GTFs in recruitment to the promoter probably occurs in both yeast and mammalian cells. Such a mode of assembly is consistent with the existence of a Pol II holoenzyme or transcription factories in living cells (Lee & Young, 2000; Pombo et al, 2000). A sequential recruitment mechanism is equally plausible (see for example Hatzis & Talianidis, 2002), but this would require that stable complexes form only at specific intermediates in the assembly process, and specifically a dependence on Pol II for the stable association of TFIIB.
Methods
Plasmids and proteins. G9ML, pET TFIIB, pET6His-TBP, pcDNA3TFIIB and pCMV Gal4(1–94)-VP16 have been described previously (Ha et al, 1993; Hawkes & Roberts, 1999). The TFIIB derivatives were produced by site-directed mutagenesis using the Stratagene Quikchange kit (Stratagene, La Jolla, CA, USA). Recombinant wild-type TFIIB, TFIIB derivatives and His-tagged TBP were purified as described previously (Reece et al, 1993; Hawkes & Roberts, 1999). Recombinant Rap30, Rap74 and purified Pol II were obtained from Protein One Inc (Bethesda, MD, USA).
Cell culture. Human embryonic kidney 293 (HEK 293) cells were maintained and transfected as described previously (Hawkes & Roberts, 1999). For production of stable cell lines, AdMLluc and pcDNA3 were transfected into 293 cells followed by selection with 1 mg/ml G418. After 7 days, stably transfected clones were selected, expanded and tested for reporter construct activity. For ablation of expression of endogenous TFIIB and replacement by smTFIIB, in conjunction with a reporter assay, HEK 293 cells were transfected twice in the following way: initially, cells were transfected with 2 μg of pSUPER-RNAi, 2 μg of each smTFIIB derivative and 0.2 μg of GAL4-VP16. After 48 h, cells were transfected with 2 μg of pSUPER-RNAi, 2 μg of each smTFIIB derivative and 2 μg of G9MLCAT. Cells were then collected at 96 h in PBS. CAT assays were performed and quantified by phosphorimager analysis. Total RNA preparation and detection of CAT RNA were as described (Hawkes & Roberts, 1999). Luciferase activity was measured with a kit (Promega, Madison, WI, USA).
Complex assembly and protein interaction assays. Band-shift assays were performed as described previously (Maldonado et al, 1990). GST–Rap30 and GST–Rap74 preparation, binding assays and immunoprecipitation were performed as described previously (Hawkes & Roberts, 1999). Immunoprecipitation was performed with anti-RNA Pol II antibody (Santa Cruz, CA, USA) or anti-promyelocytic leukaemia (PML) antibody (Santa Cruz), and the incubation was continued overnight. A 50 μl portion of a 50% slurry of Protein A agarose was then added to the samples and the samples were incubated at 4°C with rocking for 2 h. The beads were then washed four times in immunoprecipitation buffer and bound proteins eluted with SDS–PAGE loading dye.
Chromatin immunoprecipitation. 293 cells were treated with 1% formaldehyde for 10 min at 20°C before quenching with 0.125 M glycine for 5 min. Cells were collected in ice-cold PBS with complete protease inhibitors (Roche, Basel, Switzerland) and washed sequentially with Buffer I (10 mM HEPES pH 6.5, 0.5 mM EGTA, 10 mM EDTA, 0.25% Triton X-100) and Buffer II (10 mM HEPES pH 6.5, 0.5 mM EGTA, 1 mM EDTA, 200 mM NaCl) and then resuspended in SDS lysis buffer (50 mM Tris pH 8.1, 10 mM EDTA, 1% SDS). Lysates were sonicated on ice to yield 200–800 bp DNA fragments. One half of a 6 cm dish was used per immunoprecipitation, diluted 1/10 in immunoprecipitation dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl pH 8.1, 167 mM NaCl) and incubated overnight at 4°C with either 1 μg of T7 antibody (Novagen, Charlottesville, VA, USA) or 1 μg of nonspecific IgG (Upstate, Darmstadt, Germany). Immunocomplexes were precipitated by incubation for 30 min with protein G-conjugated magnetic beads (Dynal, Carlsbad, CA, USA) that had been pre-blocked by incubation with 10 μg of salmon sperm DNA. Immunoprecipitates were washed sequentially with TSEI (20 mM Tris pH 8.1, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% SDS), TSEII (20 mM Tris pH 8.1, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% SDS), Buffer III (10 mM Tris pH 8.1, 0.25 M LiCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate) and Tris–EDTA before eluting in 1% SDS/0.1 M NaHCO3. Crosslinks were reversed by heating to 65°C overnight and then treating with proteinase K for 1 h at 45°C. Chromatin was cleaned using QiaQuick PCR clean-up columns (Qiagen, Hilden, Germany). Quantitative PCR was performed with Quantitect SYBR green reagent (Qiagen) using primers specific to human GAPDH promoter (forward, AAAAGCGGGGAGAAAGTAGG; reverse, GCTGCGGGCTCAATT TATAG) or a non-promoter region in intron 3 of the SRF gene (forward, GCCACAGGGCAGTAGATGTT; reverse, TCAGGCC CAAGTATCCACTC).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Fig S1
Acknowledgments
We thank R. Reece, P. Shore and members of the lab for comments on the manuscript. This work was funded by the Wellcome Trust. L.M.E. is a Wellcome Prize student. L.M.G. is funded by the BBSRC. S.G.E.R. is a Wellcome Trust Senior Research Fellow.
References
- Bagby S, Kim S, Maldonado E, Tong KI, Reinberg D, Ikura M (1995) Solution structure of the C-terminal core domain of human TFIIB: similarity to cyclin A and interaction with TATA-binding protein. Cell 82: 857–867 [DOI] [PubMed] [Google Scholar]
- Buratowski S, Zhou H (1993) Functional domains of transcription factor TFIIB. Proc Natl Acad Sci USA 90: 5633–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell DA, Westover KD, Davis RE, Kornberg RD (2004) Structural basis of transcription: an RNA Polymerase II–TFIIB cocrystal at 4.5 Angstroms. Science 303: 983–988 [DOI] [PubMed] [Google Scholar]
- Butler JE, Kadonaga JT (2002) The RNA polymerase II core promoter: a key component in the regulation of gene expression. Genes Dev 16: 2583–2592 [DOI] [PubMed] [Google Scholar]
- Chen H-T, Hahn S (2003) Binding of TFIIB to RNA polymerase II: mapping the binding site for the TFIIB zinc ribbon domain within the preinitiation complex. Mol Cell 12: 437–447 [DOI] [PubMed] [Google Scholar]
- Chen H-T, Hahn S (2004) Mapping the location of TFIIB within the RNA polymerase II transcription preinitiation complex: a model for the structure of the PIC. Cell 119: 169–180 [DOI] [PubMed] [Google Scholar]
- Deng W, Roberts SGE (2005) A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev 19: 2418–2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans R, Fairley JA, Roberts SGE (2001) Activator-mediated disruption of sequence-specific DNA contacts by the general transcription factor TFIIB. Genes Dev 15: 2945–294911711430 [Google Scholar]
- Fairley JA, Evans R, Hawkes NA, Roberts SGE (2002) Core promoter-dependent TFIIB conformation and a role for TFIIB conformation in transcription start site selection. Mol Cell Biol 22: 6697–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faitar SL, Brodie SA, Ponticelli AS (2001) Promoter-specific shifts in transcription initiation conferred by yeast TFIIB mutations are determined by the sequence in the immediate vicinity of the start sites. Mol Cell Biol 21: 4427–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang SM, Burton ZF (1996) RNA polymerase II-associated protein (RAP) 74 binds transcription factor (TF) IIB and blocks TFIIB-RAP30 binding. J Biol Chem 271: 11703–11709 [DOI] [PubMed] [Google Scholar]
- Gill G (2001) Regulation of the initiation of eukaryotic transcription. Essays Biochem 37: 33–43 [DOI] [PubMed] [Google Scholar]
- Glossop JA, Dafforn TR, Roberts SGE (2004) A conformational change in TFIIB is required for activator-mediated assembly of the preinitiation complex. Nucleic Acids Res 32: 1829–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Couto E, Klages N, Strubin M (1997) Synergistic and promoter-selective activation of transcription by recruitment of transcription factors TFIID and TFIIB. Proc Natl Acad Sci USA 94: 8036–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha I, Roberts S, Maldonado E, Sun X, Kim L-U, Green MR, Reinberg D (1993) Multiple functional domains of general transcription factor IIB: distinct interactions with two general transcription factors and RNA polymerase II. Genes Dev 7: 1021–1032 [DOI] [PubMed] [Google Scholar]
- Hahn S (2004) Structure and mechanism of the RNA polymerase II transcription machinery. Nat Stuct Mol Biol 11: 394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzis P, Talianidis I (2002) Dynamics of enhancer–promoter communication during differentiation-induced gene activation. Mol Cell 10: 1467–1477 [DOI] [PubMed] [Google Scholar]
- Hawkes NA, Roberts SGE (1999) The role of human TFIIB in transcription start site selection in vitro and in vivo. J Biol Chem 274: 14337–14343 [DOI] [PubMed] [Google Scholar]
- Hawkes NA, Evans R, Roberts SGE (2000) The conformation of TFIIB modulates the response to transcriptional activators in vivo. Curr Biol 10: 273–276 [DOI] [PubMed] [Google Scholar]
- Kuras L, Struhl K (1999) Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399: 609–613 [DOI] [PubMed] [Google Scholar]
- Lagrange T, Kapanidis AN, Tang H, Reinberg D, Ebright RH (1998) New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev 12: 34–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Young RA (2000) Transcription of eukaryotic protein-coding genes. Annu Rev Genet 34: 77–137 [DOI] [PubMed] [Google Scholar]
- Li XY, Virbasius A, Zhu X, Green MR (1999) Enhancement of TBP binding by activators and general transcription factors. Nature 399: 605–609 [DOI] [PubMed] [Google Scholar]
- Maldonado E, Ha I, Cortes P, Weis L, Reinberg D (1990) Role of transcription factors IIA, IID and IIB during formation of a transcription competent complex. Mol Cell Biol 10: 6335–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov D, Chen BH, Halay ED, Usheva AA, Hisatake K, Lee DK, Roeder RG, Burley SK (1995) Crystal structure of a TFIIB–TBP–TATA-element ternary complex. Nature 377: 119–128 [DOI] [PubMed] [Google Scholar]
- Pinto I, Wu W-H, Na JG, Hampsey M (1994) Characterization of sua7 mutations defines a domain of TFIIB involved in transcription start site selection in yeast. J Biol Chem 269: 30569–30573 [PubMed] [Google Scholar]
- Pombo A, Jones E, Iborra FJ, Kimura H, Sugaya K, Cook PR, Jackson DA (2000) Specialized transcription factories within mammalian nuclei. Crit Rev Eukaryot Gene Expr 10: 21–29 [PubMed] [Google Scholar]
- Qureshi SA, Jackson SP (1998) Sequence-specific DNA binding by the S. shibatae TFIIB homolog, TFB, and its effect on promoter strength. Mol Cell 1: 389–400 [DOI] [PubMed] [Google Scholar]
- Ranish JA, Yudkovsky N, Hahn S (1999) Intermediates in formation and activity of the RNA polymerase II preinitiation complex: holoenzyme recruitment and a postrecruitment role for the TATA box and TFIIB. Genes Dev 13: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece RJ, Rickles RJ, Ptashne M (1993) Overproduction and single-step purification of GAL4 fusion proteins from Escherichia coli. Gene 126: 105–107 [DOI] [PubMed] [Google Scholar]
- Tubon TC, Tansey WP, Herr W (2004) A nonconserved surface of the TFIIB zinc ribbon domain plays a direct role in RNA polymerase II recruitment. Mol Cell Biol 24: 2863–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Zeng Q, Colangelo CM, Lewis M, Summers MF, Scott RA (1996) The N-terminal domain of TFIIB from Pyrococcus furiosus forms a zinc ribbon. Nat Struct Biol 3: 122–124 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Fig S1