

Abstract
Although neurotrophins have been postulated to have antidepressant properties, their effect on anxiety is not clear. We find that transgenic overexpression of the neurotrophin BDNF has an unexpected facilitatory effect on anxiety-like behavior, concomitant with increased spinogenesis in the basolateral amygdala. Moreover, anxiogenesis and amygdalar spinogenesis are also triggered by chronic stress in control mice but are occluded by BDNF overexpression, thereby suggesting a role for BDNF signaling in stress-induced plasticity in the amygdala. BDNF overexpression also causes antidepressant effects, because transgenic mice exhibit improved performance on the Porsolt forced-swim test and an absence of chronic stress-induced hippocampal atrophy. Thus, structural changes in the amygdala and hippocampus, caused by genetic manipulation of the same molecule BDNF, give rise to contrasting effects on anxiety and depressive symptoms, both of which are major behavioral correlates of stress disorders.
Keywords: amygdala, chronic stress, hippocampus, morphology, dendritic spines
Prolonged and severe stress is associated with various cognitive and affective behavioral abnormalities. The hippocampus, which plays an important role in the formation of declarative memories, has been the primary focus of research on the cognitive effects of chronic stress (1). From a cell biological perspective, a reduction in the length and complexity of dendritic arbors of hippocampal CA3 pyramidal neurons is a well characterized morphological feature of various rodent models of repeated stress (2). This stress-induced dendritic remodeling, in turn, has been hypothesized to underlie the hippocampal volume loss (3, 4) seen in patients suffering from various stress-related disorders, including major depression, Cushing’s syndrome, and posttraumatic stress disorder (5–7). Recent evidence has pointed to neurotrophins as a cause for these changes and have led to the “neurotrophic hypothesis,” which states that symptoms associated with depression, both pathological and behavioral, are a result of decreased neurotrophic support, and conversely, that increasing neurotrophic support would lead to the resolution of these symptoms (8, 9).
Although hippocampal plasticity has, over the years, provided a useful framework for studying the cognitive effects of stress at multiple levels of neural organization, the affective aspects of stress disorders, such as increased anxiety, are likely to involve the amygdala, which plays a pivotal role in processing aversive experiences (10, 11). Interestingly, in contrast to the hippocampus, which inhibits the hypothalamic–pituitary–adrenal (HPA) axis, the amygdala stimulates it (12). In addition, pathological anxiety disorders are associated with an increase in amygdalar volume and output (13–16). Analogously, in rodents, the same chronic immobilization stress (CIS) that causes hippocampal atrophy can elicit dendritic growth in the basolateral amygdala (BLA), as well as enhanced anxiety-like behavior (17–19). These crucial differences between the hippocampus and amygdala indicate that previous studies examining the hippocampus have provided an incomplete view of stress-related disorders. In contrast to the hippocampus, the roles of neurotrophins in chronic stress-induced modulation of amygdalar pyramidal cell structure and anxiety-like behavior have not been resolved. Hence, we sought to extend the neurotrophic hypothesis into anxiety disorders, which share considerable comorbidity with depression (14, 20, 21). To this end, we used genetically modified mice overexpressing BDNF in excitatory neurons of the forebrain (22), including the hippocampus, cortex, and amygdala. Because many antidepressants are also effective in reducing anxiety (23), we predicted that genetic overexpression of BDNF would also protect against chronic stress-induced anxiety.
Results
Transgenic Mice Overexpress BDNF in the Hippocampus and Amygdala.
To verify that BDNF-overexpressing transgenic mice have up-regulated BDNF levels in the hippocampus and basolateral amygdala (BLA), we conducted ELISA analysis on tissue isolated from the two regions. In parallel, Western blot analysis using antibodies against the BDNF receptor trkB was also performed to determine whether up-regulation of BDNF led to any compensatory changes in trkB. There were significantly higher levels of BDNF in both the hippocampus (controls, 0.43 ± 0.03 pg/μg protein; transgenics, 3.4 ± 0.4 pg/μg protein; n = 6; P < 0.01) and BLA (controls, 0.29 ± 0.04 pg/μg protein; transgenics, 3.6 ± 1.5 pg/μg protein; n = 6; P < 0.01) of transgenic animals. Although there was a trend toward higher trkB levels in the hippocampus, there was no statistically significant difference between trkB levels in the control and transgenic animals in either the hippocampus (controls, 1.0 ± 0.2; transgenics, 1.5 ± 0.3; data normalized to control animals; n = 6, P > 0.2) or the BLA (controls, 1.0 ± 0.2; transgenics, 1.1 ± 0.1; data normalized to control animals; n = 6, P > 0.2). Thus, BDNF levels were higher, without compensatory decreases in trkB levels, in both the hippocampus and the BLA of the transgenic mice.
BDNF-Overexpressing Transgenic Mice Show Increased Anxiety.
As mentioned earlier, BDNF has been identified as a candidate antidepressant, and although depression and anxiety share considerable comorbidity, and many antidepressants also act as anxiolytics, the effect of BDNF on anxiety has not been determined. Thus, we sought to determine the effect of enhanced BDNF signaling on anxiety using chronic stress models known to facilitate anxiety-like behavior in rodents (17, 24–26). To this end, we first examined anxiety-like behavior of control B6 mice exposed to CIS (2 h per day for 10 days), along with unstressed littermate mice, by subjecting them to the open-field test. As shown in Fig. 1a Left, control stressed animals spent significantly less time in the center of the apparatus, confirming data from the literature that chronic stress leads to increased anxiety. To our surprise, levels of anxiety displayed by unstressed transgenic animals were statistically indistinguishable from those of the stressed control animals (Fig. 1a Left). Further, exposure to CIS failed to trigger any additional statistically significant increase in anxiety in the transgenic mice. When we analyzed the behavior of the mice during the course of the entire 10-min session in the open-field test, we found that the unstressed control mice spent increasingly more time in the center of the arena. By contrast, the stressed control and transgenic mice, irrespective of whether they were stressed, occupied primarily the periphery of the arena during the first few minutes and continued to do so during the 10-min session (Fig. 1a Right). Importantly, although CIS increased locomotor activity consistent with data from the literature (27), our results are not likely to be caused by such changes in locomotor activity, because the stressed control and transgenic animals did not display hyperactivity after the first 3 min of the test (Fig. 1b Right), whereas the decreased occupancy of the center of the apparatus by the stressed control, unstressed transgenic, and stressed transgenic mice occurred primarily after those first 3 min had elapsed (Fig. 1a Right).
Fig. 1.

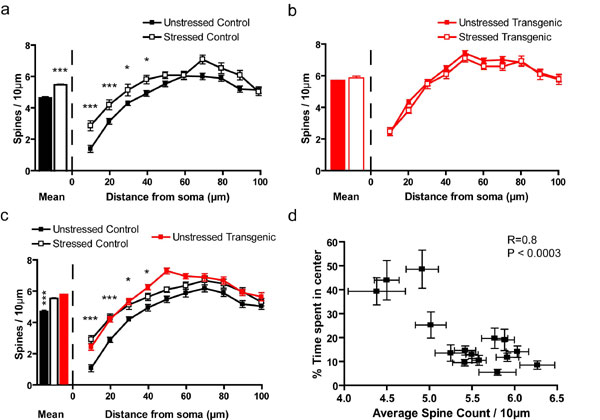
Forebrain BDNF overexpression increases anxiety and spine density in the BLA. (a) CIS causes a decrease in amount of time spent in the center of the open-field apparatus. This is seen both in mean time spent in the center of the apparatus (Left) and when the behavior of the animals is quantified during the entire 10 min of the test (Right). Transgenic mice show the same decrease in time spent in the center of the open field and have no further decrease in time spent in the center when subjected to CIS. (b) CIS causes an increase in locomotor activity, but there is no genotype effect. Furthermore, after 3 min of the test, there is no difference in total movement time between any of the groups, indicating that changes in locomotor activity are not responsible for the differences in anxiety between groups seen in a. (c) Elevated-plus maze data show that transgenic mice spend significantly less time in the open arm compared to control animals (Left). There is no change in activity levels as measured by number of closed-arm entries (Right). (d) Representative photomicrographs of apical dendritic spines from BLA pyramidal neurons of control and transgenic mice, with and without stress. (Scale bar, 10 μm.) (e) In control animals, CIS causes a significant increase in apical spine density, when the mean spine density (Left) along the whole dendrite is considered, or in the distal portion of the dendrite, when segmental analysis is performed (Right). (f) In transgenic animals, there is no significant change in spine density, either in overall mean values (Left) or when segmental analysis is performed (Right). (g) BDNF overexpression leads to the same increase in spine-density as chronic stress. This is shown in both overall mean spine density and segmental analysis (*, P < 0.05; **, P < 0.01; ***, P < 0.001; at the right in a, b, and g, when any statistical significance is noted, it is between the unstressed control group and the least different of the stressed control group and unstressed transgenic group, which are statistically indistinguishable from each other). (h) Correlation analysis shows that mean spine density is strongly correlated with anxiety, as measured by time spent in the center of the open field (R = 0.7, P < 0.01).
As a further confirmation that the transgenic mice exhibited greater anxiety even without exposure to chronic stress, control and transgenic mice were subjected to the elevated plus-maze test. We found that the transgenic mice spent less time in the open arm as compared to the controls (Fig. 1c Left). Importantly, there was no difference in the number of closed-arm entries (Fig. 1c Right), indicating that differences in locomotor activity were not responsible for the observed differences between the two groups. Thus, results obtained from the open-field and the elevated plus-maze paradigms both demonstrated greater anxiety in the BDNF-overexpressing mice relative to control animals. Importantly, the BDNF-overexpressing animals had the same level of anxiety as stressed control animals and were not susceptible to any further increase in anxiety upon exposure to chronic stress.
BDNF-Overexpressing Transgenic Mice Show Increased BLA Spine Density.
We next sought to elucidate the cellular substrate behind the enhanced anxiety. As mentioned earlier, the amygdala, a critical component of the neural circuitry underlying fear, is activated by chronic stress. Furthermore, previous studies have reported that chronic stress-induced anxiety is accompanied by increases in dendritic area and spine density in the BLA (17–19, 28). Because BDNF promotes dendritic and spine growth (29), we postulated that BDNF overexpression may mimic the effects of chronic stress on amygdalar neuronal morphology, and that this structural remodeling may serve as a substrate for the enhanced anxiety displayed by the transgenic animals. To quantify the structural changes, we performed morphometric analysis of Golgi-impregnated (30) spiny pyramidal neurons (Fig. 1d) of the BLA. Compared to unstressed control mice, there was a significant increase in BLA apical dendritic spine density in stressed control mice (Fig. 1e Left). A more detailed segmental analysis (Fig. 1e Right) showed a trend toward higher spine density along the entire length of the apical dendrite in stressed animals, with statistically significant changes in the distal portion of the dendrite. There was also a significant increase in basal dendrite spine density. Segmental analysis showed an increase in spine density in BLA pyramidal neurons derived from stressed animals along the entire length of the basal dendrite (Fig. 4a, which is published as supporting information on the PNAS web site), with statistically significant changes in the proximal portion of the dendrite. This confirms previous results, obtained in rats, on enhanced synaptic connectivity of excitatory neurons located at the input interface of the amygdala as a result of chronic stress (28).
However, the BDNF-overexpressing transgenic mice showed no chronic stress-induced changes in apical dendritic spine density either in overall mean spine density or when more detailed segmental analysis of dendrites was used (Fig. 1f). There was also no difference between the unstressed and stressed transgenic animals with respect to spine density of the basal dendrites (Fig. 4b). Interestingly, the spine density of both the unstressed and stressed transgenic animals was the same as that of the stressed control animals, when both overall mean spine density (Fig. 1g Left) and individual segments were examined, in the apical (Fig. 1g Right) as well as the basal dendrites (Fig. 4c). Thus, paralleling our observations on anxiety-like behavior described earlier, both chronic stress and BDNF overexpression led to enhanced spine density in BLA pyramidal neurons, such that BDNF overexpression occluded any additional stress-induced increase in BLA spine density. This is consistent with the view that chronic stress mediates its effects on anxiety by BLA spinogenesis, and that this process shares common molecular mechanism with the BDNF pathway.
If BLA spinogenesis is indeed the substrate underlying enhanced anxiety seen in the stressed control and BDNF-overexpressing animals, a comparison of the spine density measured in individual animals with the anxiety exhibited by them should reflect a correlation. Behavioral and cellular data obtained from animals in all four experimental groups displayed a strong correlation between mean spine density on BLA apical dendrites and behavioral anxiety (Fig. 1h), supporting a role for increased spine density in the facilitation of anxiety triggered by both chronic stress and BDNF overexpression. There was also a strong correlation of anxiety with overall mean spine density of the basal dendrites (Fig. 4d).
In contrast, there was no difference in dendritic arborization of BLA spiny pyramidal neurons between the four groups; neurons derived from unstressed control, stressed control, unstressed transgenic, and stressed transgenic animals had the same total dendritic length and number of branch points along the entire length of the apical dendrite (Fig. 5 a–c, which is published as supporting information on the PNAS web site). This result is consistent with a recent report in rats demonstrating that stress can lead to spinogenesis, as well as enhanced anxiety, even in the absence of dendritic remodeling (28).
Anxiogenic Effect of BDNF Overexpression Is Not the Result of Increased Stress Response.
We next decided to examine the mechanism by which BDNF mimics the facilitatory effects of chronic stress on anxiety and BLA spine formation. Specifically, two alternate hypotheses were tested. First, in view of the potent role of the HPA axis and glucocorticoids in stress-induced modulation of neuronal morphology and behavior (31), it is possible that the role of BDNF is to increase the stimulatory effect of the amygdala on the HPA axis, thereby increasing the secretion of stress hormones and causing chronic stress-induced changes throughout the brain. The alternate explanation would entail a role for BDNF that is downstream of glucocorticoid secretion, thereby playing a more direct role within the amygdala.
We differentiated between these two hypotheses by measuring, 1 day after the end of the 10-day CIS protocol, the blood plasma levels of adrenocorticotropic hormone (ACTH) and corticosterone, two common indicators of HPA axis activation. Fig. 2a shows that there was no significant difference in ACTH concentration between unstressed control and unstressed transgenic animals. CIS increased the levels of ACTH in both control and transgenic animals, but there was no significant effect of genotype. Although corticosterone levels were slightly, but significantly, lower in unstressed transgenic animals as compared to unstressed control animals, there was no difference between stressed control and stressed transgenic mice (Fig. 2b). Thus, BDNF overexpression in forebrain excitatory cells has no effect on stress hormone levels in animals that have undergone chronic stress.
Fig. 2.

Forebrain BDNF overexpression does not affect commonly used endocrine indicators of stress level in response to CIS. (a and b) Forebrain BDNF overexpression does not affect chronic stress-induced increases in blood plasma concentrations of ACTH (a) and corticosterone (b). (a) In unstressed animals, there is no significant difference between control and transgenic animals in ACTH concentration. Chronically stressed animals have a significant increase in ACTH concentration compared to unstressed animals, but again there is no significant genotype difference. (b) In unstressed animals, there is a significant difference between control and transgenic animals in corticosterone levels. Chronically stressed animals have a significant increase in corticosterone concentration, but there is no genotype effect. (c) There is no difference in body weight between unstressed control and unstressed transgenic mice. (d and e) Forebrain BDNF overexpression does not affect chronic stress-induced weight loss. Unstressed control and transgenic animals show no change in weight, whereas stressed control and transgenic animals show significant weight loss at both 6 days (d) and 11 days (e) after the onset of chronic stress. There is no genotype effect in either unstressed or stressed animals (*, P < 0.05 between unstressed and stressed animals; ‡, P < 0.05 in a comparison between unstressed control and unstressed transgenic animals).
The stress hormone data indicate that the increase in anxiety and BLA spine density observed in the transgenic mice is not caused by an increase in the levels of stress due to hyperactivity of the HPA axis. This conclusion was strengthened by analysis of body weight, because loss in body weight is another key measure of stress. We observed no difference in body weight between the transgenic and control mice in the absence of stress (Fig. 2c). When we analyzed the temporal profile of weight loss that the animals undergo during the course of 10 days of CIS, a significant decrease in the body weight of both control and transgenic animals was evident both 6 and 11 days after the beginning of the stress protocol (Fig. 2 d and e). However, there was no statistically significant genotype effect observed in the experiment. Thus, these data, taken together with the neuroendocrine data, indicate that the anxiogenic effects of BDNF in the transgenic mice are not due to variations in stress levels between mice of the two genotypes, as measured by common indicators of stress.
BDNF Overexpression Prevents Chronic Stress-Induced CA3 Dendritic Atrophy.
Our results described above showed that genetic overexpression of BDNF can mimic the effects of repeated behavioral stress, which is in stark contrast to the neurotrophic hypothesis (8, 9) that predicts that BDNF overexpression should counteract the effects of chronic stress. Because the neurotrophic hypothesis was formulated with a focus on depressive symptoms and chronic stress-induced hippocampal damage, rather than anxiety behavior and amygdalar hypertrophy, we wanted to examine whether the original neurotrophic hypothesis still holds in our BDNF-overexpressing transgenic mice. For this purpose, we focused on a well established cellular correlate of stress-induced plasticity in the hippocampus, dendritic remodeling of CA3 pyramidal neurons after exposure to chronic stress (2). We subjected the brains from unstressed control, stressed control, unstressed transgenic, and stressed transgenic animals to Golgi–Cox histology (30) 1 day after the end of the CIS protocol. As depicted in Fig. 3a, and in agreement with earlier studies in rats (2), CIS induced a significant atrophy of the apical dendritic tree of CA3 pyramidal cells in control animals, when total dendritic length (Fig. 3b Left) or number of branch points (Fig. 3c Left) was quantified. Scholl’s analysis indicated, as shown in Fig. 3 b Right and c Right, that the decrease in dendritic length and number of branch points in stressed control animals compared to unstressed control animals were most pronounced only in apical dendritic segments >50 μm away from the soma, corresponding to the stratum radiatum of area CA3. In contrast, there was no change in either the dendritic length or number of branch points in the basal dendrites, which is in agreement with earlier studies using rats (2).
Fig. 3.

Forebrain BDNF overexpression prevents chronic stress-induced dendritic atrophy in the hippocampus and reduces immobility in the Porsolt forced-swim test. (a) Representative camera lucidatracings of CA3 pyramidal neurons from control and BDNF-overexpressing transgenic mice with and without stress. (Scale bar, 50 μm.) (b and c) Effects of CIS on CA3 apical dendritic morphology in control mice. (Left) Mean values for dendritic length (b) and number of branch points (c) in a 50-μm shell as a function of the radial distance from the soma. (Right) These mean values are computed by averaging across incremental steps of 50 μm along the apical dendrites using Sholl’s analysis. (*, P < 0.05; **, P < 0.01; ***, P < 0.001). (d and e) Effects of CIS on CA3 apical dendritic morphology in transgenic mice. (Left) Mean values for dendritic length (d) and number of branch points (e) in a 50-μm shell as a function of the radial distance from the soma. (Right) These mean values are computed by averaging across incremental steps of 50 μm along the apical dendrites using Sholl’s analysis. (f) Forebrain BDNF overexpression improves performance on the Porsolt forced-swim test. On both day 1 and day 2, the transgenic animals show less immobility as compared to control littermates (*, P < 0.05 between control and transgenic animals for each day; †, P < 0.01 between days 1 and 2 for control animals; ‡, P < 0.01 between days 1 and 2 for transgenic animals). (g) Forebrain BDNF overexpression improves performance in the Porsolt forced-swim test when the difference in immobility between days 2 and 1 is considered. CTL, control; TG, transgenic (*, P < 0.05).
Having established the efficacy of the CIS paradigm in eliciting dendritic atrophy in control mice, we next analyzed its impact on littermate transgenic mice. In agreement with the original neurotrophic hypothesis, we found no differences between the morphology of CA3 pyramidal cells in unstressed and stressed transgenic animals (Fig. 3 a, d Left, and e Left), either in dendritic length or number of branch points. Sholl’s analysis also confirmed there was no change in dendritic length or number of branch points anywhere along the length of the dendrite (Fig. 3 d and e).
BDNF-Overexpressing Transgenic Mice Exhibit Improved Performance on Porsolt Forced-Swim Test.
Because we obtained evidence that BDNF overexpression prevents chronic stress-induced hippocampal damage, we next examined whether BDNF overexpression would also improve a behavioral symptom of depression, another key facet of the neurotrophic hypothesis (8, 9). To this end, we used the Porsolt forced-swim test that has been a commonly used behavioral paradigm for depression in rodents, and one that has been shown to be robust in the C57/BL6 strain of mice (32), in which background the transgenic mice were engineered (22). In this test, rodents exposed to an inescapable container of water display an immobile posture after initial escape-oriented behavior. This immobility increases during reexposure to the same container 24 h later. This enhanced immobility, which can be blocked by antidepressant treatment, is thought to represent a state of behavioral despair that is characteristic of depression (32, 33). As shown in Fig. 3f, compared to control mice, the transgenic mice exhibited significantly decreased immobility both on day 1 and day 2 of the forced-swim test. Furthermore, the difference in immobility between the 2 days was significantly lower in the transgenic animals as compared to the control mice (Fig. 3g). There was no difference in locomotor activity between control and transgenic mice (Fig. 1b), indicating that the decreased immobility displayed by the mice was not due to increased hyperactivity. Thus, the decreased immobility shown by the transgenic mice is consistent with an antidepressant effect of BDNF, as proposed by the neurotrophic hypothesis. Hence, increasing BDNF function seems to be able to prevent symptoms associated with depression at both the cellular and behavioral levels.
Discussion
BDNF Overexpression, Amygdalar Structural Plasticity, and Facilitated Anxiety.
Using genetically engineered mice, we have shown that BDNF overexpression can lead to increased anxiety, in a manner mimicking and occluding chronic stress-induced anxiety. Furthermore, this increase in anxiety is paralleled by an increase in BLA spine density, also similar to and occluding the effects of chronic stress. Although our data do not exclude the possibility that the occlusion of chronic stress-induced anxiogenesis may be caused by a “ceiling” effect, whereby no further increase in anxiety is seen after chronic stress in the transgenic animals simply because the animals are incapable of expressing any more anxiety, the strong correlation between spine density and anxiety lends support to our interpretation of the data that anxiety is mediated by increased spine density in the amygdala. Furthermore, this hypothesis is supported by recent evidence from the literature correlating changes in amygdalar spine density with changes in anxiety in rats (28). This increase in amygdalar spine density may be a cause of the increased amygdalar size and functional output, which has been reported by several investigators studying depression and anxiety in humans (13–16).
It has been suggested that the hippocampus might mediate anxiety-like behaviors (34, 35). Because overexpression of BDNF counteracts the effects of chronic stress in the hippocampus, whereas it facilitates anxiety even in the absence of stress, it is unlikely that hippocampal atrophy is the cellular substrate for the anxiety seen in the transgenic mice. In addition, some manipulations that reverse hippocampal atrophy, such as antidepressant treatment and stress-free recovery, fail to prevent stress-induced anxiety and structural plasticity in the amygdala (19, 24). Mice overexpressing BDNF specifically in the hippocampus or amygdala will be useful to gain further insight into this issue. Interestingly, it was recently found that BDNF mRNA is elevated transiently in the basolateral amygdala 2 h after cued fear conditioning (36). This temporally restricted elevation of BDNF mRNA level and BDNF signaling in the amygdala observed after the formation of a cue-specific fear is in contrast to the chronic up-regulation of BDNF protein exhibited by the transgenic mice used here, which may underlie the cue-nonspecific and generalized fear, or anxiety, observed in our transgenic mice.
BDNF Overexpression and the Neurotrophic Hypothesis.
We have also demonstrated in this report that up-regulating BDNF can prevent chronic stress-induced atrophy of hippocampal CA3 pyramidal cells, which has been postulated as the underlying cause of the smaller hippocampal volumes seen in patients suffering from posttraumatic stress disorder, Cushing’s syndrome, and major depression (3–7, 9). In addition, these mice exhibited decreased immobility in the forced-swim test, suggesting they are less susceptible to depression (32, 33). Thus, our results support the neurotrophic hypothesis by demonstrating that BDNF, elevated chronically through genetic manipulation, can act as an antidepressant and protect against stress-induced hippocampal atrophy. In addition, our data provide genetic evidence linking structural plasticity in the hippocampus with depressive behavior.
Interestingly, anxiety disorders and depression share considerable comorbidity, and several antidepressants have been used in the treatment of anxiety. Thus, although the neurotrophic hypothesis was formulated with a focus on depression, it was thought that neurotrophin up-regulation could also be used in the treatment of anxiety. However, our data indicate that, whereas BDNF can ameliorate depressive symptoms, it increases anxiety-like symptoms. Thus, we suggest that BDNF plays different roles in depression and anxiety, namely, locally in the hippocampus to inhibit depressive symptoms and locally in the amygdala to facilitate anxiety-like symptoms. These results with BDNF suggest that, unless antidepressants have a differential effect in the hippocampus and amygdala, they would not be expected to be effective anxiolytic drugs. Consistent with this view, some clinical reports indicate that anxiety actually increases upon initiation of certain types of antidepressant treatments (37). Based on our cellular data, we suggest that differences in the action of such drugs on depression versus anxiety are a consequence of their differential actions on the hippocampus and amygdala.
Forebrain BDNF Overexpression and Its Effects on the HPA Axis.
What are the functional implications of BDNF overexpression with respect to the response to chronic stress? We found there was no difference in HPA axis regulation in the transgenic mice, except for a small decrease in corticosterone levels in unstressed transgenic animals. This indicates that the role of BDNF in ameliorating the debilitating effects of stress is downstream of the stress hormones, and thus the impact of elevated stress hormones can be reversed without affecting the normal endocrine response to stress. This is further supported by the data that body-weight loss, which is governed by the hypothalamus, occurs to a similar extent in both control and transgenic animals. Interestingly, similar to the neuroendocrine and body-weight changes that occur in response to chronic stress, chronic stress-induced increases in locomotor activity also occur in both control and transgenic animals, suggesting that the up-regulation of locomotor activity by chronic stress occurs independently of the morphological changes that take place in the forebrain due to BDNF overexpression. This functional specificity of BDNF’s effects, consistent with the regional specificity of the pattern of overexpression in the transgenic mice, may be important therapeutically, because modulation of stress hormones is required for normal cognitive function (2). In other words, therapeutic interventions that act downstream of stress hormones, such as the BDNF pathway, may allow for a more attractive strategy for resolving depressive symptoms without adversely affecting cognition. However, as our results show, not all of these pathways may be therapeutically useful, because they may have deleterious effects, such as increased anxiety. It is desirable to identify components further downstream of BDNF that are involved more specifically in depression or anxiety.
In conclusion, our data, obtained from a single genetically engineered mouse strain, provide evidence of BDNF-induced facilitation of amygdalar spinogenesis and promotion of anxiety. In addition, our results establish a correlation among chronic BDNF up-regulation at the molecular level, prevention of stress-induced hippocampal atrophy at the cellular level, and improvement in the resolution of depressive symptoms at the behavioral level, thereby providing comprehensive evidence spanning multiple levels of neural organization in support of the neurotrophic hypothesis of depression (8, 9). Our results point to the inherent difference between the hippocampus and amygdala in manifesting the diverse emotional symptoms related to mood disorders. Therefore, elucidation of the mechanisms behind the differential effects of chronic stress and molecules such as BDNF on hippocampal and amygdalar plasticity would provide further insights into novel therapeutic targets that selectively ameliorate the varied symptoms of affective disorders.
Methods
CIS.
Male transgenic and control littermates were divided randomly into stressed and unstressed groups. CIS was performed on the stressed group by using rodent immobilization bags. Blood for hormone level quantification was obtained on day 11, and ACTH and corticosterone measurements were performed by Analytics (Bethesda, MD). All procedures were performed in accordance with National Institutes of Health guidelines and after approval by the Massachusetts Institute of Technology Committee for Animal Care.
Morphological Analysis.
Mice were killed under deep anesthesia and processed for Golgi staining technique as described (38, 39). Ten pyramidal neurons from the CA3 region and five or six pyramidal cells from the BLA were selected from each animal for analysis based on established morphological criteria (40). Sholl’s analysis used a segment diameter of 50 μm for CA3 pyramidal neurons and 20 μm for BLA neurons. For segmental analysis of amygdalar spine density, 8-μm segments were used (39, 41).
Behavior.
In the Porsolt forced-swim test, mice were forced to swim for 5 min on 2 consecutive days in a beaker of water (diameter, 13 cm; temperature, 23–25°C). Mice were marked as immobile if they performed the minimal amount of work required to float for at least 1 sec. For the open-field test, the mice were tested for 10 min by using the Accuscan system (Dayton, OH) according to the manufacturer’s instructions. The elevated-plus maze apparatus consisted of two open arms (30 × 5 cm) and two closed arms (wall height, 10 cm) elevated 60 cm in the air. The test lasted 10 min. Experiments in which control animals exhibited open-arm time < 30% were discarded. At least 10 animals of each genotype were used for each behavioral test, and no animal underwent more than one behavioral test.
ELISA and Western Blot Analysis.
For ELISA analysis of BDNF levels, the hippocampus and amygdala were dissected from mice on day 11 and processed for BDNF ELISA by using the BDNF ELISA Kit according to the manufacturer’s instructions (Promega, Madison, WI). Western blot analysis was performed as described (42).
Statistical Analysis.
For hormone analysis, weight measurements, and pooled morphometric analysis, two-way ANOVA was used with the Tukey post hoc test. For time-based analysis and segmental/Sholl’s analysis, a three-way ANOVA was performed with the time/segment factor used as a within-subject repeated measure. For the ELISA data, Porsolt forced-swim test, and elevated-plus maze tests, a Mann–Whitney test was used. Statistics were performed by using SPSS software (SPSS, Chicago, IL).
Additional Methods.
For additional methods, see Supporting Text, which is published as supporting information on the PNAS web site.
Supplementary Material
Acknowledgments
We are grateful to Ronald Duman and Vidita Vaidya for helpful discussions. We thank Lorene Leiter and Frank Bushard for technical assistance with the Porsolt test, Thomas McHugh for critical reading of the manuscript, and members of the Tonegawa and Chattarji laboratories for helpful discussions. A.G. is a recipient of a Howard Hughes Medical Institute (HHMI) Predoctoral Fellowhip. S.C. is supported by an International Senior Research Fellowship from the Wellcome Trust. This research was supported by HHMI and the RIKEN–MIT Neuroscience Research Center (S.T.) and by the National Center for Biological Sciences (S.C.).
Abbreviations
- CIS
chronic immobilization stress
- BLA
basolateral amygdala
- HPA
hypothalamic–pituitary–adrenal
- ACTH
adrenocorticotropic hormone.
Footnotes
Conflict of interest statement: No conflicts declared.
References
- 1.McEwen B. S., Sapolsky R. M. Curr. Opin. Neurobiol. 1995;5:205–216. doi: 10.1016/0959-4388(95)80028-x. [DOI] [PubMed] [Google Scholar]
- 2.McEwen B. S. Annu. Rev. Neurosci. 1999;22:105–122. doi: 10.1146/annurev.neuro.22.1.105. [DOI] [PubMed] [Google Scholar]
- 3.Sapolsky R. M. Proc. Natl. Acad. Sci. USA. 2001;98:12320–12322. doi: 10.1073/pnas.231475998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sapolsky R. M. Nat. Neurosci. 2002;5:1111–1113. doi: 10.1038/nn1102-1111. [DOI] [PubMed] [Google Scholar]
- 5.Bremner J. D. Curr. Psychiatry Rep. 2002;4:254–263. doi: 10.1007/s11920-996-0044-9. [DOI] [PubMed] [Google Scholar]
- 6.Sheline Y. I., Mittler B. L., Mintun M. A. Eur. Psychiatry. 2002;17(Suppl. 3):300–305. doi: 10.1016/s0924-9338(02)00655-7. [DOI] [PubMed] [Google Scholar]
- 7.Starkman M. N., Giordani B., Gebarski S. S., Berent S., Schork M. A., Schteingart D. E. Biol. Psychiatry. 1999;46:1595–1602. doi: 10.1016/s0006-3223(99)00203-6. [DOI] [PubMed] [Google Scholar]
- 8.Duman R. S., Heninger G. R., Nestler E. J. Arch. Gen. Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 9.Nestler E. J., Barrot M., DiLeone R. J., Eisch A. J., Gold S. J., Monteggia L. M. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- 10.LeDoux J. Cell Mol. Neurobiol. 2003;23:727–738. doi: 10.1023/A:1025048802629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis M., Whalen P. J. Mol. Psychiatry. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- 12.Herman J. P., Cullinan W. E. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- 13.Anand A., Shekhar A. Ann. N.Y. Acad. Sci. 2003;985:370–388. doi: 10.1111/j.1749-6632.2003.tb07095.x. [DOI] [PubMed] [Google Scholar]
- 14.Rattiner L. M., Davis M., Ressler K. J. Neuroscientist. 2005;11:323–333. doi: 10.1177/1073858404272255. [DOI] [PubMed] [Google Scholar]
- 15.MacMillan S., Szeszko P. R., Moore G. J., Madden R., Lorch E., Ivey J., Banerjee S. P., Rosenberg D. R. J. Child Adolesc. Psychopharmacol. 2003;13:65–73. doi: 10.1089/104454603321666207. [DOI] [PubMed] [Google Scholar]
- 16.Tebartz van Elst L., Woermann F., Lemieux L., Trimble M. R. Neurosci. Lett. 2000;281:103–106. doi: 10.1016/s0304-3940(00)00815-6. [DOI] [PubMed] [Google Scholar]
- 17.Vyas A., Chattarji S. Behav. Neurosci. 2004;118:1450–1454. doi: 10.1037/0735-7044.118.6.1450. [DOI] [PubMed] [Google Scholar]
- 18.Vyas A., Mitra R., Shankaranarayana Rao B. S., Chattarji S. J. Neurosci. 2002;22:6810–6818. doi: 10.1523/JNEUROSCI.22-15-06810.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vyas A., Pillai A. G., Chattarji S. Neuroscience. 2004;128:667–673. doi: 10.1016/j.neuroscience.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Mineka S., Watson D., Clark L. A. Annu. Rev. Psychol. 1998;49:377–412. doi: 10.1146/annurev.psych.49.1.377. [DOI] [PubMed] [Google Scholar]
- 21.McEwen B. S. Ann. N.Y. Acad. Sci. 2004;1032:1–7. doi: 10.1196/annals.1314.001. [DOI] [PubMed] [Google Scholar]
- 22.Huang Z. J., Kirkwood A., Pizzorusso T., Porciatti V., Morales B., Bear M. F., Maffei L., Tonegawa S. Cell. 1999;98:739–755. doi: 10.1016/s0092-8674(00)81509-3. [DOI] [PubMed] [Google Scholar]
- 23.Strohle A., Holsboer F. Pharmacopsychiatry. 2003;36(Suppl. 3):S207–S214. doi: 10.1055/s-2003-45132. [DOI] [PubMed] [Google Scholar]
- 24.Conrad C. D., LeDoux J. E., Magarinos A. M., McEwen B. S. Behav. Neurosci. 1999;113:902–913. doi: 10.1037//0735-7044.113.5.902. [DOI] [PubMed] [Google Scholar]
- 25.Adamec R. E., Burton P., Shallow T., Budgell J. Physiol. Behav. 1999;65:723–737. doi: 10.1016/s0031-9384(98)00226-1. [DOI] [PubMed] [Google Scholar]
- 26.Mechiel Korte S., De Boer S. F. Eur. J. Pharmacol. 2003;463:163–175. doi: 10.1016/s0014-2999(03)01279-2. [DOI] [PubMed] [Google Scholar]
- 27.Strekalova T., Spanagel R., Bartsch D., Henn F. A., Gass P. Neuropsychopharmacology. 2004;29:2007–2017. doi: 10.1038/sj.npp.1300532. [DOI] [PubMed] [Google Scholar]
- 28.Mitra R., Jadhav S., McEwen B. S., Vyas A., Chattarji S. Proc. Natl. Acad. Sci. USA. 2005;102:9371–9376. doi: 10.1073/pnas.0504011102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McAllister A. K., Katz L. C., Lo D. C. Annu. Rev. Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 30.Pasternak J. F., Woolsey T. A. J. Comp. Neurol. 1975;160:307–312. doi: 10.1002/cne.901600304. [DOI] [PubMed] [Google Scholar]
- 31.McEwen B. S. Biol. Psychiatry. 2000;48:721–731. doi: 10.1016/s0006-3223(00)00964-1. [DOI] [PubMed] [Google Scholar]
- 32.Cryan J. F., Mombereau C. Mol. Psychiatry. 2004;9:326–357. doi: 10.1038/sj.mp.4001457. [DOI] [PubMed] [Google Scholar]
- 33.Porsolt R. D. Rev. Neurosci. 2000;11:53–58. doi: 10.1515/revneuro.2000.11.1.53. [DOI] [PubMed] [Google Scholar]
- 34.Bannerman D. M., Rawlins J. N., McHugh S. B., Deacon R. M., Yee B. K., Bast T., Zhang W. N., Pothuizen H. H., Feldon J. Neurosci. Biobehav. Rev. 2004;28:273–283. doi: 10.1016/j.neubiorev.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 35.Leonardo E. D., Hen R. Annu. Rev. Psychol. 2006;57:117–137. doi: 10.1146/annurev.psych.57.102904.190118. [DOI] [PubMed] [Google Scholar]
- 36.Rattiner L. M., Davis M., French C. T., Ressler K. J. J. Neurosci. 2004;24:4796–4806. doi: 10.1523/JNEUROSCI.5654-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masand P. S., Gupta S. Harv. Rev. Psychiatry. 1999;7:69–84. [PubMed] [Google Scholar]
- 38.Shankaranarayana Rao B. S., Raju T. R. In: Brain and Behavior. Raju T. R., Kutty B. M., Shankaranarayana Rao B. S., editors. Bangalore, India: National Institute of Mental Health and Neurosciences; 2004. pp. 108–111. [Google Scholar]
- 39.Shankaranarayana Rao B. S., Govindaiah, Laxmi T. R., Meti B. L., Raju T. R. Neuroscience. 2001;102:319–327. doi: 10.1016/s0306-4522(00)00462-0. [DOI] [PubMed] [Google Scholar]
- 40.McDonald A. J. J. Comp. Neurol. 1982;212:293–312. doi: 10.1002/cne.902120307. [DOI] [PubMed] [Google Scholar]
- 41.Shankaranarayana Rao B. S., Madhavi R., Sunanda, Raju T. R. Curr. Sci. 2001;80:653–659. [Google Scholar]
- 42.Kelleher R. J., III, Govindarajan A., Jung H. Y., Kang H., Tonegawa S. Cell. 2004;116:467–479. doi: 10.1016/s0092-8674(04)00115-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}