

Introduction
In 1967, Earl Sutherland described the simplest possible model for signalling through heptahelical receptors when he wrote, “It seems likely that in most, and perhaps all, tissues the β receptor and adenylyl cyclase are the same” (Robison et al, 1967). Within ten years, Rodbell and Gilman had disproved this model by showing that heterotrimeric guanine nucleotide-binding proteins (G proteins) were transducers of the receptor-mediated signal (Rodbell et al, 1971; Ross & Gilman, 1977). During the past 30 years, our knowledge of the structure, function and regulation of these G-protein-coupled receptors (GPCRs) has continued to grow in size, scope and complexity.

This Keystone symposium on G-Protein-Coupled Receptors: Evolving Concepts and New Techniques took place between 12 and 17 February 2006, in Keystone, Colorado, and was organized by P. Insel, G. Milligan and D. Perez.
Today, more than 800 genes (3% of the human genome) have been shown to encode heptahelical receptors for various extracellular stimuli, including hormones, neurotransmitters, chemokines, light, odours and tastes, and they are the therapeutic targets for half of all prescribed drugs. Although the predominant role of these receptors is G-protein activation, GPCRs are increasingly being recognized as scaffolds for the formation and localization of signalling complexes in the cell. Indeed, the magnitude and manner of the receptor response is determined by the complex relationship between the ligand, receptor, G protein and other associated proteins.
This Keystone meeting focused on recent advances in our understanding of the structure, function and regulation of GPCRs: in particular, the functional consequences of receptor oligomerization, the identification of ligands for orphan receptors, and the characterization of desensitization and downstream signalling pathways. In this report, we highlight recent scientific accomplishments in the field of GPCR function as well as the novel technologies and strategies that enabled them to be made.
Structural basis of receptor activation
Each member of the GPCR superfamily has a similar structural design with seven transmembrane-spanning α-helices, an extracellular amino-terminus, intracellular carboxy-terminus, and three interhelical loops on each side of the membrane. In 2000, Palczewski and colleagues solved the first crystal structure of a GPCR: rhodopsin bound to the inverse agonist 11-cis-retinal (Palczewski et al, 2000). Despite extensive efforts by many investigators, rhodopsin remains the only GPCR for which atomic resolution is available. Although this structure has provided a basis for current models of ligand binding and receptor activation, many questions remain about ligand interactions in other receptor families.
To study the structural features of ligand binding in the M1-muscarinic receptor, E. Hulme (London, UK) used alanine-scanning mutagenesis interpreted in the context of a rhodopsin-based homology model. These studies have indicated that closure of aromatic residues around the tetramethylammonium headgroup of acetylcholine might be a fundamental component of the activation mechanism. However, additional understanding of ligand binding in this receptor will require moving beyond the rhodopsin homology model to crystal structures. Now, Hulme and colleagues are developing M1-muscarinic receptor mutants that have increased stability in the short-chain detergents and that are most compatible with the formation of a crystal lattice.
Extending the use of rhodopsin homology models beyond the sub-family of rhodopsin-like receptors to those GPCRs with different modes of ligand binding, such as the peptide-binding cholecystokinin receptors, has proven to be more difficult. Beginning with a rhodopsin homology model for these receptors, L. Miller's group (Scottsdale, AZ, USA) used site-directed mutagenesis, photoaffinity labelling and structure–activity relationships to understand the basis of ligand–receptor interactions. Interestingly, these studies showed that the mode of ligand binding and activation differs between the closely related type A and type B cholecystokinin receptors. Activation of these receptors is also sensitive to the membrane lipid composition. Alterations in the cholesterol levels reduced agonist binding and receptor signalling, and sphingolipid depletion resulted in abnormal receptor transport, which might correlate with the clinical manifestations of dyslipidaemia and obesity, such as gallstone formation.
W. Hendrickson (New York, NY, USA) presented another mechanism of receptor activation that he has deduced using crystallographic studies of the extracellular domain of the follicle stimulating hormone receptor and its ligand, as well as for the 5HT2C receptor. Hendrickson showed that the extracellular domain of the follicle stimulating hormone receptor is a solenoid that binds to its ligand in a perpendicular manner, orientating it so that the α-tip of the ligand can interact with the extracellular surface of the receptor, thus causing activation. The crystal structure of the ligand–receptor complex revealed dimers of the ligand-binding domain, which also form in solution (Fan & Hendrickson, 2005). The functional role of receptor dimerization (or oligomerization) was investigated further using the 5HT2C receptor. Using cysteine mutagenesis on the extracellular portions of the transmembrane helices, Hendrickson and colleagues were able to trap dimers of the receptor with disulphide bonds. This cross-linking showed that the interface occurred between transmembrane helices IV and V and was modulated by 5HT2C binding to the receptor. When either of the receptors in the dimer was mutated to block 5HT2C binding, the extent of dimerization decreased. However, a receptor-Gα fusion was able to promote full dimerization depending on which member of the dimer contained the fusion protein and on the length of the linker between the receptor and the G protein. These studies are broadly compatible with the functional non-equivalence of hormone-binding sites within dimers of both categories of receptor that leads to half-site binding and cis-activation.
After receptor activation, the α-subunit of the G protein binds to the receptor (Fig 1), which results in a conformational change that decreases the affinity of the G protein for its bound guanosine diphosphate (GDP; Hamm, 1998). The mechanism underlying the activation step of the G protein—that is, the exchange of GDP for guanosine triphosphate (GTP)—has remained elusive due to the technological limitations of attaining crystal structures of the receptor–G-protein complex. W. Oldham (Nashville, TN, USA) and colleagues are addressing this question with the use of site-directed spin labelling and electron paramagnetic resonance spectroscopy to observe conformational changes directly in the Gα-subunit leading to the release of GDP in the receptor-bound state. This approach has enabled the direct observation of this complex and has begun to answer the crucial question of how G proteins are activated by their receptors. With the use of double electron-electron resonance spectroscopy, it should be possible to improve on current models of receptor–G-protein complex formation until such time as the crystal structure is solved.
Figure 1.
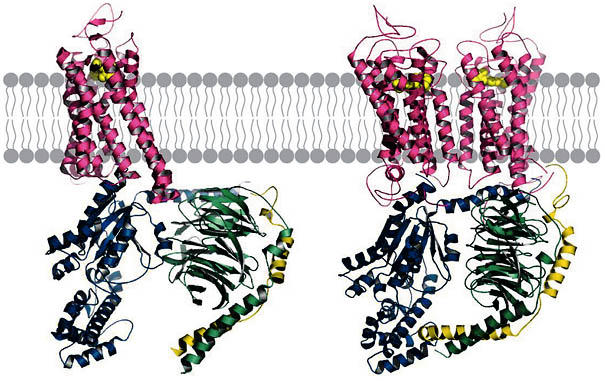
Models of the receptor–G-protein complex. Two representations of receptor–G-protein complexes are shown. On the left (R1·Gα(O) βγ complex) is a representative model of classical GPCR–G-protein interactions in which one GPCR interacts with one G protein. On the right is a representation of an alternative model that depicts the ability of a heterotrimeric G protein to interact with a GPCR dimer complex (R2·Gα(O) βγ complex) on the basis of coordinates generously provided by K. Palczewski (Filipek et al, 2004).
GPCR dimerization and therapeutics
GPCRs were first observed to form both homo- and heterodimers several years ago (Fig 1). Initial structural and functional studies identified a potential for differential signalling among GPCR family members on the basis of their tissue distribution, dimerization profile and mode of activation. Taking advantage of what is known about monomeric receptor signalling, numerous laboratories have sought to address the mechanisms by which heterodimers can function to cross-regulate a signalling cascade whereby the regulation of one receptor might result in modulation of the activity of another GPCR.
Two clever experimental approaches were presented by J.-P. Pin (Montpelier, France) and J.L. Banères (Montpelier, France) to address the contributions of each monomer to the heterodimer complex. Pin showed that the quality control system of the γ-aminobutyric acid B (GABAB) receptor can be used to enhance the cell-surface expression of a specific pair of metabotropic glutamate receptors. When only one member of the dimer can bind to glutamate, the dimer cannot fully activate the G protein. In truncated receptors lacking the extracellular domain, the recently identified positive and negative allosteric modulators of metabotropic glutamate receptors function as agonists and inverse agonists, respectively. Although two negative allosteric modulators are necessary to inhibit receptor activity, only one positive allosteric modulator is required to enhance signalling. A construct in which a negative allosteric modulator targeted at a heptahelical domain disabled for G-protein signalling by a mutation in the i3 loop actually became a positive allosteric modulator of the non-disabled subunit, which suggests that activation of only one helical domain occurs before G protein activation. Banères presented a method by which different tags are used to isolate leukotriene B4 receptor BLT1 and BLT2 heterodimers. Through the creation of a fluorescent probe with site-specific mutations of residues to a 5-hydroxytryptophan, different excitation spectra for each receptor were generated showing that ligand binding to one receptor causes a conformational change in the other receptor. In addition, full G-protein activation was achieved with one ligand per dimer (Fig 1). Banères also showed that agonist-free binding of G protein to the second receptor results in a restriction of the full, activating conformational change of the receptor.
Another example of heterodimer regulation was presented by L. Devi (New York, NY, USA), who discussed the mechanism by which opioid receptor dimerization modulates analgesic effects. Using bioluminescence resonance energy transfer, Devi showed that the μ and δ opioid receptors interact. Furthermore, co-immunoprecipitation experiments and antibodies targeted to the μδ heterodimer revealed that the receptors dimerize in specific locations in the brain (prefrontal cortex, nucleus accumbens and the ventral tegmental area). Devi and colleagues were then able to show that antagonizing the δ receptor resulted in potentiation of the μ-induced analgesia. μδ heterodimers showed a prolonged agonist-induced phosphorylation of extracellular-signal-regulated kinase (ERK) in the cytosol as well as recruitment of β-arrestin to the receptor complex compared with μ alone. These studies delineate a specific heterodimer target for increased analgesia, possibly without the addictive properties observed in current therapeutic targets for the attenuation of chronic pain.
A novel type of functional selectivity elicited by heterodimerization was described by S. George (Toronto, Canada). Her group is trying to determine how dopamine is able to induce Ca2+ changes in some neuronal systems when the individual receptor subtypes alone are unable to elicit this physiological correlate—D2, D3 and D4 decrease cAMP, whereas D1 and D5 increase cAMP. Through both co-immunoprecipitation and fluorescence resonance energy transfer, George and colleagues were able to show that the D1 and D2 receptors co-localize in the same complex in close proximity to one another. Interestingly, agonists to both receptors co-expressed in cells resulted in an intracellular Ca2+ transient not observed with agonists to either receptor alone. George also showed that pharmacological inhibitors of phospholipase C—but not of protein kinase A or C—resulted in inhibition of the Ca2+ transient. Furthermore, co-internalization was promoted by agonist occupation of either D1 or D2. The data presented indicate clearly that heterodimerization can induce a functional switch in signalling, which is not recapitulated by either receptor alone.
J. Whistler (San Francisco, CA, USA) discussed how functional selectivity results in differential conformations of active μ-opioid receptor. DAMGO, the μ-selective agonist, induced a conformational change in the μ-receptor such that its C-terminal tail could bind to arrestin, thus facilitating endocytosis. Morphine was unable to induce this conformational change, resulting in a longer activation time for each receptor coupled to a decrease in receptor endocytosis. Whistler also showed that a mutant μ-receptor containing an extended C-terminal tail showed an increase in endocytosis coupled to a decrease in tolerance and compensation. These findings indicate that combination therapy, through increased endocytosis of the receptor, might lead to a decrease in pain-killer addiction by reducing the tolerance to morphine.
A functional theme that was eloquently described by Pin, Devi, Whistler and George was that to understand and thus modulate the various endogenous GPCR signalling systems, we must first identify the signalling unit that regulates the physiological responses. In some cases, each receptor acts as an individual signalling unit, in other conditions one receptor can modulate the activity of another, and in yet another case, when two receptors are dimerized, the physiological signal might be independent of what is observed when either receptor signals alone. The question of the functional unit of signalling has become more complex during the past 10 years and the identification of these signalling units will allow for a more defined approach for therapeutic intervention.
Orphan receptors
The identification of orphan receptor structure and function was a resounding theme throughout the meeting as the number of receptors identified with no known ligand has continued to expand (Civelli, 2005). S. Offermanns (Heidelberg, Germany) discussed how a recently discovered orphan receptor known as GPR109A is activated by nicotinic acid. Furthermore, he described how this receptor is coupled to the inhibition of stimulatory G (Gs) signalling by the inhibitory G (Gi) subunit in the adipocyte. GPR109A is thus able to regulate the production of circulating free fatty acids resulting in anti-lipolytic effects in adipocytes. As nicotinic acid is clinically used as an anti-dyslipidaemic drug, understanding how GPR109A functions is crucial for the development of more specific drugs with fewer side-effects. Offermans showed, through knockout studies of the mouse homologue PUMA-G, that the nicotinic acid receptor has cell-type-specific physiological effects and that the unwanted ‘flushing' effect observed after the administration of niacin is due to cutaneous vasodilation through a prostaglandin pathway (Benyo et al, 2005). Additionally, he suggested that 3-hydroxy butyrate and acetoacetate, rather than nicotinic acid, might be the endogenous agonists, and that this receptor might help to maintain homeostasis under conditions of starvation.
While investigating GPR50, an orphan receptor in the brain thought to be involved in mental disorders, R. Jockers (Paris, France) was able to determine not only that GPR50 heterodimerizes with the melatonin receptor MT, but also that this orphan receptor specifically inhibits agonist binding and G-protein signalling of MT1 but has no effect on MT2. Jockers showed that the C-terminal tail of GPR50 is responsible for the inhibition of MT1 signalling, as the truncation of 300 amino acids from the GPR50 C-terminus alleviated the observed inhibition of MT1 agonist binding and resultant cellular signalling. Jockers hypothesized that GPR50 disrupts the interaction of MT1 with regulatory proteins. As indicated earlier, oligomerization of GPCRs might result in modulation of receptor activity thus allowing for the wide array of physiological responses that are thought to be both receptor- as well as cell-specific.
A high-throughput screening approach for the identification of ligands for orphan receptors was used by G. Tsujimoto (Kyoto, Japan) to study free fatty-acid receptors. By expressing GPR120 in HEK 293 cells, Tsujimoto was able to determine that long chain-free fatty acids (especially linolenic acid) induced internalization of the receptor as well as Ca2+ mobilization, ERK activation and secretion of glucagon-like peptide 1 (GLP1). Through expression of orphan receptors in a cell-based system of physiological markers, Tsujimoto has shown the usefulness of applying high-throughput screening to identify endogenous ligand–receptor interactions.
GPCR signalling and desensitization
Receptor desensitization is of great interest to clinical researchers trying to identify how receptor signalling is regulated by both endogenous as well as exogenous agonist-mediated activation. The meeting had many insightful talks focusing on this topic. J. Benovic (Philadelphia, PA, USA) opened the session by discussing the regulation of β-arrestin 1 (ARRB1 and receptor endocytosis. ARRB1 undergoes a conformational change on binding to a GPCR that opens up its C-terminus. This conformational change is necessary for the interaction of ARRB1 with its numerous binding partners. Benovic also showed that inositol hexaphosphate, which binds to the N- and C-termini of ARRB1, has a much higher binding affinity to ARRB1 than does phosphatidylinositol bisphosphate, which can only bind to its C-terminus. Phosphatidylinositol phosphate binding to ARRB1 induces oligomerization as well as localization of ARRB1. Although binding to inositol hexaphosphate does not interfere with ARRB1 oligomerization or its binding to other partners such as clathrin, adaptin and ERK, inositol hexaphosphate inhibits ARRB1 binding to GPCRs as it becomes localized to the nucleus instead of the plasma membrane where the GPCR resides. Therefore, binding to phosphatidylinositol bisphosphate is crucial for the normal localization of the ARRB1 to the GPCR on the cell surface.
Perspectives on GPCR structure and function
This meeting provided an opportune time to reflect on current progress in both the mechanism of receptor activation and the functional consequences of this activation. Many advances have been made in identifying the mode of receptor activation through novel and innovative techniques. Additionally, we will most likely see articles published on the crystal structures of GPCRs other than rhodopsin in the near future, as well as a structure of the GPCR–G-protein interface. Numerous talks focused on how GPCRs signal through both homo- and heterodimers and that this dimerization results in more complex signalling than previously thought. Finally, great strides have been made towards identifying the ligands and signalling pathways of several orphan receptors with the aid of new technologies such as high-throughput screening coupled to the more traditional approaches of transgenic and knockout studies in the mouse. These advances will allow for a greater understanding that should enable the development of the next generation of therapeutics for the treatment of disease.

William Oldham, Heidi E. Hamm & Michael Holinstat
Acknowledgments
We thank the organizers for making this symposium possible and acknowledge all of the participants who shared their work and ideas but were omitted from this review due to space constraints.
References
- Benyo Z, Gille A, Kero J, Csiky M, Suchankova MC, Nusing RM, Moers A, Pfeffer K, Offermanns S (2005) GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest 115: 3634–3640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civelli O (2005) GPCR deorphanizations: the novel, the known and the unexpected transmitters. Trends Pharmacol Sci 26: 15–19 [DOI] [PubMed] [Google Scholar]
- Fan QR, Hendrickson WA (2005) Structure of human follicle-stimulating hormone in complex with its receptor. Nature 433: 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek S, Krzysko KA, Fotiadis D, Liang Y, Saperstein DA, Engel A, Palczewski K (2004) A concept for G protein activation by G protein-coupled receptor dimers: the transducin/rhodopsin interface. Photochem Photobiol Sci 3: 628–638 [DOI] [PubMed] [Google Scholar]
- Hamm HE (1998) The many faces of G protein signaling. J Biol Chem 273: 669–672 [DOI] [PubMed] [Google Scholar]
- Palczewski K et al. (2000) Crystal structure of rhodopsin: a G protein-coupled receptor. Science 289: 739–745 [DOI] [PubMed] [Google Scholar]
- Robison GA, Butcher RW, Sutherland EW (1967) Adenyl cyclase as an adrenergic receptor. Ann NY Acad Sci 139: 703–723 [DOI] [PubMed] [Google Scholar]
- Rodbell M, Birnbaumer L, Pohl SL, Krans HM (1971) The glucagon-sensitive adenyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem 246: 1877–1882 [PubMed] [Google Scholar]
- Ross EM, Gilman AG (1977) Resolution of some components of adenylate cyclase necessary for catalytic activity. J Biol Chem 252: 6966–6969 [PubMed] [Google Scholar]