

Abstract
Obesity endangers the lives of millions of people worldwide, through comorbidities such as heart disease, cancers, type 2 diabetes, stroke, arthritis, and major depression. New approaches to control body weight remain a high priority. Vaccines traditionally have been used to protect against infectious diseases and, more recently, for unconventional targets such as drug addiction. Methodologies that could specifically modulate the bioavailability of an endogenous molecule that regulates energy balance might provide a new foundation for treating obesity. Here we show that active vaccination of mature rats with ghrelin immunoconjugates decreases feed efficiency, relative adiposity, and body weight gain in relation to the immune response elicited against ghrelin in its active, acylated form. Three active vaccines based on the 28-aa residue sequence of ghrelin, a gastric endocrine hormone, were used to immunize adult male Wistar rats (n = 17). Synthetic ghrelin analogs were prepared that spanned residues 1–10 [ghrelin (1–10) Ser-3(butanoyl) hapten, Ghr1], 13–28 [ghrelin (13–28) hapten, Ghr2], and 1–28 [ghrelin(1–28) Ser-3(butanoyl) hapten, Ghr3], and included n-butanoyl esters at Ser-3. Groups immunized with Ghr1 or Ghr3 showed greater and more selective plasma binding capacity for the active, Ser-3-(n-octanoyl) form of ghrelin as compared with Ghr2 or keyhole limpet hemocyanin vaccinated controls. Accordingly, they gained less body weight, with sparing of lean mass and preferential reduction of body fat, consistent with reduced circulating leptin levels. The ratio of brain/serum ghrelin levels was lower in rats with strong anti-ghrelin immune responses. Effects were not attributable to nonspecific inflammatory responses. Vaccination against the endogenous hormone ghrelin can slow weight gain in rats by decreasing feed efficiency.
Keywords: feed efficiency, ghrelin, obesity, body weight regulation, vaccine
Approximately 1 billion people worldwide are overweight or obese (body mass index = 25–29.9 or ≥30 kg/m2, respectively), with disproportionately higher prevalence rates in affluent countries (1). For example, in the United States, the National Health and Nutrition Examination Survey (NHANES) found that, in 2003–2004, ≈66% of all American adults 20 years of age or older were overweight or obese. Almost 4 of every 5 adult men aged 40–59 were so classified (2). Even in children and adolescents between the ages of 6–11 and 12–19, 19% and 17%, respectively, were overweight (2). Alarmingly, the prevalence of obesity has tripled for adolescents in the past two decades. The increase in the number of people who are overweight or obese cuts across all ages, racial and ethnic groups, and both genders (2), and is increasingly global (3, 4). For example, the prevalence of obesity in urban preschoolers in China climbed >8-fold between 1989 and 1997 (5), and the rate of obesity in British adults rose almost 3-fold from 1980–2002 (6). In 2000, >110,000 deaths in the United States were associated with obesity, as shown by confound-adjusted analysis of three NHANES cohorts (7), and the economic cost of obesity in the United States was estimated to be $117 billion (8).
At this time, available nonsurgical treatments for obesity, including drugs, are palliative and effective only while treatment is maintained. When treatments are discontinued, weight gain inevitably results. For obesity treatments to work, they must affect energy intake, absorption, expenditure, or storage. Although many drugs have been marketed or are currently under investigation for the treatment of obesity, several have adverse side effects, including insomnia, asthenia, fecal incontinence, hypertension, tachychardia, valvular heart abnormalities, and even death. Accordingly, several weight loss drugs have been banned by the Food and Drug Administration, including the first one approved for this indication, desoxyephedrine (1946) and, more recently, d-fenfluramine/fenfluramine (September 1997) and ephedrine alkaloids (April 2004) (9, 10).
Fortunately, research over the past 15 years has revolutionized understanding of the molecular mechanisms that homeostatically control body weight and fat. Accumulated findings support a lipostatic hypothesis of energy homeostasis, in which the brain seeks to retain stored energy constant over long periods as adipose tissue. Accordingly, a highly integrated, redundant neurohumoral energy homeostasis feedback system, through behavioral and metabolic mechanisms, serves to minimize the impact of short-term fluctuations in energy balance, and especially negative energy balance, on fat mass. The identification of genes whose loss-of-function mutations result in monogenic obesity syndromes or confer resistance to obesity in humans or rodents have provided critical genetic entry points for characterizing the interconnected pathways that regulate energy homeostasis (11–14). The identification of these receptors and ligands has led to new therapeutic efforts to target signals at both ends of the energy spectrum.
In this context, ghrelin was identified in 1999 as an endogenous ligand for the growth hormone secretagogue receptor (ghrelin receptor), previously localized to peripheral tissues and hypothalamic nuclei that control energy homeostasis (15, 16). Human studies found a preprandial rise and postprandial decline in plasma ghrelin levels of putative gastric origin, suggesting that ghrelin might play a physiological role in energy homeostasis (17–19). Ghrelin is orexigenic when administered to humans or sated rodents with a likely central site of action (20, 21). Ghrelin also promotes weight gain and adiposity through its metabolic actions, decreasing both energy expenditure and fat catabolism (21, 22). Relevant to a possible tonic role for ghrelin in longer term energy homeostasis, circulating ghrelin levels are persistently increased during weight loss and suppressed in obese states (23). Consistent with a hypothesized anabolic role for endogenous ghrelin in energy balance, mice deficient for ghrelin or its receptor store less of their consumed food and resist accumulating body weight and fat on energy-dense diets (22, 24). Ghrelin knockout mice also expend more energy and locomote more (22), and ghrelin receptor deficient mice show increased whole-body fat oxidation (24).
Ghrelin is the first peptide isolated from animal sources with the posttranslational modification of octanoylation. Specifically, the hydroxyl group of a Ser residue (Ser-3) is acylated by n-octanoic acid (Fig. 1). Octanoylation is essential for the growth hormone-releasing activity of ghrelin (15, 25). Moreover, short peptides that encompass the first 4–5 residues of ghrelin activate the growth hormone secretagogue receptor as efficiently as full-length ghrelin, suggesting that the N-terminal Gly-Ser-Ser(n-octanoyl)-Phe segment constitutes the requisite core for receptor binding and activation (25). The present study sought to develop and test three haptenic structures, ghrelin (1–10) Ser-3(butanoyl) hapten (Ghr1), ghrelin (13–28) hapten (Ghr2), and ghrelin(1–28) Ser-3(butanoyl) hapten (Ghr3), respectively (Fig. 1), as potential anti-ghrelin vaccines. Relatedly, the work was founded on the hypotheses that immunologically specific effects could be imposed to slow weight gain, either through an action on food intake or feed efficiency.
Fig. 1.

Ghrelin hapten immunoconjugates Ghr1, Ghr2, and Ghr3 used to generate ghrelin-specific immune responses. Each asterisk denotes the site of conjugation with KLH or BSA.
Results
Vaccine Responses.
As shown in Table 1, both Ghr1-keyhole limpet hemocyanin (KLH) and Ghr3-KLH ghrelin immunoconjugates produced good immune responses (increased vaccine antibody titers) by week 9 or after the fourth immunization. Responses were of appropriate specificity, because antibodies elicited by Ghr1-KLH did not cross-react with Ghr2-BSA, and antibodies elicited by Ghr2-KLH likewise had low affinity for Ghr1-BSA (data not shown). Although each immunization group raised an immune response against the immunoconjugate with which they were vaccinated, only rats from groups immunized with Ghr1-KLH or Ghr3-KLH developed good plasma binding affinity for n-octanoylated ghrelin, the putatively active circulating form. Plasma from rats vaccinated with Ghr1-KLH or Ghr3-KLH was not potently bound by the inactive, des-octanoyl form of ghrelin that predominates in circulation (at least 1.8:1 ratio) (26) and might otherwise compete for neutralizing antibody, resulting in greater specificity for acyl vs. des-acyl ghrelin (Table 1).
Table 1.
Plasma vaccine antibody titers and ghrelin binding affinity in vaccinated rats
| Immunization group | Vaccine titers (×100) |
Plasma affinity (KD-app) after 13 weeks for |
Selectivity ratio(KD-app-des-octanoyl/KD-app-n-octanoyl) |
|||
|---|---|---|---|---|---|---|
|
n-octanoyl ghrelin |
des-octanoyl ghrelin ELISA, nM |
|||||
| 9 weeks | 13 weeks | ELISA, nM | Equilibrium dialysis, nM | |||
| Ghr1-KLH (n = 5) | 11 ± 3 | 34 ± 12 | 1,461 ± 569 | 45 ± 22 | 47,655 ± 235 | 98 ± 48 |
| Ghr2-KLH (n = 4) | 23 ± 12 | 13 ± 3 | >50,000 | >20,000 | 14,308 ± 11,916 | <0.3 ± 0.2 |
| Ghr3-KLH (n = 5) | 56 ± 15 | 137 ± 51 | 619 ± 173 | 243 ± 192 | 26,010 ± 10,201 | 41 ± 12 |
| KLH (n = 3) | 2 ± 1 | 2 ± 1 | >50,000 | >20,000 | >50,000 | N.D. |
Data are expressed as mean ± SEM. Immunized mature, male Wistar rats raised antibodies against the immunoconjugate with which they were vaccinated, seen as increased vaccine titers 1 week after the fourth and fifth immunization (weeks 9 and 13, respectively). However, only rats immunized with the N-terminal inclusive hapten immunoconjugates, Ghr1-KLH and Ghr3-KLH, developed plasma affinity for the putatively active, n-octanoylated form of ghrelin, shown by apparent equilibrium constants of dissociation (KD-app) in competition ELISA and equilibrium dialysis analysis. Their plasma did not comparably bind the putatively inactive, des-octanoylated form of ghrelin, resulting in two orders greater selectivity of binding for the acylated vs. des-acyl form of ghrelin compared with Ghr2-KLH immunized rats. Competition ELISA and selectivity ratio results reflect all subjects. Equilibrium dialysis, which yielded similar results, was performed with plasma from rats from each group that showed high vaccine titers to compare subjects that mounted stronger immune responses. Titers and plasma affinity were log-transformed to calculate averages, selectivity ratios, and for statistical analysis. Values reflect antilog transformations.
Postimmunization Weight Gain, Food Intake, and Feed Efficiency.
Fig. 2 shows the corresponding rate of body weight gain, daily food intake, and feed efficiency of immunized rats as a function of the vaccine that subjects received, once titers of several rats had increased into the 1:10,000 range. Rats that received the Ghr1-KLH and Ghr3-KLH immunoconjugates gained less weight per day than did rats immunized with Ghr2-KLH or KLH across the 7-day observation period after the fourth immunization (Fig. 2A). This reduction in weight gain occurred despite rats eating (Fig. 2B) and drinking (data not shown) normally, indicating a reduced feed efficiency of Ghr1-KLH and Ghr3-KLH-vaccinated subjects (Fig. 2C). Before this time, during weeks 5–8, when titers were modest and still rising, vaccine-related differences in daily weight gain (KLH: 1.22 ± 0.32 vs. Ghr3-KLH: 0.92 ± 0.12 and Ghr1-KLH: 1.01 ± 0.15 g/day) and feed efficiency (KLH: 44.4 ± 5.5 vs. Ghr3-KLH: 28.5 ± 3.0 and Ghr1-KLH: 33.6 ± 4.4 mg/kcal) were in the same direction but correspondingly smaller, supporting the proposed dependence on anti-ghrelin plasma binding affinity.
Fig. 2.
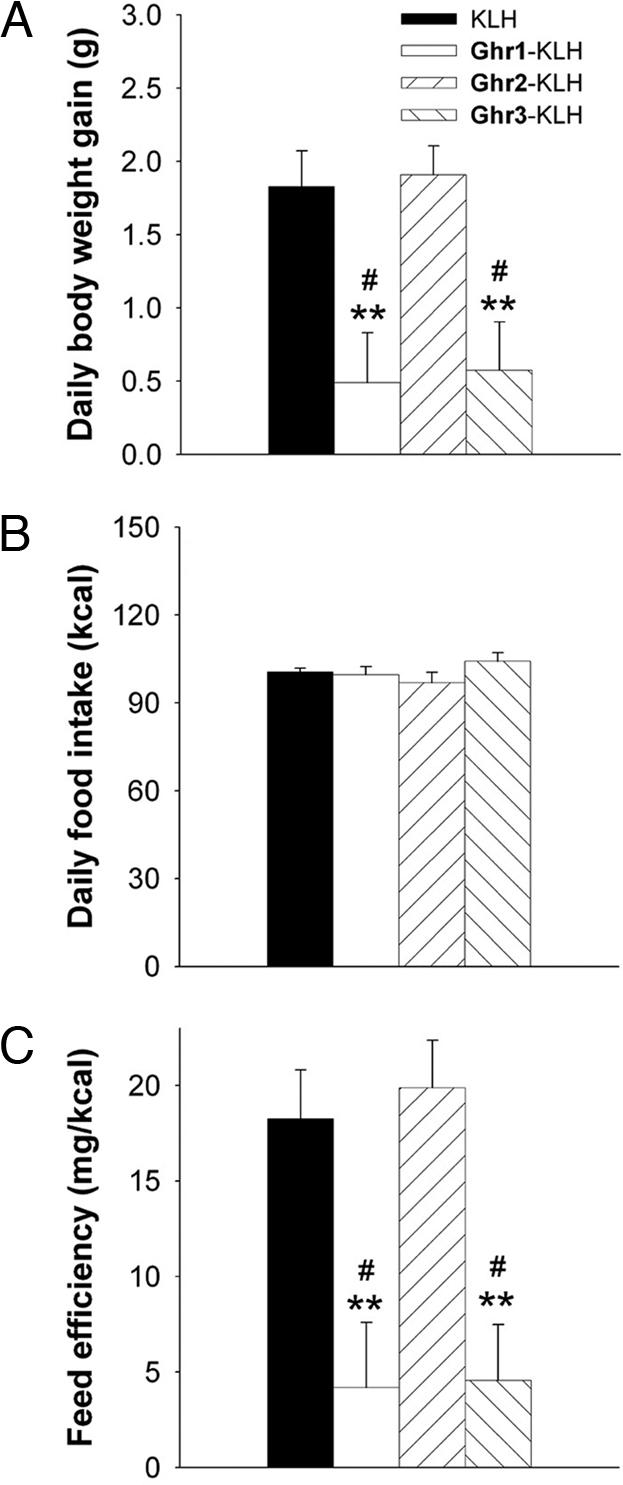
Effects of vaccination with ghrelin hapten immunoconjugates on whole-body energy homeostasis. Data express mean (+SEM) daily body weight gain (A), food intake (B), and feed efficiency (C) (body weight gained per unit energy intake) in Wistar rats previously immunized with a series of ghrelin hapten-keyhole limpet hemocyanin (KLH) immunoconjugates. Rats vaccinated using N-terminal inclusive ghrelin haptens (Ghr1-KLH and Ghr3-KLH; n = 5 per group) showed slower body weight gain and reduced feed efficiency compared with those immunized with Ghr2-KLH (n = 4) or KLH only (n = 3). Food intake and body weight were measured daily throughout the week after the fourth immunization, when vaccine antibody titers had increased. Rats were matched for baseline age and body weight and, as expected, had not differed reliably in rate of body weight accrual or food intake before the fourth immunization, when titers were still low. #, P < 0.05 vs. KLH-only treated group; ∗∗, P < 0.01 vs. Ghr2-KLH treated group [Fisher’s post hoc protected least significant difference tests, after significant one-way ANOVAs for body weight gain, F(3, 13) = 6.02 and P = 0.008, and feed efficiency, F(3, 13) = 7.28 and P = 0.004].
Relation of Antioctanoylated Ghrelin Titers to Vaccine Outcomes of Leanness and Slowed Weight Gain.
To test directly the hypothesized relation of circulating anti-ghrelin binding capacity to the attenuation of body weight gain, subjects were divided into those that developed high or low antibody titers to Ghr3-BSA (Fig. 3A) and compared in the weeks after the fourth and fifth immunizations. The high titer group was selected to exhibit mean anti-Ghr3-BSA titers >1:10,000, a level that is functionally and categorically meaningful for influencing brain bioavailability of circulating molecules in other active immunization paradigms (27–29). Each subject with high Ghr3-BSA titers also exhibited good plasma affinity for n-octanoyl ghrelin per competition ELISA analysis [KD-app (apparent dissociation constant) <650 nM], more than one order greater on average than those with lower Ghr3-BSA titers (Fig. 3B). Those rats with higher plasma ghrelin binding capacity gained ≈0.5 g less weight per day than those with lower plasma affinity for ghrelin (Fig. 3C), despite normal food intake (Fig. 3D), again indicating decreased feed efficiency (Fig. 3E), with the same profile of results observed after each immunization. Even rats with lower, but measurable, plasma ghrelin binding capacity exhibited reduced feed efficiency compared with KLH controls (Fig. 3E). Carcass analysis showed that rats with strong anti-ghrelin responses gained fat-free dry mass preferentially over fat mass, as compared with those with weaker anti-ghrelin immune responses, and the samples did not differ in water content (Fig. 3F and Table 2). Consistent with the relative decrease in fattiness, terminal plasma levels of the adipocyte hormone leptin were reduced in rats with strong plasma affinity for ghrelin (Fig. 3G). Circulating insulin levels did not differ reliably as a function of the plasma ghrelin binding affinity achieved (mean ± SEM: high, 0.8 ± 0.2 vs. low, 1.3 ± 0.2 ng/ml). Possibly consistent with a reduced passage of circulating ghrelin to the central compartment, the ratio of brain/plasma levels of total ghrelin was reduced in rats in relation to their plasma anti-ghrelin binding capacity (Fig. 3H).
Fig. 3.

Relation of plasma anti-ghrelin binding capacity to whole-body energy homeostasis, adiposity, and brain/plasma ghrelin ratios. Rats were grouped as those that developed “high” or “low” anti-ghrelin specificity (n = 5 and 9, respectively) in response to vaccination with ghrelin hapten immunoconjugates, as defined by Ghr3-BSA antibody titers (A) and validated independently by competition ELISA analysis of plasma affinity for Ser-3 n-octanoylated ghrelin (B). Rats with high anti-ghrelin binding capacity gained weight more slowly (C) despite eating similarly (D) compared not only with KLH controls but also with hapten-vaccinated rats that acquired less anti-ghrelin affinity. Slower weight gain despite normal food intake indicated reduced feed efficiency, present in relation to the development of anti-ghrelin affinity (E). Rats with strong anti-ghrelin responses had reduced adiposity, measured as a relative decrease in fattiness by chemical carcass composition analysis (F) and reduced circulating levels of the adipocyte hormone leptin (G). The ratio of brain/plasma ghrelin levels decreased in relation to the strength of the plasma anti-ghrelin response (G). Food intake and body weight were measured daily throughout the weeks after the fourth and fifth immunizations. Data are expressed as mean + SEM. #, P < 0.05 vs. low Ghr3-BSA titers group; ∗, P < 0.05 vs. KLH immunized control group [Fisher’s post hoc protected least significant difference tests, after significant one-way ANOVAs for Ghr3-BSA titers, F(2, 14) = 42.88, P < 0.0001; plasma ghrelin binding, F(2, 14) = 10.66, P = 0.001; and brain/plasma ghrelin ratio, F(2, 14) = 25.52, P = 0.00002; Welch’s corrected t tests for other measures, which had unequal variance between groups].
Table 2.
Relation of anti-ghrelin immune response to terminal carcass composition in vaccinated rats
| Group Ghr3-BSA | Fat-free dry mass, % |
Fat mass, % | Water, % | ||
|---|---|---|---|---|---|
| Protein | Ash | Total | |||
| High titers (n = 5) | 22.9 ± 0.2* | 2.9 ± 0.1 | 25.8 ± 0.2* | 13.6 ± 1.6# | 60.6 ± 1.4 |
| Low titers (n = 9) | 22.0 ± 0.4 | 2.8 ± 0.1 | 24.8 ± 0.4 | 17.1 ± 1.3 | 58.2 ± 1.0 |
Data are expressed as mean ± SEM. Carcass composition of previously vaccinated mature male Wistar rats showed that rats developed stronger anti-ghrelin binding capacity, as defined by high Ghr3-BSA titers and also reflected in high plasma ghrelin binding capacity (Table 1), and had greater relative fat-free dry mass and less relative fat mass compared with those with weaker anti-ghrelin binding capacity. Chemical carcass analysis was performed 1 week after the fifth vaccination with ghrelin hapten immunoconjugates. ∗, P < 0.05; #, P = 0.06 vs. low Ghr3-BSA titer group, Welch’s corrected t test.
Anti-Ghrelin Vaccine Did Not Elicit Systemic Inflammatory Response.
To examine the possibility that vaccination effects might result from a nonspecific systemic immune response, proinflammatory mediators, including IL-1β, IL-6, tumor necrosis factor-α, monocyte chemoattractant protein-1, and total plasminogen activator inhibitor type-1, were measured in vaccinated rats. Plasma levels were low on an absolute basis across treatment groups and unrelated to plasma specificity for acylated ghrelin (see Supporting Information, which is published on the PNAS web site).
Discussion
Diverse neurotransmitters, neuropeptides, hormones, and their targets offer potential inroads to understand and combat medicinally the growing problem of obesity. One classic neuropharmacological approach to understand the function of such molecules and their pathways has involved the administration of direct/indirect agonists, antagonists, or inverse antagonists or chemical or neuroanatomical lesions. Genetic manipulations, including selective breeding, study of inbred lines and their intercrosses, and manipulation of specific genes by means of molecular biologic approaches or mutagenesis, also have provided useful insights. An alternative strategy, termed immunopharmacotherapy, has been highly touted in other fields (30), including the field of drug abuse (31); however, until now, it has not been applied to the problem of obesity.
Recently, we used immunopharmacotherapy in developing therapeutic vaccine(s) against exposure to drugs of abuse (31). More specifically, the methodology sought to trigger an active (unnatural host immune system response) or passive (administration of a monoclonal or phage-displayed antibody) immunosequestration of the small molecule drug of abuse before it could access its target(s) in the CNS to produce psychoactive effects. We and others have used both active and passive vaccines therapeutically in animal models of cocaine, nicotine, and, most recently, tetrahydrocannabinol (marijuana) dependence, and clinical trials are ongoing in humans (31).
The present study analogously extended this approach to determine whether active vaccination against the endogenous centrally acting, predominantly stomach-synthesized (80%), anabolic feedback peptide hormone ghrelin could slow body weight gain and fat accrual of mature male rats. Toward this goal, the following physiological/chemical issues had to be addressed: (i) Rat vs. human ghrelin differs by two residues (K-A vs. R-V at positions 11 and 12), a potential obstacle to generalizing haptens across species; (ii) the lipophilic Ser-3 octanoic ester, although important for bioactivity, would cause micelle formation or solubility problems during hapten preparation; and (iii) little is known about how the distinct circulating forms of ghrelin (i.e., acylated or des-acyl ghrelin) interact with other molecules in vivo. Thus, to identify key elements of a potential therapeutic anti-ghrelin vaccine, three ghrelin haptens were studied (Fig. 1). Ghr1 contained N-terminal amino acids 1–10 of ghrelin, Ghr2 contained C-terminal amino acids 13–28, and Ghr3 contained the full-length rat ghrelin sequence (1–28). Ghr1 and Ghr2 were designed to bypass the species specificity of ghrelin’s sequence (i.e., rat vs. human), investigate epitope requirements, and identify any role of protein-bound circulation of ghrelin. In lieu of the native Ser-3 n-octanoyl fatty acid side chain, Ghr1 and Ghr3 used a Ser-3 butanoyl ester to increase hapten solubility and reduce micelle formation. Although this approach might be thought to compromise antibody recognition of ghrelin in its active n-octanoyl form, we have shown previously that truncation of lipid side chains actually improved immunogenicity for lipid A hapten vaccination (32). Finally, a cysteine residue was appended to the hapten C terminus (Ghr1 and Ghr3) or N terminus (Ghr2) to permit chemoselective conjugation chemistries (33) (Fig. 1).
Importantly, vaccine efficacy was related to the conjugate’s ability to induce high, specific plasma binding affinity for n-octanoyl as opposed to des-octanoyl ghrelin. Both the time course and magnitude of the vaccine’s action to slow weight gain correlated with the development of anti-acyl-ghrelin titers above ≈1:10,000, similar to results observed for vaccines against other circulating molecules whose activity depends on access to the brain (27–29, 34, 35). Hence, even though strong titers were obtained to Ghr2, this acyl ester-lacking “epitope” of ghrelin was unable to stimulate an immune system response that could sequester functionally relevant ghrelin isoforms, thereby compromising the vaccine’s ability to impact energy homeostasis. Accordingly, hapten design appears to be critical, similar to findings observed for effective antinicotine and anticocaine vaccines (27–29, 34, 35). In accord with active vaccination studies against drugs of abuse, effective anti-ghrelin immunization resulted in a decreased brain/plasma ratio of ghrelin. At this juncture, high, specific plasma binding affinity for acylated ghrelin appears critical for vaccine efficacy, and this finding may explain why the N-terminal inclusive ghrelin (Ser-3-n-butanoylated) haptens (Ghr1 and Ghr3) more effectively slowed weight gain. In total, the results demonstrate a proof of principle that active immunization against ghrelin, and possibly other anabolic regulatory molecules, can be used to control weight gain and adiposity in mammals.
Immunized rats ate normally but, once titers increased, accrued less body weight and fat, indicating reduced energy thrift. The findings implicate a metabolic (22, 24), rather than ingestive (21), mechanism in the vaccines’ efficacy, perhaps because of differential sensitivity or compensation of these dissociable modes of ghrelin action. Accordingly, exogenous ghrelin administration is known to decrease energy expenditure, brown adipose tissue mitochrondrial uncoupling protein-1 activity, and fat catabolism (21, 22). Although Sun and colleagues (36) initially observed that constitutive ghrelin deficiency did not alter food intake, weight accrual, or body composition of mice, more recent literature has supported an endogenous role for ghrelin and its receptor in the control of body weight and metabolism (22, 24, 37). For example, both ghrelin and ghrelin receptor knockout mice were subsequently found to be less thrifty with food that they ate and to resist accumulating body weight and fat on energy-dense diets to a degree greater than could be explained by energy intake differences (22, 24). These findings reflected that ghrelin knockout mice expend more energy and show increased locomotor activity (22), and that ghrelin receptor deficient mice show increased whole-body utilization of fat as an energy substrate (24). De Smet and colleagues (37) also recently reported metabolic data that indicate that ghrelin null mice are leaner and exhibit less de novo lipogenesis during fed states than wild-type mice, again despite similar energy intake. That anti-ghrelin vaccines also decreased feed efficiency and adiposity harmonizes with reviewed results and supports the proposed endogenous role for ghrelin in metabolism. Further immunization studies can help specify the precise physiologic role of ghrelin-induced changes in resting metabolic rate, whole-body, or non-exercise activity-induced energy expenditure or relative fuel substrate utilization by discrete tissues in the vaccine’s efficacy. Importantly, the vaccines may offer a means to separate these metabolic actions of ghrelin from potentially confounding, phasic changes in food intake that have been seen with other approaches (21, 22, 24).
The effective active ghrelin vaccines, Ghr1 and Ghr3, did not alter spontaneous daily food intake under the current experimental conditions, a finding that contrasts with reports that intracerebroventricular administration with anti-ghrelin antibodies of uncertain isoform specificity acutely reduce food intake (38–40). These results indicate that sequestration of central ghrelin curbs food intake, whereas peripheral active immunization does not produce anorexia yet still reduces weight gain. The dissociation may reflect an orexigenic role for centrally synthesized ghrelin in appetite neurocircuitry. Alternatively, it is conceivable that appetite is less sensitive than metabolism to immunization-induced decreases in the levels of ghrelin that can reach the central compartment. On the other hand, several caveats bear mentioning. First, higher circulating titers or a larger sample size might have uncovered an endogenous action of ghrelin on intake. Second, rats were consistently fed in ad libitum conditions. However, ghrelin may play a greater role in controlling food intake during sustained energy insufficiency, during which ghrelin levels rise, putatively to motivate meal initiation and compensatory hyperphagia. Thus, the vaccine might be able to blunt restriction-induced, if not ad libitum, feeding, a relevant question for vaccine efficacy during low calorie diet-induced weight loss. Third, we cannot discount the possibility that the induced immune response is (partly) directed to alkanoylated serine residues in structurally and functionally related signaling peptides other than ghrelin, and/or esterification signals contained in other peptides. Fourth, perhaps redundant, compensatory orexigenic mechanisms were recruited that offset any direct action of the anti-ghrelin vaccine to reduce feeding, as has been proposed for chow-fed ghrelin null mutant models (36, 37). Fifth, perhaps phasic changes in ghrelin levels, which would be relatively unaffected by vaccination, control food intake from meal-to-meal, whereas longer term, tonic changes in ghrelin levels (e.g., obesity and starvation) control longer term energy homeostasis through metabolic mechanisms. Finally, rats were fed low-fat chow; however, anorectic effects of ghrelin receptor deficiency have only been reported in high-fat diet-fed mice, whereas chow-fed female growth hormone secretagogue receptor knockout mice exhibit reduced body weight and feed efficiency, but not intake, similar to what was observed here (24). Thus, perhaps diet composition modulates the regulatory role of ghrelin on food intake, and an anti-ghrelin vaccine might similarly reduce intake of high-fat diets. Studies of the effects of central vs. peripheral anti-ghrelin immunization on appetite-regulating neurocircuitry in the mediobasal hypothalamus and caudal hindbrain will help further clarify ghrelin’s biological significance in controlling food intake.
A limitation of the study concerns the possibility of type II errors (“false negatives”) because of insufficient power from the current sample sizes. For example, although insulin levels did not differ significantly between rats according to their vaccine response, rats that developed strong anti-ghrelin responses did tend to show lower (≈40%) circulating insulin levels, which would be consistent with their reduced adiposity and plasma leptin levels. Perhaps a larger sample size would have revealed an effect. On the other hand, the lack of decrease in insulin levels may reflect the direct action of ghrelin to suppress insulin secretion (41). From this perspective, the ghrelin vaccine would proximately increase insulin levels, thereby offsetting any decrease related to increased leanness.
Finally, the vaccines’ therapeutic potential in models of overeating and obesity are of interest. Rats in the current study were fed low-energy, low-fat, and relatively less palatable chow diets and were comparatively lean. Whether active immunization against ghrelin would help prevent the development of obesity attributable to energy-dense, palatable high-fat “Western” diets or would facilitate weight loss once obesity is established are uncertain. For example, circulating ghrelin levels are decreased during obese states, which might suggest a reduced physiological relevance of blocking ghrelin action (23). On the other hand, ghrelin levels rise with attempted or successful weight loss and strongly predict subsequent weight regain (23), so an anti-ghrelin vaccine might be especially helpful in opposing this counterregulatory mechanism that defends an elevated body weight set point. Clarifying these and potential unintended effects of active vaccines on additional actions of ghrelin (42, 43) will help define the therapeutic potential of N-terminal inclusive ghrelin hapten vaccines to control body weight. In summary, active vaccination against ghrelin, and potentially other anabolic molecules, appears to be a viable method to slow weight gain and fat accrual in mammals. Additional studies with these active vaccines and passive antibodies, including catalytic antibodies to ghrelin (M.M.M. and K.D.J., unpublished data), will further define the role of immunopharmacotherapy in perturbing energy homeostasis and ultimately in combating obesity.
Methods
Ghrelin Hapten Immunoconjugate Synthesis.
Ghr1-Ghr3 were synthesized and coupled to the carrier protein KLH, yielding immunoconjugates Ghr1-KLH, Ghr2-KLH, and Ghr3-KLH. For peptide synthesis, all haptens and substrates were prepared on a 1.0-mmol scale as C-terminal amides by using custom-written diisopropyl carbodiimide/hydroxybenzotriazole (DIC/HOBt) protocols for fluorenylmethyloxycarbonyl/tert-butyl solid phase peptide synthesis (Fmoc/tBu SPPS) on a CS Bio 136 automated peptide synthesizer. For experimental details, see Supporting Information, which supplies the synthetic details and HPLC chromatograms and electrospray ionization-MS spectra of RP-HPLC purified synthetic wild-type rat ghrelin and haptens Ghr1, Ghr2, and Ghr3.
Ghr1.
Electrospray ionization-MS: Theory, MW = 1260.4; M1+ = 1261.4, M2+ = 631.2. Observed, M1+ = 1261.6, M2+ = 631.5.
Ghr2.
Electrospray ionization-MS: Theory, MW = 2021.4; M2+ 1011.7, M3+ = 674.8. Observed, M2+ = 1011.8, M3+ = 675.0
Ghr3.
Electrospray ionization-MS: Theory, MW = 3360.9; M2+ = 1681.5, M3+ = 1121.3. Observed, M2+ = 1681.4, M3+ = 1121.3.
Subjects.
Mature male Wistar rats (n = 17, mean + SEM: 413 + 8 g at onset, 502 + 12 g by fourth immunization) (Charles River, Hollister, CA) were individually housed in a 12-h:12-h lit (0600 h lights on), humidity-controlled (60%), and temperature-controlled (22°C) vivarium with continuous access to chow and water. The pelleted chow diet (LM-485 Diet 7012; Harlan Teklad, Madison, WI) is a corn-based, extruded cereal composed of 65% carbohydrate, 13% fat, 21% protein, and metabolizable energy of 3.41 kcal/g. Procedures adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and The Principles of Laboratory Animal Care and were approved by the Institutional Animal Care and Use Committee of The Scripps Research Institute.
Active Immunization.
Age- and weight-matched mature rats were immunized by using protocols of our laboratory (44) involving five immunizations over 12 weeks. Age- and weight-matched rats received immunizations (0.4 ml i.p.) 90 min before the dark cycle on experimental days 0, 21, 35, 56, and 84. The first three immunizations consisted of Ribi emulsion adjuvant containing monophosphorylated lipid A and trehalose dimycolate (Ribi Immunochemical Research, Inc., Hamilton, MT) containing 250 μg of Ghr1-KLH, Ghr2-KLH, Ghr3-KLH, or KLH in 100 mM PBS at pH 7.4. The last two used alum (Pierce, Rockford, IL) as the adjuvant. Tail blood was collected 1 week after immunization and centrifuged, and plasma was analyzed for antibody titers and ghrelin binding affinity.
Body Weight, Food Intake, and Feed Efficiency.
Body weight and food intake were determined daily (0.1 g precision), 2 h before the onset of the dark cycle during the weeks after the fourth and fifth immunization, the time at which antibody titers maximized. Feed efficiency was calculated as body weight gained per unit energy intake (milligrams per kilocalories).
Carcass Analysis.
The gastrointestinal tract was removed from thawed (25°C) carcasses. The carcass was then dried (70°C) to constant mass to determine water content and then extracted with petroleum ether in a Soxhlet apparatus to determine fat-free dry mass, composed of both protein and ash, as compared with fat mass (45).
Statistics.
Analyses of variance (ANOVAs) were group differences. Fisher’s protected least significant difference tests were used for post hoc comparisons. Welch’s t tests, corrected for multiple comparisons, were used to compare groups with unequal variance. To reduce heterogeneity of variance and achieve a more normal distribution, data for IL-6, TNF-α, total plasminogen activator inhibitor type-1, monocyte chemoattractant protein-1, and plasma antibody titers, apparent ghrelin binding affinity constants of plasma and brain/plasma total ghrelin ratios were first log-transformed for statistical analysis, including calculation of titer selectivity ratios. Corresponding values presented in tables or figures represent antilog transformations. χ2 analysis was used to test whether subjects differed in the frequency of having detectable circulating cytokine levels. The software package was Systat 11.0 (SPSS, Chicago, IL).
Supplementary Material
Acknowledgments
We thank Tim Nagy and the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Program Project funded by the University of Alabama Clinical Nutrition Research Unit, Small Animal Phenotyping Core (Birmingham, AL) for the body composition analysis and Diane Kubitz for technical assistance. The work was supported by NIDDK Grants DK64871 and DK72169.
Abbreviations:
- Ghr1
ghrelin(1–10) Ser-3(butanoyl) hapten
- Ghr2
ghrelin(13–28) hapten
- Ghr3
ghrelin(1–28) Ser-3(butanoyl) hapten
- KLH
keyhole limpet hemocyanin.
Footnotes
Conflict of interest statement: No conflicts declared.
See Commentary on page 12961.
References
- 1.Yach D., Stuckler D., Brownell K. D. Nat. Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 2.Ogden C. L., Carroll M. D., Curtin L. R., McDowell M. A., Tabak C. J., Flegal K. M. J. Am. Med. Assoc. 2006;295:1549–1555. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 3.Silventoinen K., Sans S., Tolonen H., Monterde D., Kuulasmaa K., Kesteloot H., Tuomilehto J. Int. J. Obes. Relat. Metab. Disord. 2004;28:710–718. doi: 10.1038/sj.ijo.0802614. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y., Monteiro C., Popkin B. M. Am. J. Clin. Nutr. 2002;75:971–977. doi: 10.1093/ajcn/75.6.971. [DOI] [PubMed] [Google Scholar]
- 5.Luo J., Hu F. B. Int. J. Obes. Relat. Metab. Disord. 2002;26:553–558. doi: 10.1038/sj.ijo.0801944. [DOI] [PubMed] [Google Scholar]
- 6.Rennie K. L., Jebb S. A. Obes. Rev. 2005;6:11–12. doi: 10.1111/j.1467-789X.2005.00164.x. [DOI] [PubMed] [Google Scholar]
- 7.Flegal K. M., Graubard B. I., Williamson D. F., Gail M. H. J. Am. Med. Assoc. 2005;293:1861–1867. doi: 10.1001/jama.293.15.1861. [DOI] [PubMed] [Google Scholar]
- 8.U.S. Department of Health and Human Services. The Surgeon General’s Call to Action to Prevent and Decrease Overweight and Obesity. Washington, DC: U.S. Govt. Print. Office; 2001. Available at www.surgeongeneral.gov/topics/obesity. [Google Scholar]
- 9.Colman E. Ann. Intern. Med. 2005;143:380–385. doi: 10.7326/0003-4819-143-5-200509060-00013. [DOI] [PubMed] [Google Scholar]
- 10.Knight J. Nature. 2004;427:90. doi: 10.1038/427090b. [DOI] [PubMed] [Google Scholar]
- 11.Farooqi I. S., O’Rahilly S. Annu. Rev. Med. 2005;56:443–458. doi: 10.1146/annurev.med.56.062904.144924. [DOI] [PubMed] [Google Scholar]
- 12.Halaas J. L., Gajiwala K. S., Maffei M., Cohen S. L., Chait B. T., Rabinowitz D., Lallone R. L., Burley S. K., Friedman J. M. Science. 1995;269:543–546. doi: 10.1126/science.7624777. [DOI] [PubMed] [Google Scholar]
- 13.Huszar D., Lynch C. A., Fairchild-Huntress V., Dunmore J. H., Fang Q., Berkemeier L. R., Gu W., Kesterson R. A., Boston B. A., Cone R. D., et al. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 14.Lee G. H., Proenca R., Montez J. M., Carroll K. M., Darvishzadeh J. G., Lee J. I., Friedman J. M. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- 15.Kojima M., Hosoda H., Date Y., Nakazato M., Matsuo H., Kangawa K. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 16.Mori K., Yoshimoto A., Takaya K., Hosoda K., Ariyasu H., Yahata K., Mukoyama M., Sugawara A., Hosoda H., Kojima M., et al. FEBS Lett. 2000;486:213–216. doi: 10.1016/s0014-5793(00)02308-5. [DOI] [PubMed] [Google Scholar]
- 17.Kamegai J., Tamura H., Shimizu T., Ishii S., Sugihara H., Oikawa S. Endocrinology. 2001;142:4154–4157. doi: 10.1210/endo.142.9.8492. [DOI] [PubMed] [Google Scholar]
- 18.Shiiya T., Nakazato M., Mizuta M., Date Y., Mondal M. S., Tanaka M., Nozoe S., Hosoda H., Kangawa K., Matsukura S. J. Clin. Endocrinol. Metab. 2002;87:240–244. doi: 10.1210/jcem.87.1.8129. [DOI] [PubMed] [Google Scholar]
- 19.Tschop M., Wawarta R., Riepl R. L., Friedrich S., Bidlingmaier M., Landgraf R., Folwaczny C. J. Endocrinol. Invest. 2001;24:RC19–RC21. doi: 10.1007/BF03351037. [DOI] [PubMed] [Google Scholar]
- 20.Druce M. R., Neary N. M., Small C. J., Milton J., Monteiro M., Patterson M., Ghatei M. A., Bloom S. R. Int. J. Obes. 2006;30:293–296. doi: 10.1038/sj.ijo.0803158. [DOI] [PubMed] [Google Scholar]
- 21.Tschop M., Smiley D. L., Heiman M. L. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- 22.Wortley K. E., Del Rincon J. P., Murray J. D., Garcia K., Iida K., Thorner M. O., Sleeman M. W. J. Clin. Invest. 2005;115:3573–3578. doi: 10.1172/JCI26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kojima M., Kangawa K. Physiol. Rev. 2005;85:495–522. doi: 10.1152/physrev.00012.2004. [DOI] [PubMed] [Google Scholar]
- 24.Zigman J. M., Nakano Y., Coppari R., Balthasar N., Marcus J. N., Lee C. E., Jones J. E., Deysher A. E., Waxman A. R., White R. D., et al. J. Clin. Invest. 2005;115:3564–3572. doi: 10.1172/JCI26002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bednarek M. A., Feighner S. D., Pong S. S., McKee K. K., Hreniuk D. L., Silva M. V., Warren V. A., Howard A. D., Van Der Ploeg L. H., Heck J. V. J. Med. Chem. 2000;43:4370–4376. doi: 10.1021/jm0001727. [DOI] [PubMed] [Google Scholar]
- 26.Sato T., Fukue Y., Teranishi H., Yoshida Y., Kojima M. Endocrinology. 2005;146:2510–2516. doi: 10.1210/en.2005-0174. [DOI] [PubMed] [Google Scholar]
- 27.de Villiers S. H., Lindblom N., Kalayanov G., Gordon S., Johansson A. M., Svensson T. H. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004;370:299–304. doi: 10.1007/s00210-004-0960-3. [DOI] [PubMed] [Google Scholar]
- 28.de Villiers S. H., Lindblom N., Kalayanov G., Gordon S., Malmerfelt A., Johansson A. M., Svensson T. H. Respiration. 2002;69:247–253. doi: 10.1159/000063628. [DOI] [PubMed] [Google Scholar]
- 29.Hieda Y., Keyler D. E., Ennifar S., Fattom A., Pentel P. R. Int. J. Immunopharmacol. 2000;22:809–819. doi: 10.1016/s0192-0561(00)00042-4. [DOI] [PubMed] [Google Scholar]
- 30.Gupta S. K., Chakravarty S., Kadunganattil S. Chem. Immunol. Allergy. 2005;88:98–108. doi: 10.1159/000087823. [DOI] [PubMed] [Google Scholar]
- 31.Meijler M. M., Matsushita M., Wirsching P., Janda K. D. Curr. Drug Discov. Technol. 2004;1:77–89. doi: 10.2174/1570163043484851. [DOI] [PubMed] [Google Scholar]
- 32.Jones L. H., Altobell L. J., 3rd, MacDonald M. T., Boyle N. A., Wentworth P., Jr., Lerner R. A., Janda K. D. Angew. Chem. Int. Ed. Engl. 2002;41:4241–4244. doi: 10.1002/1521-3773(20021115)41:22<4241::AID-ANIE4241>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 33.Peeters J. M., Hazendonk T. G., Beuvery E. C., Tesser G. I. J. Immunol. Methods. 1989;120:133–143. doi: 10.1016/0022-1759(89)90298-6. [DOI] [PubMed] [Google Scholar]
- 34.Carrera M. R., Ashley J. A., Parsons L. H., Wirsching P., Koob G. F., Janda K. D. Nature. 1995;378:727–730. doi: 10.1038/378727a0. [DOI] [PubMed] [Google Scholar]
- 35.Martell B. A., Mitchell E., Poling J., Gonsai K., Kosten T. R. Biol. Psychiatry. 2005;58:158–164. doi: 10.1016/j.biopsych.2005.04.032. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y., Ahmed S., Smith R. G. Mol. Cell. Biol. 2003;23:7973–7981. doi: 10.1128/MCB.23.22.7973-7981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Smet B., Depoortere I., Moechars D., Swennen Q., Moreaux B., Cryns K., Tack J., Buyse J., Coulie B., Peeters T. L. J. Pharmacol. Exp. Ther. 2006;316:431–439. doi: 10.1124/jpet.105.091504. [DOI] [PubMed] [Google Scholar]
- 38.Bagnasco M., Tulipano G., Melis M. R., Argiolas A., Cocchi D., Muller E. E. Regul. Pept. 2003;111:161–167. doi: 10.1016/s0167-0115(02)00283-5. [DOI] [PubMed] [Google Scholar]
- 39.Nakazato M., Murakami N., Date Y., Kojima M., Matsuo H., Kangawa K., Matsukura S. Nature. 2001;409:194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- 40.Shuto Y., Shibasaki T., Otagiri A., Kuriyama H., Ohata H., Tamura H., Kamegai J., Sugihara H., Oikawa S., Wakabayashi I. J. Clin. Invest. 2002;109:1429–1436. doi: 10.1172/JCI13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y., Asnicar M., Saha P. K., Chan L., Smith R. G. Cell. Metab. 2006;3:379–386. doi: 10.1016/j.cmet.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 42.Diano S., Farr S. A., Benoit S. C., McNay E. C., da Silva I., Horvath B., Gaskin F. S., Nonaka N., Jaeger L. B., Banks W. A., et al. Nat. Neurosci. 2006;9:381–388. doi: 10.1038/nn1656. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J. V., Ren P. G., Avsian-Kretchmer O., Luo C. W., Rauch R., Klein C., Hsueh A. J. Science. 2005;310:996–999. doi: 10.1126/science.1117255. [DOI] [PubMed] [Google Scholar]
- 44.Qi L., Yamamoto N., Meijler M. M., Altobell L. J., 3rd, Koob G. F., Wirsching P., Janda K. D. J. Med. Chem. 2005;48:7389–7399. doi: 10.1021/jm050442r. [DOI] [PubMed] [Google Scholar]
- 45.Dobush G. R., Ankney C. D., Krementz D. G. Can. J. Zool. 1985;63:1917–1920. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.