

Abstract
The efficient functioning of the endoplasmic reticulum (ER) is essential for most cellular activities and survival. Conditions that interfere with ER function lead to the accumulation and aggregation of unfolded proteins. ER transmembrane receptors detect the onset of ER stress and initiate the unfolded protein response (UPR) to restore normal ER function. If the stress is prolonged, or the adaptive response fails, apoptotic cell death ensues. Many studies have focused on how this failure initiates apoptosis, as ER stress-induced apoptosis is implicated in the pathophysiology of several neurodegenerative and cardiovascular diseases. In this review, we examine the role of the molecules that are activated during the UPR in order to identify the molecular switch from the adaptive phase to apoptosis. We discuss how the activation of these molecules leads to the commitment of death and the mechanisms that are responsible for the final demise of the cell.
Keywords: apoptosis, BCL2 family, ER stress, unfolded protein response
Introduction
The endoplasmic reticulum (ER) is primarily recognized as the site of synthesis and folding of secreted, membrane-bound, and some organelle-targeted proteins. Several factors are required for optimum protein folding, including ATP, Ca2+ and an oxidizing environment to allow disulphide-bond formation (Gaut & Hendershot, 1993). As a consequence of this specialist environment, the ER is highly sensitive to stresses that perturb cellular energy levels, the redox state or Ca2+ concentration. Such stresses reduce the protein folding capacity of the ER, which results in the accumulation and aggregation of unfolded proteins—a condition referred to as ER stress. Protein aggregation is toxic to cells and, consequently, numerous pathophysiological conditions are associated with ER stress, including ischaemia, neurodegenerative diseases and diabetes (Kaufman, 2002).
To combat the deleterious effects of ER stress, cells have evolved various protective strategies, collectively termed the unfolded protein response (UPR). This concerted and complex cellular response is mediated through three ER transmembrane receptors: pancreatic ER kinase (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1). In resting cells, all three ER stress receptors are maintained in an inactive state through their association with the ER chaperone, GRP78. On accumulation of unfolded proteins, GRP78 dissociates from the three receptors, which leads to their activation and triggers the UPR. The UPR is a pro-survival response to reduce the accumulation of unfolded proteins and restore normal ER functioning (Schroder & Kaufman, 2005; Fig 1). However, if protein aggregation is persistent and the stress cannot be resolved, signalling switches from pro-survival to pro-apoptotic. The molecular mechanisms that facilitate this switch are now emerging. This review explores the mechanisms through which UPR-mediated signals might elicit apoptosis by discussing the three distinct phases of the process: initiation, commitment and execution.
Figure 1.
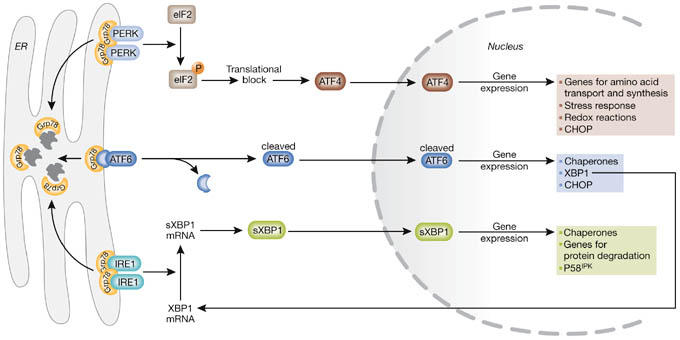
The unfolded protein response. On aggregation of unfolded proteins, GRP78 dissociates from the three endoplasmic reticulum (ER) stress receptors, pancreatic ER kinase (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6) and inositol-requiring enzyme 1 (IRE1), allowing their activation. The activation of the receptors occurs sequentially, with PERK being the first, rapidly followed by ATF6, whereas IRE1 is activated last. Activated PERK blocks general protein synthesis by phosphorylating eukaryotic initiation factor 2α (eIF2α). This phosphorylation enables translation of ATF4, which occurs through an alternative, eIF2α-independent translation pathway. ATF4, being a transcription factor, translocates to the nucleus and induces the transcription of genes required to restore ER homeostasis. ATF6 is activated by limited proteolysis after its translocation from the ER to the Golgi apparatus. Active ATF6 is also a transcription factor and it regulates the expression of ER chaperones and X box-binding protein 1 (XBP1), another transcription factor. To achieve its active form, XBP1 must undergo mRNA splicing, which is carried out by IRE1. Spliced XBP1 protein (sXBP1) translocates to the nucleus and controls the transcription of chaperones, the co-chaperone and PERK-inhibitor P58IPK, as well as genes involved in protein degradation. This concerted action aims to restore ER function by blocking further build-up of client proteins, enhancing the folding capacity and initiating degradation of protein aggregates. CHOP, C/EBP homologous protein.
Initiation phase of ER stress-induced apoptosis
Given the central role of PERK, ATF6 and IRE1 in UPR signalling, it is likely that these UPR mediators are also fundamental to ER stress-induced apoptosis. The roles of each UPR mediator and their possible links to apoptotic signalling are discussed below.
PERK. Dissociation of GRP78 from PERK initiates the dimerization and autophosphorylation of the kinase and generates active PERK. Once activated, PERK phosphorylates eukaryotic initiation factor 2 (eIF2), which leads to inhibition of general (cap- or eIF2α-dependent) protein translation. Inhibition of protein translation aids cell survival by decreasing the load of nascent proteins arriving at the ER. In fact, Perk−/− mouse embryonic fibroblasts, when challenged with ER stress-inducing agents, failed to block protein translation and exhibited increased cell death. Inhibition of translation with cycloheximide reduced ER stress-induced cell death, confirming that blocking the accumulation of unfolded nascent proteins is crucial for cell survival (Harding et al, 2000). However, this attenuation of translation is not absolute; genes carrying certain regulatory sequences in their 5′ untranslated regions—for example, the internal ribosomal entry site (IRES)—can bypass the eIF2α-dependent translational block (Schroder & Kaufman, 2005). The most studied of these genes is ATF4, which encodes a cAMP response element-binding transcription factor (C/EBP). ATF4 promotes cell survival by inducing genes involved in amino-acid metabolism, redox reactions, stress response and protein secretion (Harding et al, 2003). However, not all the genes induced by ATF4 are anti-apoptotic. The transcription factor C/EBP homologous protein (CHOP), whose induction strongly depends on ATF4, is well known to promote apoptotic cell death (the mechanisms are detailed as part of the commitment phase; Fig 1). In conclusion, activation of PERK is initially protective and crucial for survival during even mild stress. However, activation of PERK also leads to the induction of CHOP, which, as detailed later, is an important element of the switch from pro-survival to pro-death signalling.
ATF6. After the dissociation of GRP78, ATF6 translocates to the Golgi apparatus where it is cleaved into its active form by site-1 and site-2 proteases. Active ATF6 then moves to the nucleus and induces genes with an ER stress response element (ERSE) in their promoter (Schroder & Kaufman, 2005). So far, the identified targets of ATF6 include ER chaperone proteins such as GRP78, GRP94, protein disulphide isomerase, and the transcription factors CHOP and X box-binding protein 1 (XBP1). XBP1 is important in IRE1 signalling and is discussed below (Fig 1). Although ATF6 can induce CHOP mRNA expression, no reports have linked ATF6 to ER stress-induced apoptosis; therefore, it seems that ATF6-mediated signals are purely pro-survival and aim to counteract ER stress.
IRE1. IRE1 is a dual-activity enzyme, having a serine–threonine kinase domain and an endoribonuclease domain. On activation, the endonuclease activity of IRE1 removes a 26-nucleotide intron from the XBP1 mRNA, previously induced by ATF6. The generated frameshift splice variant (sXBP1) encodes a stable, active transcription factor (Yoshida et al, 2001). sXBP1 has diverse targets, including ER chaperones and the HSP40 family member P58IPK (Lee et al, 2003; Fig 1). P58IPK binds and inhibits PERK, thereby providing a negative feedback loop that relieves the PERK-mediated translational block (Yan et al, 2002). Upregulation of P58IPK is not an immediate event, as its induction only occurs several hours after the phosphorylation of PERK and eIF2α. It is possible that by relieving the translational block, P58IPK induction represents the termination of the UPR. At this point, if the UPR has been successful, the ER returns to normal functioning and the cell survives; however, if the stress persists, relieving the translational block by P58IPK might allow the synthesis of pro-apoptotic proteins. The pro-apoptotic nature of P58IPK was not confirmed in P58IPK-deficient mice, as these mice—similar to Perk−/− mice—showed increased apoptosis of pancreatic islet cells and developed diabetes (Ladiges et al, 2005). Although the mechanism of pancreatic islet cell apoptosis was not examined in P58IPK−/− mice, it is known that owing to the fluctuating demand on insulin and glucagon synthesis, β-cells are the most affected by deregulated UPR in the body. A lack of P58IPK can result in prolonged PERK activity after ER stress, blocking protein translation for a considerable time. This can lead to a deficit in vital proteins produced through cap-dependent translation as well as overexpression of proteins produced by the cap-independent pathway. Indeed, enhanced expression of both ATF4 and CHOP was detected after P58IPK gene silencing (van Huizen et al, 2003). Whether the increased pancreatic β-cell apoptosis detected in the P58IPK-deficient mice was due to increased CHOP expression or deregulated general protein synthesis is not yet known.
Although the IRE1–XBP1 axis seems to have pro-survival effects through the induction of ER chaperones and P58IPK, overexpression of IRE1 in HEK293T cells resulted in apoptotic cell death (Wang et al, 1998). How could IRE1 initiate cell death? The answer might lie with the activation of kinase pathways, most notably the c-Jun N-terminal kinase (JNK) pathway. Active IRE1 has been shown to recruit the adaptor molecule TNF-receptor-associated factor 2 (TRAF2). The IRE1–TRAF2 complex formed during ER stress can recruit the apoptosis-signal-regulating kinase (ASK1), which is a mitogen-activated protein kinase kinase kinase (MAPKKK) that has been shown to relay various stress signals to the downstream MAPKs JNK and p38 (Nishitoh et al, 1998). Overexpression of ASK1 induced apoptosis in several cell types, whereas neurons from Ask1−/− mice exhibited resistance to lethal ER stress, illustrating the importance of ASK1 in ER stress-induced cell death (Hatai et al, 2000; Nishitoh et al, 2002). Activation of JNK has also been reported in response to ER stress and was shown to be IRE1- and TRAF2-dependent (Urano et al, 2000). Activation of JNK is a common response to many forms of stress and is known to influence the cell-death machinery through the regulation of BCL2 family proteins (Davis, 2000). For example, phosphorylation of BCL2 by JNK, which occurs primarily at the ER, suppresses the anti-apoptotic activity of BCL2. Besides BCL2, JNK also phosphorylates BH3 (Bcl-2 homology domain 3)-only members of the BCL2 family such as Bim, which enhances their pro-apoptotic potential (Fig 2).
Figure 2.
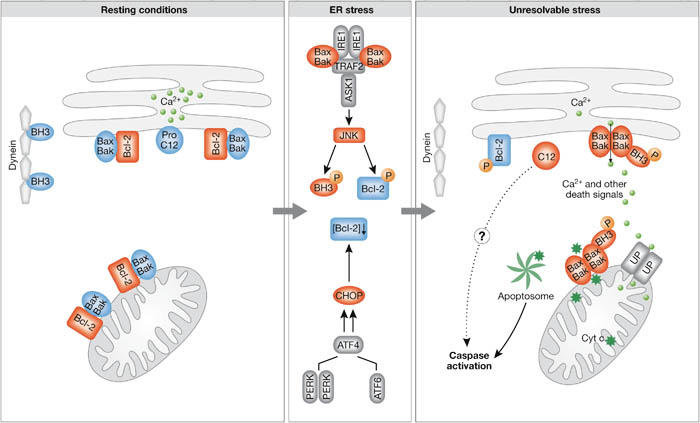
The BCL2 family of proteins in resting cells and in endoplasmic reticulum stress conditions. In resting conditions, the pro-apoptotic Bax and Bak (Bax/Bak) are kept inactive by interaction with BCL2 both on the mitochondrial and endoplasmic reticulum (ER) membranes, whereas Bim (BH3) is inhibited by binding to cytoskeletal dynein. Severe ER stress leads to activation of c-Jun N-terminal kinase (JNK) and induction of C/EBP homologous protein (CHOP; initiation phase). Both JNK and CHOP eliminate the anti-apoptotic effect of BCL2; CHOP blocks expression of BCL2, whereas JNK phosphorylates it. JNK also phosphorylates Bim, which leads to its release from the cytoskeleton and to its activation (commitment phase). Collectively, these changes allow activation of Bax and Bak, transmission of the signal from the ER to the mitochondria and execution of death (execution phase). Caspases are activated possibly on the ER membrane itself, as well as in the apoptosome, after transmission of the death signal to mitochondria and the release of cytochrome c. Blue labels show inactive molecules, whereas red labels indicate active molecules, with the rounded shapes representing the pro-apoptotic molecules and rectangles representing the anti-apoptotic molecules. ATF6, activating transcription factor 6; IRE1, inositol-requiring enzyme 1; PERK, pancreatic ER kinase (PKR)-like ER kinase; TRAF2, TNF-receptor-associated factor 2; UP, uniporter.
If IRE1 has both pro- and anti-apoptotic functions, how are these two opposing functions separated? Yeast two-hybrid screens searching for IRE1-interacting proteins identified c-Jun N-terminal inhibitory kinase (JIK) and Jun activation domain-binding protein 1 (JAB1) as molecules that interact with IRE1 (Oono et al, 2004; Yoneda et al, 2001). The ability of JIK to bind both IRE1 and TRAF2 was also shown, indicating a potential role for JIK in regulating the recruitment of TRAF2 and thus activation of the MAPK pathway. The other IRE1 binding partner, JAB1, was shown to bind to IRE1 during resting conditions. Mild ER stress enhanced this reaction, whereas strong ER stress diminished it. Thus, JAB1 might regulate the choice between the UPR and apoptosis by association with or dissociation from IRE1. Whether JIK or JAB1 provide the switch between pro-survival and pro-apoptotic IRE1 signalling requires more research.
On the basis of these studies, IRE1 seems to be important for the initiation of pro-apoptotic signals. Interestingly, IRE1 is thought to be the last arm of the UPR to be activated, with PERK being the first, closely followed by ATF6. Perhaps the PERK- and ATF6-mediated pathways attempt to resolve the stress before activation of Ire1. Once activated, IRE1 initially aids the UPR by splicing XBP1, but ultimately terminates it by relieving the translational inhibition by inducing P58IPK. At this point, either the cell returns to normal functioning or, if the stress persists, IRE1 triggers apoptosis by recruiting ASK1 and JNK.
Commitment phase of ER stress-induced apoptosis
Signalling through PERK, ATF6 and IRE1 can trigger pro-apoptotic signals during prolonged ER stress. However, they do not directly cause cell death but rather initiate the activation of downstream molecules such as CHOP or JNK, which further push the cell down the path of death. This section discusses the commitment phase of ER stress-induced apoptosis, focusing on how CHOP, JNK and BCL2 family proteins relay the pro-apoptotic signal to the final execution phase.
CHOP. CHOP, also known as growth-arrest- and DNA-damage-inducible gene 153 (GADD153), was originally identified in response to DNA damage. However, CHOP induction is probably most sensitive to ER stress conditions (Zinszner et al, 1998). During ER stress, all three arms of the UPR induce transcription of CHOP. However, to upregulate CHOP protein expression the PERK–eIF2α–ATF4 branch of the UPR is essential. In addition to being controlled at the level of transcription and translation, CHOP is also regulated post-translationally by phosphorylation on serine residues 78 and 81 by p38 MAPK, which increases its activity. This is interesting, given that p38 is a substrate of ASK1, which is recruited to the IRE1–TRAF2 complex on ER stress. Thus, during prolonged stress, the PERK and the IRE1 pathways might converge on CHOP, possibly increasing each other's effect. The role of CHOP in ER stress-induced apoptosis has been illustrated in Chop−/− mice. These mice are born at the expected frequency and appear phenotypically normal. However, a study of Chop−/− mouse embryonic fibroblasts revealed that CHOP deficiency provides partial resistance to ER stress-induced apoptosis (Zinszner et al, 1998). Studies examining the mechanism of CHOP-induced apoptosis identified numerous target genes including BCL2, GADD34, endoplasmic reticulum oxidoreductin 1 (ERO1α) and Tribbles-related protein 3 (TRB3). Although CHOP mainly induces gene expression, BCL2 is an example of a gene that is downregulated by CHOP. How this might impinge on the regulation of apoptosis is discussed later in this section.
GADD34 is a protein phosphatase 1 (PP1)-interacting protein that causes PP1 to dephosphorylate eIF2α and thus release the translational block (Brush et al, 2003). Expression of GADD34 correlates with apoptosis induced by various signals, and its overexpression can initiate or enhance apoptosis (Adler et al, 1999). The mechanism by which GADD34 promotes apoptosis is unknown, although several theories exist. For example, restoration of protein synthesis owing to induction of GADD34 might allow the synthesis of pro-apoptotic proteins. Indeed, compared with wild-type mice, GADD34ΔCΔC mice (deficient in eIF2α phosphatase activity) display milder renal toxicity after the injection of tunicamycin, which causes ER stress through the inhibition of protein glycosylation. Moreover, inhibition of eIF2α phosphatases (with salubrinal) has been reported to reduce ER stress-induced apoptosis (Boyce et al, 2005; Marciniak et al, 2004). Although GADD34 removes the PERK-mediated translational block in a manner similar to P58IPK, deficiency of GADD34 and P58IPK has seemingly opposing effects. This could be due to as yet unexplored functions of the two proteins, or the possible different efficiency with which they alter the protein translation machinery.
TRB3 is another gene induced by CHOP. Knockdown of TRB3 through RNA interference in HEK293T and HeLa cells confers resistance against tunicamycin (Ohoka et al, 2005). Previous studies examining the role of TRB3, although not in ER stress, have indicated that it might promote apoptosis by binding to the pro-survival serine/threonine kinase Akt, thereby preventing its phosphorylation and reducing its kinase activity (Du et al, 2003). A few studies have examined the role of Akt in ER stress-induced apoptosis. Hu and colleagues recently reported transient activation of Akt during ER stress, induced by the sarcoplasmic reticulum calcium pump (SERCA) inhibitor thapsigargin or tunicamycin in MCF-7 cells (Hu et al, 2004). Blocking Akt activity sensitized MCF-7 cells to ER stress-induced apoptosis, suggesting that Akt activation is a pro-survival pathway activated during ER stress. It has also been proposed that, during transient ER stress, TRB3 feeds back onto CHOP, blocking its pro-apoptotic activity and thus allowing the cell to return to normal functioning. However, during prolonged ER stress, TRB3 induction is much more robust and is thought to act as an Akt inhibitor, therefore pushing the cell in the direction of apoptosis (Ohoka et al, 2005). Further studies are required to clarify the exact role of TRB3 in ER stress-induced apoptosis.
BCL2 family. Members of the BCL2 family of proteins are important regulators of apoptotic cell death. Until recently, BCL2 proteins were thought to exclusively regulate the mitochondrial-mediated apoptotic pathway. The first reports linking ER stress-induced cell death to the BCL2 family of proteins showed that overexpression of BCL2, or deficiency of Bax and Bak, conferred protection against lethal ER stress (Distelhorst & McCormick, 1996; Wei et al, 2001). Further reports showed that overexpression of the mitochondrion-localized viral mitochondrial inhibitor of apoptosis (vMIA) or ER-targeted BCL2 (BCL2/cb5) blocked ER stress-induced cytochrome c release and apoptosis. By contrast, expression of ER-targeted Bak (Bak/cb5) in Bax/Bak double-knockout cells induced apoptosis. These findings indicate that stress signals are relayed from the ER to mitochondria, and that ER stress-induced apoptosis, similar to mitochondrial-mediated apoptosis, is also regulated by the BCL2 family of proteins (Boya et al, 2002; Hacki et al, 2000; Zong et al, 2003).
Although the involvement of BCL2 proteins in ER stress-induced cell death is clear, how they are regulated by ER stress is less well understood. So far, UPR-mediated activation of CHOP and JNK has been shown to be involved. CHOP is known to repress BCL2 gene expression, which increases the proportion of pro-apoptotic BCL2 proteins in the cell and enables their activation. Overexpression of CHOP has been shown to induce apoptosis, which was associated with the activation and mitochondrial translocation of Bax. In this model, overexpression of BCL2 could block CHOP-induced apoptosis (Matsumoto et al, 1996; McCullough et al, 2001). As discussed in the section on the initiation phase, JNK is activated by the IRE1–TRAF2–ASK1 branch of the UPR. JNK is known to regulate BCL2 proteins by phosphorylation. First of all, JNK is able to phosphorylate BCL2 localized to the ER. This has a knock-on pro-apoptotic effect, as phosphorylated BCL2 is unable to sequester and inhibit pro-apoptotic BH3-only proteins and cannot control ER Ca2+ fluxes (Bassik et al, 2004; Fig 2). JNK can also target the BH3-only members of the BCL2 family. Induction and/or post-translational modification of BH3-only proteins have a central role in setting the apoptotic cascade in motion. Of the BH3-only subfamily, p53-upregulated modulator of apoptosis (PUMA), Noxa and Bim have been reported to have a role in ER stress. Bim exists in several isoforms, including a short form (BimS) and two longer forms—BimL and BimEL—with the two longer forms being constitutively expressed. The pro-apoptotic effects of BimL and BimEL are restrained in unstressed cells by their binding to the dynein motor complex. Phosphorylation by JNK releases Bim from this inhibitory association and allows it to exert its pro-apoptotic effects (Lei & Davis, 2003; Fig 2). Supporting the role of Bim in ER stress, Morishima and colleagues reported that Bim translocates from the cytoskeleton to the ER in C2C12 cells exposed to tunicamycin, whereas we detected strong induction of Bim in PC12 cells treated with thapsigargin (Morishima et al, 2004; E.S., K.R. Herbert, E. Kavanagh, A.S. and A.G., unpublished data). Together, these data suggest that JNK activated by ER stress targets BCL2 proteins, which would allow the activation of Bax and Bak leading to the execution of apoptosis. Interestingly, a new publication by Hetz and colleagues reports a reverse interaction between JNK and Bax/Bak (Hetz et al, 2006). This report shows that in Bax/Bak double-knockout mice, tunicamycin fails to induce sXBP1 and JNK phosphorylation. Moreover, Bax and Bak were found to interact directly with IRE1 during ER stress. Reconstitution of Bak expression in the Bax/Bak double-knockout mouse embryonic fibroblast cells restored tunicamycin-induced JNK phosphorylation, suggesting a direct, although unexpected, connection between the UPR and the apoptotic machinery.
Another two BH3-only proteins are regulated by ER stress. Microarray analysis of tunicamycin-treated SH-SY5Y neuroblastoma cells reported an upregulation of PUMA (Reimertz et al, 2003). Similarly, another study examining the expression of BH3-only proteins in response to either thapsigargin or tunicamycin showed upregulation of both PUMA and Noxa in a p53-dependent manner (Li et al, 2006). The question of how p53 is activated during the UPR is yet to be answered. Collectively, the regulation of BCL2 family proteins, whether by JNK, CHOP or by the upregulation of BH3-only proteins, activates Bax and Bak leading to caspase activation and, ultimately, cell death (Fig 2).
The execution phase of ER stress-induced apoptosis
All upstream signals, such as the activation of transcription factors, kinase pathways and the regulation of BCL2 family members, ultimately lead to caspase activation, resulting in the ordered and sequential dismantling of the cell.
Caspase activation. Both the death-receptor- and mitochondrial-mediated apoptotic pathways are well-characterized processes with specific subsets of caspases associated with each pathway. The cohort of caspases linked to ER stress-induced apoptosis has not yet been conclusively established. Processing of caspases 12, 3, 6, 7, 8 and 9 has been observed in different studies of ER stress. Although caspase activation is required for the apoptotic process, the identity of the apical caspase is subject to debate. Caspase 12 has been proposed as a key mediator of ER stress-induced apoptosis (Szegezdi et al, 2003). Caspase 12 is expressed only in rodents; its human homologue has been silenced by several mutations during evolution. Caspase 4 has been proposed to fulfil the function of caspase 12 in humans, but this is under debate. Caspase 12−/− mouse embryonic fibroblast cells have been reported to exhibit partial resistance specifically against ER stress-inducing agents, suggesting an important role for this caspase (Nakagawa et al, 2000). However, recent work published by Saleh and colleagues, also using caspase 12−/− mouse embryonic fibroblasts—although from a different source—observed no resistance to ER stressors such as thapsigargin (Saleh et al, 2006). If caspase 12 functions as an initiator caspase during ER stress as proposed, it should target downstream caspases; however, little consistent data linking caspase 12 to downstream caspase activation is available. Moreover, no definitive substrates or human orthologue for caspase 12 have yet been identified, making it difficult to establish a common role for this caspase in ER stress-induced apoptosis.
Conclusion
ER stress conditions have been observed in numerous diseases including Alzheimer disease, Creutzfeldt–Jakob disease, Huntington disease as well as cardiovascular diseases, indicating that ER stress-induced apoptosis is an important factor in pathophysiological conditions. To be able to treat or halt the progress of such conditions, a firm understanding of the mechanisms mediating ER stress-induced apoptosis is essential. Research so far has identified many candidates involved in orchestrating the switch from the protective UPR signalling to pro-apoptotic signalling. Some of these genes, such as P58IPK, GADD34 and TRB3, are involved in switching off the PERK-mediated pathway. Blocking this protective pathway can be a central element of the switch from adaptation to suicide. These proteins, however, seem to affect sensitivity to ER stress in opposing ways. In addition, P58IPK also acts as an Akt inhibitor. Are there additional functions for GADD34 and TRB3? Are these the real orchestrators of life-or-death decisions during ER stress? It is hard to tell. Besides these proteins with negative-feedback functions, two additional, reasonably potent, pro-apoptotic molecules are activated during the UPR: CHOP and ASK1. Both overexpression and knockout experiments have confirmed the pro-apoptotic role of these proteins in ER stress. Both molecules can target the BCL2 family and are able to set the death machinery in motion. Furthermore, activation of ASK1 might be regulated by stress-sensitive adaptor proteins such as JAB1, offering another possible mechanism to switch from an adaptive response to cell suicide.
It is also worth mentioning that, dissimilar to other stress responses, the mediators of ER stress are well defined and are specific to ER stress; therefore, they could be useful targets for therapy. Inhibition of the IRE1–TRAF2 interaction by small chemicals or by antagonistic IRE1-interacting adaptor proteins is a plausible strategy. Such an inhibition might not interfere with XBP1 splicing but could prevent the activation of the pro-apoptotic processes.
Many unanswered questions remain about ER stress-induced apoptosis. Is there one defining pro-apoptotic event that ultimately controls all others, or do several pro-apoptotic pathways simultaneously function to commit the cell to death? Which caspases are associated with ER stress-induced cell death? What defines the point of no return for the cell? ER stress-induced cell death is a new, exciting apoptotic pathway, the full impact of which, especially in the pathology of diseases, remains undetermined. Continued research in this field is necessary to tease out the complexities of this cell-death pathway.
Acknowledgments
We thank U. Fitzgerald and G. Molnar for critical reading of the manuscript. We apologize to colleagues whose work has not been cited due to space limitations. Our work is supported by grants from Higher Education Ireland (HEA) and Science Foundation Ireland (SFI).
References
- Adler HT, Chinery R, Wu DY, Kussick SJ, Payne JM, Fornace AJ Jr, Tkachuk DC (1999) Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol Cell Biol 19: 7050–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassik MC, Scorrano L, Oakes SA, Pozzan T, Korsmeyer SJ (2004) Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J 23: 1207–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G (2002) Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ 9: 465–467 [DOI] [PubMed] [Google Scholar]
- Boyce M et al. (2005) A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 307: 935–939 [DOI] [PubMed] [Google Scholar]
- Brush MH, Weiser DC, Shenolikar S (2003) Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 α to the endoplasmic reticulum and promotes dephosphorylation of the α subunit of eukaryotic translation initiation factor 2. Mol Cell Biol 23: 1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252 [DOI] [PubMed] [Google Scholar]
- Distelhorst CW, McCormick TS (1996) Bcl-2 acts subsequent to and independent of Ca2+ fluxes to inhibit apoptosis in thapsigargin- and glucocorticoid-treated mouse lymphoma cells. Cell Calcium 19: 473–483 [DOI] [PubMed] [Google Scholar]
- Du K, Herzig S, Kulkarni RN, Montminy M (2003) TRB3: a tribbles homolog that inhibits Akt/PKB activation by insulin in liver. Science 300: 1574–1577 [DOI] [PubMed] [Google Scholar]
- Gaut JR, Hendershot LM (1993) The modification and assembly of proteins in the endoplasmic reticulum. Curr Opin Cell Biol 5: 589–595 [DOI] [PubMed] [Google Scholar]
- Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19: 2286–2295 [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D (2000) Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell 5: 897–904 [DOI] [PubMed] [Google Scholar]
- Harding HP et al. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633 [DOI] [PubMed] [Google Scholar]
- Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K (2000) Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J Biol Chem 275: 26576–26581 [DOI] [PubMed] [Google Scholar]
- Hetz C et al. (2006) Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1α. Science 312: 572–576 [DOI] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Exton JH (2004) Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem 279: 49420–49429 [DOI] [PubMed] [Google Scholar]
- Kaufman RJ (2002) Orchestrating the unfolded protein response in health and disease. J Clin Invest 110: 1389–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG (2005) Pancreatic β-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54: 1074–1081 [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23: 7448–7459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei K, Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc Natl Acad Sci USA 100: 2432–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS (2006) Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem 281: 7260–7270 [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S (1996) Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Lett 395: 143–147 [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ (2001) Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21: 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E (2004) Translocation of Bim to the endoplasmic reticulum (ER) mediates ER stress signaling for activation of caspase-12 during ER stress-induced apoptosis. J Biol Chem 279: 50375–50381 [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J (2000) Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 403: 98–103 [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Saitoh M, Mochida Y, Takeda K, Nakano H, Rothe M, Miyazono K, Ichijo H (1998) ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 2: 389–395 [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H (2002) ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev 16: 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H (2005) TRB3, a novel ER stress-inducible gene, is induced via ATF4–CHOP pathway and is involved in cell death. EMBO J 24: 1243–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oono K et al. (2004) JAB1 participates in unfolded protein responses by association and dissociation with IRE1. Neurochem Int 45: 765–772 [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH (2003) Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol 162: 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, Ulevitch RJ, Green DR, Nicholson DW (2006) Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440: 1064–1068 [DOI] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ (2005) The mammalian unfolded protein response. Annu Rev Biochem 74: 739–789 [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A (2003) Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann NY Acad Sci 1010: 186–194 [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D (2000) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: 664–666 [DOI] [PubMed] [Google Scholar]
- van Huizen R, Martindale JL, Gorospe M, Holbrook NJ (2003) P58IPK, a novel endoplasmic reticulum stress-inducible protein and potential negative regulator of eIF2α signaling. J Biol Chem 278: 15558–15564 [DOI] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, Jolicoeur EM, Kuroda M, Ron D (1998) Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J 17: 5708–5717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG (2002) Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc Natl Acad Sci USA 99: 15920–15925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M (2001) Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. J Biol Chem 276: 13935–13940 [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K (2001) XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107: 881–891 [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]