

Abstract
Ubiquitination of proliferating-cell nuclear antigen (PCNA) at K164 by RAD6/RAD18 has a key role in DNA damage tolerance in yeast. Here, we report on the first genetic study of this modification in a vertebrate cell. As in yeast, mutation of K164 of PCNA to arginine in the avian cell line DT40 results in sensitivity to DNA damage but, by contrast, the DT40 pcnaK164R mutant is more sensitive than the rad18 mutant. Consistent with this, we show the presence of residual ubiquitination of PCNA at K164 in the absence of functional RAD18, suggesting the presence of an alternate PCNA ubiquitinating enzyme in DT40. Furthermore, RAD18 and PCNA K164 have non-overlapping roles in the suppression of sister chromatid exchange in DT40, showing that RAD18 has other functions that do not involve the ubiquitination of PCNA.
Keywords: DNA damage tolerance, PCNA, RAD18, translesion synthesis, ubiquitination
Introduction
The mechanisms that allow cells to bypass damage during DNA replication involve the finely choreographed recruitment of a large number of proteins to the replication fork. DNA damage bypass can be effected either through replacement of the stalled replicative polymerases by specialized translesion polymerases or by a relatively poorly understood recombinational mode of bypass in which DNA synthesis switches to use an alternative undamaged template. At the heart of replication lies the DNA sliding clamp proliferating-cell nuclear antigen (PCNA). Recent work in the budding yeast Saccharomyces cerevisiae has shown a network of post-translational modifications of PCNA by ubiquitin and small ubiquitin-related modifier (SUMO) that orchestrate the main pathways of replicative DNA damage bypass (Hoege et al, 2002; Stelter & Ulrich, 2003; Haracska et al, 2004; Papouli et al, 2005; Pfander et al, 2005).
In budding yeast, SUMOylation of PCNA at K164 and K127 is seen constitutively during replication (Hoege et al, 2002). SUMO-modified PCNA recruits the helicase SRS2, which prevents unwanted recombination during replication (Papouli et al, 2005; Pfander et al, 2005). In response to DNA damage, PCNA can also be monoubiquitinated at K164 by RAD6/RAD18 (Hoege et al, 2002). This single ubiquitin promotes translesion synthesis (TLS) by interacting with the recently identified ubiquitin-binding motifs in the TLS polymerases (Bienko et al, 2005). RAD5/UBC13/MMS2 can extend the ubiquitin at K164 with a non-canonical K63-linked polyubiquitin chain (Hoege et al, 2002). The precise function of this modification is unclear, but it is genetically linked to an error-free mode of bypass, thought to be a form of template switch (Higgins et al, 1976).
We and others have previously reported that the role of RAD18 in vertebrates might not be as central to the control DNA damage bypass as it is in yeast (Okada et al, 2002; Ross et al, 2005; Simpson & Sale, 2005). To address this issue further, we created a genetic system, using the DT40 chicken cell line, that allowed the study of mutations in PCNA and which we used to examine the relationship between RAD18 and PCNA ubiquitination.
Results And Discussion
To create a general system for studying mutations in PCNA, we adopted a strategy in which we deleted the endogenous PCNA loci in DT40 cells with rescue by the expression of human wild-type or mutant PCNA (see supplementary Fig S1 online). We created lines carrying human PCNA (hPCNA) and hPCNAK164R, in which the conserved acceptor lysine for ubiquitination is disrupted. We also created a line carrying hPCNAK138R. K138 is the closest positioned lysine in PCNA to the auxilliary SUMOylation site in budding yeast POL30 at K127 (supplementary Fig S2 online), although unlike K127 of POL30, K138 of vertebrate PCNA is not within a canonical UBC9 acceptor sequence (ψ-K-x-D/E, where ψ is a hydrophobic residue, K is the acceptor lysine, x is any amino acid and D/E are acidic residues).
All the mutants grew with the same kinetics as wild-type DT40 (Fig 1A) and, consistent with this, mutation of K164 or K138 did not affect the recruitment of PCNA to replication foci (Fig 1B). However, the pcna:hPCNA line showed a slight increase in sensitivity to DNA damage compared with wild-type DT40 (Fig 1C), which might reflect that the hPCNA transgene is not controlled by the natural PCNA promoter.
Figure 1.

Proliferation of hPCNA DT40 and hPCNA mutants. (A) Growth of pcna:hPCNA cells and mutants compared with wild-type (WT) DT40. (B) Normal recruitment of hPCNA, and the K164R and K138R mutant PCNAs to replication foci. In each case, more than 95% of PCNA foci colocalized with BrdU foci. (C) DNA damage sensitivity of WT DT40 (black) compared with pcna:hPCNA DT40 (red). BrdU, 5-bromodeoxyuridine; hPCNA, human PCNA; MMS, methylmethane sulphonate; PCNA, proliferating-cell nuclear antigen.
To assess the overall contribution of PCNA K164 and K138 to DNA damage tolerance, we examined the sensitivity of the pcna:hPCNA cells (which we abbreviate to hPCNA) and hPCNA mutant lines to genotoxic stress. All clones were matched for expression of hPCNA (supplementary Fig S1C online). Compared with the wild-type hPCNA control, hpcnaK164R cells showed marked sensitivity to ultraviolet light, methyl methanesulphonate (MMS) and cisplatin (CDDP; Fig 2A), but not to X-rays (data not shown). In parallel, we examined the sensitivity of rad18 mutants created on the background of the hPCNA lines. In marked contrast to S. cerevisiae, the DT40 hPCNA/rad18 mutant was less sensitive than the pcnaK164R mutant. However, a pcnaK164R/rad18 double mutant was not significantly more sensitive than the pcnaK164R single mutant. This suggests that the function of RAD18 in DNA damage tolerance is dependent on K164 of PCNA, but that K164 has a role independent of RAD18. The sensitivity of the pcnaK138R mutant to MMS (Fig 2B), ultraviolet light and CDDP (data not shown) was comparable with that of wild type and the sensitivity of a pcnaK164R/K138R double mutant was the same as that of pcnaK164R (data not shown).
Figure 2.
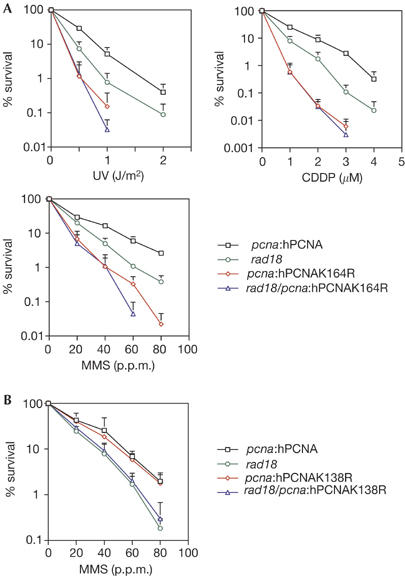
Sensitivity of hPCNA cells and mutants to DNA damage. (A) DNA damage sensitivity of pcnaK164R. (B) DNA damage sensitivity of pcnaK138R to MMS. CDDP, cisplatin; hPCNA, human proliferating-cell nuclear antigen; MMS, methylmethane sulphonate.
To examine this further, we monitored PCNA ubiquitination in DT40 by western blot of whole-cell lysates. In wild-type DT40, the ubiquitinated form of PCNA is seen constitutively and, surprisingly, is little induced by DNA damage with MMS (Fig 3A) or ultraviolet light (data not shown). In the DT40 rad18 mutant, the level of ubiquitinated PCNA is significantly reduced (Fig 3A).
Figure 3.
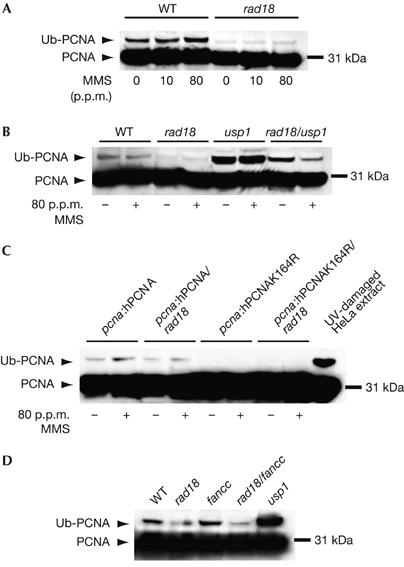
Ubiquitination of PCNA in DT40. (A) PCNA ubiquitination following DNA damage with MMS in wild-type (WT) and rad18 cells. Cells were incubated with the indicated concentration of MMS for 2 h before lysis. (B) Increased ubiquitin conjugation to PCNA following loss of USP1. Treatment with MMS (indicated by +) was carried out as in (A). (C) Ubiquitin conjugation to hPCNA in the pcna:hPCNA lines. As a comparison for size, ultraviolet light-damaged HeLa extract is shown in the right-hand lane. (D) The Fanconi anaemia core complex is not responsible for the RAD18-independent ubiquitination in DT40. These lysates are from undamaged cells. hPCNA, human proliferating-cell nuclear antigen; MMS, methylmethane sulphonate; Ub–PCNA, ubiquitin-conjugated PCNA.
As the ubiquitinated form of PCNA in DT40 seems to be present at rather lower stoichiometry than is seen in human cell lines, we took advantage of the recent identification of USP1 as the isopeptidase responsible for the removal of ubiquitin from PCNA (Huang et al, 2006) to confirm that the modification we observed was indeed ubiquitin and that PCNA was still ubiquitinated in the rad18 mutant. As expected, disruption of USP1 resulted in a significant increase in the steady-state level of the ubiquitinated form of PCNA (Fig 3B). In the rad18/usp1 mutant, the level of ubiquitinated PCNA was increased, but not to the levels seen in the usp1 single mutant. Interestingly, the level of ubiquitinated PCNA in rad18/usp1 cells was consistently reduced following DNA damage, an observation that we are investigating further.
Similar to endogenous chicken PCNA, ubiquitination of hPCNA was also seen in hPCNA DT40 and, at reduced levels, in the hPCNA/rad18 line. There was complete loss of ubiquitination in the hpcnaK164R mutant lines, confirming that K164 is the sole target for ubiquitination of PCNA (Fig 3C).
The presence of ubiquitinated PCNA in rad18 DT40 might partly explain why recruitment of translesion polymerases to the bypass of abasic sites created in the immunoglobulin loci of DT40 is, surprisingly, neither greatly decreased nor the pattern of mutation perturbed (Simpson & Sale, 2005). However, K164 of PCNA seems to be required, particularly for C/G transversions (A.L.R. & J.E.S., unpublished observations), similar to the DT40 rev1 mutant (Ross & Sale, 2006). PCNA K164 is also required for the recombination-independent mutagenic repair of DNA interstrand crosslinks (Shen et al, 2006). Thus, the DT40 rad18 and pcnaK164R mutants seem to be differentially defective in the recruitment of translesion polymerases to sites of damage bypass.
So far we have been unable to detect SUMO conjugation to PCNA in DT40. In S. cerevisiae, SUMOylated PCNA recruits the antirecombinogenic helicase SRS2 (Papouli et al, 2005; Pfander et al, 2005); however, this modification has not been detected in Schizosaccharomyces pombe. Furthermore, S. pombe SRS2 lacks the carboxy-terminal domain that is responsible for the interaction with SUMO (Frampton et al, 2006). There are also reports that SUMOylated PCNA cannot be detected in both human and Xenopus cell lines (Hoege et al, 2002; Leach & Michael, 2005; Frampton et al, 2006). Nevertheless, SUMOylation of PCNA has been clearly shown in replicating Xenopus egg extracts (Leach & Michael, 2005), but the modification was neither essential for normal replication nor affected by DNA damage. Therefore, it remains unclear what function SUMOylation of PCNA serves in organisms other than S. cerevisiae. Notably, in S. cerevisiae, SUMOylation of PCNA in the absence of ubiquitination is detrimental (Papouli et al, 2005; Pfander et al, 2005), resulting in mutation of K164 of PCNA, ameliorating the DNA damage sensitivity of the rad18 mutant, the opposite to what we observe in DT40. On the basis of the current evidence, therefore, it seems likely that the milder phenotype of a rad18 mutation in DT40 compared with a pcnaK164R mutant is due to residual ubiquitination of PCNA at K164.
We considered several possible explanations for the observation of PCNA ubiquitination in the DT40 rad18 mutant. First, RAD6 could act alone. We consider this to be unlikely, as human RAD6 is unable to monoubiquitinate PCNA on its own in vitro (Watanabe et al, 2004) and the amino-acid conservation between chicken and human RAD6A and RAD6B is 99% and 100%, respectively. Second, efficient monoubiquitination of PCNA in mouse embryonic stem (ES) cells requires the RAD6-binding region of RAD18 (Watanabe et al, 2004), a region that is highly conserved in chicken, mouse and human RAD18 (Yamashita et al, 2002).
A third possibility was that the rad18 mutant is a hypomorph, and to gain further insight into the function of RAD18 in DT40, we complemented the DT40 rad18 mutant with a series of mutant human RAD18 constructs.
The rad18 mutation in this study truncates the protein after amino acid 162, resulting in a transcript that essentially comprises an isolated RING domain (Yamashita et al, 2002). However, the phenotype of rad18 DT40 is essentially identical to rad18 murine ES cells and human fibroblasts expressing a dominant-negative form of RAD18 (C28F), namely sensitivity to a range of DNA-damaging agents, including ultraviolet light, and elevated spontaneous sister chromatid exchange (SCE; Tateishi et al, 2000, 2003; Yamashita et al, 2002). Using an approach that we have described previously (Ross et al, 2005), we expressed human RAD18 as an amino-terminal fusion with yellow fluorescent protein in rad18 DT40 (Fig 4). Expression of human RAD18 fully restored ultraviolet light sensitivity and SCE to wild-type levels. However, expression of human RAD18 (C28F), which lacks ubiquitin-ligase activity (Kannouche et al, 2004), did not restore the wild-type phenotype. Significantly, it also did not exacerbate the rad18 phenotype, as might have been expected if our rad18 allele was hypomorphic. A deletion of the entire RING domain (Δ1–64) also failed to complement the rad18 cells. However, a RAD18 construct carrying a point mutation in the central zinc-finger domain (C207F) restored the wild-type phenotype, as previously reported for murine ES cells (Watanabe et al, 2004). In addition, the localization of RAD18 and its mutants in DT40 (Fig 4) was similar to that reported in human cells (Tateishi et al, 2000). Crucially, a C-terminal deletion of RAD18, which removes the region responsible for binding to Polη and which disrupts the RAD6-interacting domain, also failed to complement the rad18 mutant, showing that the RING domain was unable to restore function without the C terminus of the protein. These data show that RAD18 functions similarly in chickens and mammals: in the mouse (Watanabe et al, 2004), both the N-terminal RING domain and C terminus of RAD18 are required. These data also confirm that the DT40 rad18 mutant is functionally null and thus PCNA K164 in DT40 must be targeted by other ubiquitin ligases that have other roles in DNA damage tolerance.
Figure 4.
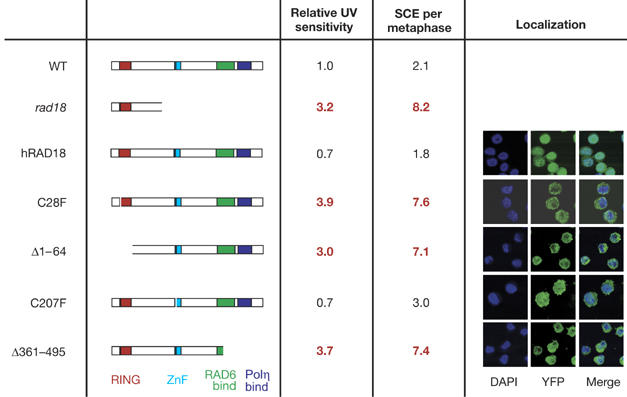
Complementation of rad18 DT40 with mutant human RAD18. The structure of the RAD18 expressed in each mutant is indicated in the left column. The transcript present in the rad18 mutant itself is also indicated. Ultraviolet light (UV) sensitivity is expressed as number of fold above the wild type (WT) and is derived from two to three independent experiments. SCE frequencies are derived from examination of at least 50 metaphases for each mutant. As previously described in mouse (Watanabe et al, 2004), RAD18 localizes predominantly in the cytoplasm, except in the C207 mutant in which it is evenly distributed between cytoplasm and nucleus. DAPI, 4,6-diamidino-2-phenylindole; SCE, sister chromatid exchange; YFP, yellow fluorescent protein.
We considered the possibility that RAD18-independent ubiquitination of PCNA is mediated by the ubiquitin-ligase function of the Fanconi anaemia (FA) complex. The known physiological role of the FA core complex is the monoubiquitination of the Fanconi D2 (FANCD2) protein (Garcia-Higuera et al, 2001). Removal of any of the components of the core complex results in its disruption and failure to monoubiquitinate FANCD2. Recently, the isopeptidase USP1 has been shown to be the common deubiquitinating enzyme for both FANCD2 and PCNA (Nijman et al, 2005; Huang et al, 2006). However, disruption of the FA core complex by mutation of FANCC does not affect the level of ubiquitin-conjugated PCNA in either the wild-type or rad18 background (Fig 3D). The identity of the ubiquitin ligase responsible for the alternate ubiquitination of PCNA therefore remains a matter of speculation.
A notable feature of rad18 cells is elevated levels of spontaneous SCE. Previously, it has been argued that this probably reflects an increased use of recombination resulting from the absence of functional post-replication repair (Yamashita et al, 2002). Therefore, on the basis of this and the data presented here, it might be anticipated that cells carrying the PCNAK164R mutation would show similar or greater levels of SCE than those deficient in RAD18; however, they do not. Undamaged hpcnaK164R cells had a level of SCE comparable with that of the wild-type control, and although they showed a greater induction of SCE than wild type after treatment with the ultraviolet light mimetic 4-nitroquinoline-1-oxide (NQO), levels of both uninduced and DNA damage-induced SCE were lower than in hPCNA/rad18 (Fig 5). Furthermore, the pcnaK164R/rad18 mutant showed a much greater level of both spontaneous and damage-induced SCE than either single mutant. This suggests that RAD18 has a function that reduces the frequency of SCE and that this is independent of ubiquitination of PCNA at K164. There is evidence that yeast RAD18 participates in double-strand break repair independently of PCNA ubiquitination: a rad18, but not pcnaK164R, strain shows sensitivity to ionizing radiation (Chen et al, 2005). We have also found that RAD18 has a role in homologous recombination in DT40 (D.S., L.J.S., S. Kabani & J.E.S., unpublished data). We believe that a proportion of the SCE in the rad18 mutant arise as a consequence of disruption of this function rather than simply from channelling of lesions from disabled post-replication repair to recombination.
Figure 5.
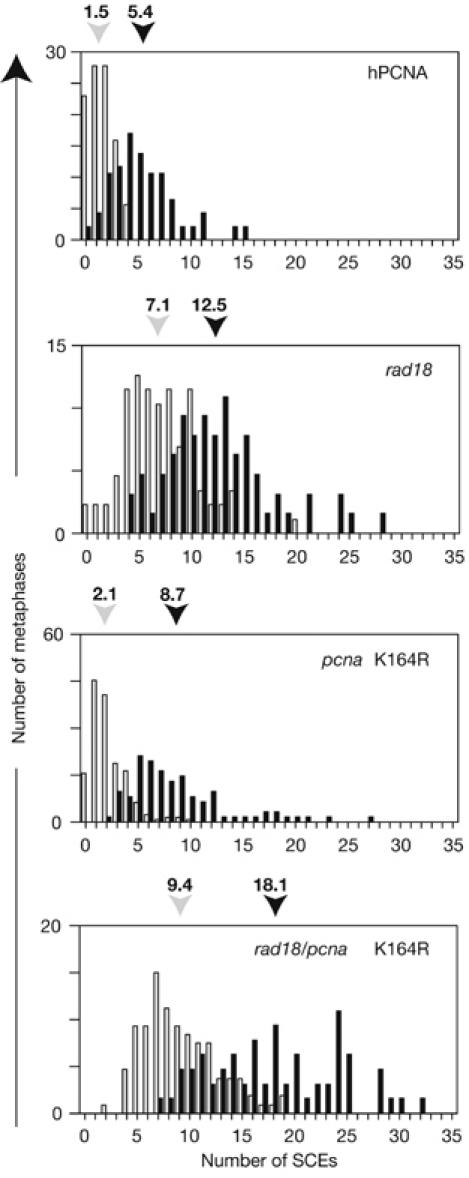
Sister chromatid exchange in rad18/pcnaK164. SCE with (black bars) and without (light grey bars) treatment with 4-nitroquinoline-1-oxide. The histogram indicates the percentage of metaphases with the number of SCE indicated on the x axis. The mean number of SCE in each case, derived from blind scoring of at least 60 metaphases, is indicated above the histogram. hPCNA, human proliferating-cell nuclear antigen; SCE, sister chromatid exchange.
Although the ability of RAD18 to monoubiquitinate PCNA at K164 seems to be conserved from yeast to vertebrates, it will be interesting to ascertain whether RAD18-independent ubiquitination of PCNA reflects a fundamental difference between birds and mammals that has arisen since their ancestors, the diapsid reptiles and synapsid reptiles, diverged some 310 million years ago (Kumar & Hedges, 1998). Although current evidence suggests that RAD18 is the dominant E3 ligase for PCNA monoubiquitination in mammals, inhibition of the ubiquitin hydrolase for monoubiquitinated PCNA, USP1 (Huang et al, 2006), in rad18 mutants of mouse or human cells will help resolve whether there is also RAD18-independent ubiquitination of PCNA in mammals.
Methods
Culture and transfection of DT40. Culture and transfection of DT40 was carried out as described previously (Simpson & Sale, 2003).
DNA constructs. Genomic DNA containing the chicken PCNA locus was isolated by high stringency screening of a chicken genomic phage λ library (Stratagene, La Jolla, CA, USA) using probes generated by PCR from the chicken genomic PCNA locus using primers based on the chicken PCNA complementary DNA sequence: 5′ probe=F1 (5′-TGCTGCTGCCGCCAAGATGTTTGAG-3′) and R4 (5′-TCACCAATGTGGCTGAGGTCTCTACAAATG- 3′); 3′ probe=F5 (5′-ACGCATTTGTAGAGACCTCAGCCACATTGG- 3′) and (5′-TCCTGCTGGTCTTCAATCTTTGGAG-3′). The arms of the construct flank the six exons of PCNA and comprised a 6 kb BamHI–EcoRI fragment as the 5′ arm and a 2.5 kb KpnI fragment as the 3′ arm (supplementary Fig S1A online). The construct was assembled in pBluescript modified to contain the multiple cloning site: SalI, BglII, EcoRI, BamHI and KpnI. Antibiotic resistance cassettes (either blasticidin or puromycin resistance) were inserted into the BamHI site. Probes for Southern blotting of NheI-digested DNA, as indicated in supplementary Fig S1A online, were prepared from λ genomic DNA fragments.
The K164R and K138R mutants were generated using Quickchange (Stratagene) site-directed mutagenesis and the following oligonucleotides: PSDMK164F (5′-GTAATTTCCTGTGCAAGAGACGGAGTGAAAT TTTCTGC-3′) a nd PSDMK164R (5′-GCAGAAAATTTCACTCCGTCTCTTGCACAGG AAATTAC-3′); and PSDMK138F (5′-GTACAGCTGTGTAGTAAGGATGCCTTCTGGT G-3′) and PSDMK138R (5′-CACCAGAAGGCATCCTTACTACACAGCTGTA C-3′). Mutagenesis was confirmed by sequencing. To rescue PCNA expression, hPCNA (or mutant hPCNA) was expressed in pIRES2–EGFP (Clontech, Palo Alto, CA, USA). Expression of green fluorescent protein (GFP) was thus operationally linked to the expression of PCNA. After targeting of the second PCNA allele, disruption was confirmed by Southern blot probing from both 5′ and 3′ ends and by reverse transcription–PCR for chicken and human PCNA (supplementary Fig S1B online). All clones used in this study were matched for expression of PCNA by fluorescence-activated cell sorting for GFP (supplementary Fig S1C online) and western blotting for PCNA.
The construct for the USP1 disruption, which removes the last exon containing a histidine domain required for enzymatic activity, will be described elsewhere (V.H.O. & K.J.P., unpublished data).
Immunocytochemistry. For immunofluorescent detection of PCNA and sites of DNA replication, cells were labelled for 10 min with 10 μM 5-bromodeoxyuridine (BrdU) and then spun down to polylysine-coated coverslips. The cells were sequentially fixed in 4% paraformaldehyde followed by −20°C methanol, treated with 4 N HCl for 30 min and blocked in PBS with 0.1% Triton X-100 and 0.02% SDS. Primary antibodies (anti-PCNA clone PC-10, Santa Cruz, CA, USA; anti-BrdU OBT0039G, Oxford Biotechnology, Kidlington, UK) and fluorescently labelled secondary antibodies (Molecular Probes, Paisley, UK) were diluted in the same buffer. Stained cells were analysed using a Bio-Rad Radiance laser confocal microscope.
Colony survival assays. Colony survival assays were performed, as described previously (Simpson & Sale, 2003). Ultraviolet light was delivered by an equilibrated 254 nm lamp (UVP Inc., Cambridge, UK) mounted in a custom-built shuttered enclosure. MMS and CDDP were obtained from Sigma.
Detection of PCNA modifications. For direct detection of PCNA modifications in DT40, 4 × 105 cells were pelleted and lysed directly in 25 μl of boiling 2 × SDS–polyacrylamide gel electrophoresis loading buffer. The lysates were boiled for 5 min with vortexing before loading onto 4–12% Bis–Tris acrylamide gels. Blots (nitrocellulose or polyvinylidene difluoride) were blocked for 2 h with 3% milk in PBS with 0.05% Tween. PCNA was detected by overnight incubation at 4°C with mouse anti-hPCNA (clone PC10, Santa Cruz) at 1:4,000, followed by rabbit anti-mouse Ig-HRP conjugate (Southern Biotechnology, Birmingham, AL, USA) at 1:10,000 for 1 h. Blots were developed with ECL Plus (Amersham, Little Chalfont, UK).
Measurement of SCE. This was performed as described previously (Simpson & Sale, 2003). For induction of DNA damage, NQO (Sigma, Poole, UK) was added to a final concentration of 0.2 ng/ml for 6 h before metaphase arrest with colcemid (Sigma).
Supplementary information is available at EMBO reports online (http://www.emboreports.org).
Supplementary Material
supplementary Figures
Acknowledgments
We thank S. Takeda for providing the RAD18 targeting constructs, H. Arakawa and J.-M. Buerstedde for discussions and sharing unpublished information, and C. Batters and C. Edmunds for helpful comments on the manuscript.
References
- Bienko M et al. (2005) Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310: 1821–1824 [DOI] [PubMed] [Google Scholar]
- Chen S, Davies AA, Sagan D, Ulrich HD (2005) The RING finger ATPase Rad5p of Saccharomyces cerevisiae contributes to DNA double-strand break repair in a ubiquitin-independent manner. Nucleic Acids Res 33: 5878–5886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, Furuya K, Watts FZ, Carr AM, Lehmann AR (2006) Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell 7: 2976–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, Prakash L (2004) Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol 24: 4267–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins NP, Kato K, Strauss B (1976) A model for replication repair in mammalian cells. J Mol Biol 101: 417–425 [DOI] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Huang TT, Nijman SM, Mirchandani KD, Galardy PJ, Cohn MA, Haas W, Gygi SP, Ploegh HL, Bernards R, D'Andrea AD (2006) Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol 8: 339–347 [DOI] [PubMed] [Google Scholar]
- Kannouche PL, Wing J, Lehmann AR (2004) Interaction of human DNA polymerase ηη with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 14: 491–500 [DOI] [PubMed] [Google Scholar]
- Kumar S, Hedges SB (1998) A molecular timescale for vertebrate evolution. Nature 392: 917–920 [DOI] [PubMed] [Google Scholar]
- Leach CA, Michael WM (2005) Ubiquitin/SUMO modification of PCNA promotes replication fork progression in Xenopus laevis egg extracts. J Cell Biol 171: 947–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D'Andrea AD, Bernards R (2005) The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 17: 331–339 [DOI] [PubMed] [Google Scholar]
- Okada T, Sonoda E, Yamashita YM, Koyoshi S, Tateishi S, Yamaizumi M, Takata M, Ogawa O, Takeda S (2002) Involvement of vertebrate polκ in Rad18-independent postreplication repair of UV damage. J Biol Chem 277: 48690–48695 [DOI] [PubMed] [Google Scholar]
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123–133 [DOI] [PubMed] [Google Scholar]
- Pfander B, Moldovan GL, Sacher M, Hoege C, Jentsch S (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436: 428–433 [DOI] [PubMed] [Google Scholar]
- Ross AL, Sale JE (2006) The catalytic activity of REV1 is employed during immunoglobulin gene diversification in DT40. Mol Immunol 43: 1587–1594 [DOI] [PubMed] [Google Scholar]
- Ross AL, Simpson LJ, Sale JE (2005) Vertebrate DNA damage tolerance requires the C-terminus but not BRCT or transferase domains of REV1. Nucleic Acids Res 33: 1280–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Jun S, O'Neal LE, Sonoda E, Bemark M, Sale JE, Li L (2006) REV3 and REV1 play major roles in recombination-independent repair of DNA interstrand cross-links mediated by monoubiquitinated proliferating cell nuclear antigen (PCNA). J Biol Chem 281: 13869–13872 [DOI] [PubMed] [Google Scholar]
- Simpson LJ, Sale JE (2003) Rev1 is essential for DNA damage tolerance and non-templated immunoglobulin gene mutation in a vertebrate cell line. EMBO J 22: 1654–1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson LJ, Sale JE (2005) UBE2V2 (MMS2) is not required for effective immunoglobulin gene conversion or DNA damage tolerance in DT40. DNA Repair (Amst) 4: 503–510 [DOI] [PubMed] [Google Scholar]
- Stelter P, Ulrich HD (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425: 188–191 [DOI] [PubMed] [Google Scholar]
- Tateishi S, Sakuraba Y, Masuyama S, Inoue H, Yamaizumi M (2000) Dysfunction of human Rad18 results in defective postreplication repair and hypersensitivity to multiple mutagens. Proc Natl Acad Sci USA 97: 7927–7932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi S, Niwa H, Miyazaki J, Fujimoto S, Inoue H, Yamaizumi M (2003) Enhanced genomic instability and defective postreplication repair in RAD18 knockout mouse embryonic stem cells. Mol Cell Biol 23: 474–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M (2004) Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 23: 3886–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita YM, Okada T, Matsusaka T, Sonoda E, Zhao GY, Araki K, Tateishi S, Yamaizumi M, Takeda S (2002) RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J 21: 5558–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Figures