

Abstract
Classical studies on protist diversity of freshwater environments worldwide have led to the idea that most species of microbial eukaryotes are known. One exemplary case would be constituted by the ciliates, which have been claimed to encompass a few thousands of ubiquitous species, most of them already described. Recently, molecular methods have revealed an unsuspected protist diversity, especially in oceanic as well as some extreme environments, suggesting the occurrence of a hidden diversity of eukaryotic lineages. In order to test if this holds also for freshwater environments, we have carried out a molecular survey of small subunit ribosomal RNA genes in water and sediment samples of two ponds, one oxic and another suboxic, from the same geographic area. Our results show that protist diversity is very high. The majority of phylotypes affiliated within a few well established eukaryotic kingdoms or phyla, including alveolates, cryptophytes, heterokonts, Cercozoa, Centroheliozoa and haptophytes, although a few sequences did not display a clear taxonomic affiliation. The diversity of sequences within groups was very large, particularly that of ciliates, and a number of them were very divergent from known species, which could define new intra-phylum groups. This suggests that, contrary to current ideas, the diversity of freshwater protists is far from being completely described.
Keywords: eukaryotes, environmental PCR, molecular ecology, protist diversity
1. Introduction
The accessibility of freshwater ecosystems has made of these environments a privileged choice for protozoological studies for more than three centuries since Leeuwenhoek's times (Finlay & Esteban 2001). Protists are key components of aquatic food webs both as members of the phytoplankton (e.g. diatoms) and as major consumers of bacterial biomass (e.g. heterotrophic flagellates and ciliates). In particular, nanoflagellates are abundant in planktonic communities, acting as the primary bacterial consumers and playing a cardinal role in nutrient cycling (Arndt et al. 2000; Weisse 2002). Aquatic ecologists have traditionally focused their attention on functional categories (trophic chain levels) rather than in species composition, although there is a recognized increasing interest in integrating the existing taxonomic data (Weisse 2002; Finlay 2004). Indeed, many species have been isolated and described, and taxonomic inventories of freshwater protists have been constructed over many years. The recurrent observation of the same morphological types in freshwater systems from all over the world has led to the idea that the global protist species richness is relatively low. Ciliates, easily distinguishable due to their complex morphology, are used as a paradigmatic example. With nearly 4000 species identified, it has been proposed that most ciliate species have already been discovered (Finlay 1998).
Molecular methods based on the amplification and sequencing of small subunit ribosomal RNA (SSU rRNA) genes have opened the possibility of studying microbial diversity independently of morphological identification and cultivation. In recent years, these methods have been applied to the study of protist diversity in a number of different ecosystems, including oceanic waters (Diez et al. 2001; López-García et al. 2001; Moon-van der Staay et al. 2001; Romari & Vaulot 2004), deep-sea vents (Edgcomb et al. 2002; López-García et al. 2003), anoxic environments (Dawson & Pace 2002; Stoeck et al. 2003), river sediment (Berney et al. 2004) and one acidic river (Amaral Zettler et al. 2002). In all these studies a large protist diversity has been revealed and, although in general they belong to well established eukaryotic kingdoms (Berney et al. 2004; Cavalier-Smith 2004), the lineages identified are very often highly divergent from known protist sequences. This suggests that a large fraction of eukaryotic microbial communities remains to be discovered (Moreira & López-García 2002). The existence of such a hidden diversity would support detractors from the above-mentioned idea that most protist species have already been described (Foissner 1999).
In order to see whether the results obtained by molecular methods fit those accumulated by classical, morphological studies, we carried out a comprehensive survey of protist small subunit ribosomal DNA (SSU rDNA) diversity from two different freshwater reservoirs. We analysed picoplanktonic and small nanoplanktonic (0.2–5 μm range), and larger planktonic (greater than 5 μm) fractions as well as sediment samples from two ponds, one oxic and one suboxic, located in the same geographic region. Our results reveal a large diversity of lineages within known eukaryotic phyla, including some phylotypes that are divergent from already known sequences. This is particularly noticeable in the case of ciliates, which dominate the protist diversity in the suboxic system. These results support the idea that the inventory of freshwater protists is far from complete.
2. Material and methods
(a) Sampling
Samples were collected in late autumn–early winter from a suboxic pond (CV1) and from an oxic pool (CH1) located a few kilometres south of Paris, France. The suboxic CV1 was an approximately 15 m diameter pond located at the campus of the Université Paris-Sud (pH 6.5, water temperature 11.5 °C). When stirred, its waters bubbled and smelled strongly of H2S and CH4, indicators of oxygen-depleted environments. ConOx probe (WTW, Weilheim) oxygen measurements yielded values ranging from 0.38 mg l−1 (bottom) to 2.4 mg l−1 (surface). The small (ca 30×50 m2), normally oxygenated (9.6 mg O2 per litre) shallow lake CH1 was located at the Chevreuse locality in the Regional Natural Park of the Haute Vallée de Chevreuse (pH 7.6, water temperature 4 °C, a thin ice layer covered the lake). In both cases, sediment and plankton were sampled. Dark brown/black sediments rich in organic detritus were collected from the bottom of the two ponds using sterile 50 ml Falcon tubes. A second, sandy, sediment sample was additionally collected from the edge of the suboxic CV1 system. Water was collected into sterile 1 l glass bottles with a sterile gauze adapted to their openings. Samples were processed (filtration and DNA extraction) immediately after collection. Water samples were passed through several layers of sterile gauze to filter out the components larger than 50–100 μm in diameter. Approximately 0.5 l water was then filtered across a 5 μm-pore diameter filter (TMTP, Millipore), and the resulting filtrate was further passed across a 0.22 μm-pore diameter filter (GTTP, Millipore). DNA was extracted from the biomass retained on the 5 μm (CV1-5 and CH1-5) and 0.22 μm (CV1-2 and CH1-2) filters.
(b) DNA extraction, PCR amplification, cloning and sequencing
The planktonic biomass retained on filters was suspended in 500 μl PBS. Cells were then lysed in the presence of 80 μg ml−1 proteinase K, 1% SDS, 1.4 M NaCl, 0.2% β-mercaptoethanol and 2% CTAB (final concentrations) at 55 °C. DNA was then extracted once with phenol–chloroform–isoamylalcohol, and once with chloroform–isoamylalcohol. Nucleic acids were concentrated by ethanol precipitation. DNA from sediments was directly extracted by using the SoilMaster DNA Extraction Kit (Epicentre) using manufacturer's instructions. Amplification of 95–98% of the total length of the SSU rDNA was done using all four combinations of the following specific eukaryotic primers: 18S–42F (CTCAARGAYTAAGCCATGCA), 18S–82F (GAAACTGCGAATGGCTC), 18S–1498R (CACCTACGGAAACCTTGTTA) and 18S–1520R (CYGCAGGTTCACCTAC). PCR reactions were carried out in 25 μl of reaction buffer containing 1 μl DNA template (10–100 ng), 1.5 mM MgCl2, dNTPs (10 nmol each), 20 pmol of each primer, 1 U Taq DNA polymerase (Promega), and sometimes 1.25 μl DMSO and 0.05% of a detergent solution. PCR reactions were performed under the following conditions: 30 cycles (denaturation at 94 °C for 15 s, annealing at 55 °C for 30 s, extension at 72 °C for 2 min) preceded by 2 min denaturation at 94 °C, and followed by 8 min extension at 72 °C. Amplicons were cloned into pCR2.1 Topo TA cloning vector (Invitrogen) and transformed into Escherichia coli TOP10′ One Shot cells (Invitrogen) according to the manufacturer's instructions. A total of 14 different libraries were constructed in this way, including duplicate libraries for both sediment and plankton samples. Clone inserts were amplified with vector T7 and M13R primers, and inserts of the expected size were sequenced directly using either specific or vector primers by Genome-Express (Meylan, France).
(c) Sequence and phylogenetic analyses
Initially, we obtained partial sequences from positive clones that were trimmed for ambiguities and used to construct a local database, which was then analysed by internal Blast to identify identical or highly similar sequences. Sequences exhibiting greater than 97% identity were considered to be single phylotypes. In total, 377 eukaryotic sequences from 7 duplicate libraries (247 sequences corresponding to CV1 and 130 to CH1) were included in our analysis (Electronic Appendix—table S1). Representative sequences of the difference (greater than 97%) identity groups were then selected and fully sequenced to obtain an almost complete SSU rDNA. Full-length sequences were checked for the presence of chimeras using Chimera_Check v. 2.7 at the Ribosomal Database Project II (Cole et al. 2003). For suspicious sequences, we looked for incongruencies in phylogenetic trees obtained from different parts of the sequence, and subsequently, multiple alignments were inspected visually to detect possible recombination sites between distant phylotypes. In addition, full sequence datasets were analysed using Bellerophon (Huber et al. 2004), and putative chimeric clones thus identified were manually checked. From the potential chimeras identified by Chimera_Check and Bellerophon, only nine chimeras were unambiguously verified (three in CV1 sample, six in CH1 sample), which were excluded from our analyses. In total, we obtained 83 almost complete eukaryotic SSU rDNA sequences, which were included in a local eukaryotic SSU rDNA database containing more than 5000 sequences. The obtained sequences ranged from 1335 bp (CV1-B2-35) to 1956 bp (CH1-S2-16). The closest Blast matches to our new sequences that were not included initially in this database were retrieved from GenBank at NCBI, aligned using ClustalX v. 1.82 (Thompson et al. 1997), and the multiple alignment edited manually. Ambiguous positions in the alignment were discarded from phylogenetic analyses. Trees were reconstructed using both Bayesian and maximum likelihood methods. Bayesian phylogenetic analyses were done with MrBayes v. 3.0b4 (Ronquist & Huelsenbeck 2003) using the GTR+Γ+I model of sequence evolution. They were repeatedly run from a random starting tree and run well beyond convergence. We used four to six hidden Markov chains, and up to 5 000 000 generations. For the reconstruction of the consensus Bayesian tree with posterior probabilities the trees before convergence were discarded. Maximum likelihood trees were constructed with Phyml v. 2.3/2.4.4 (Guindon & Gascuel 2003) using the HKY+Γ model of sequence evolution. Non-parametric bootstrapping was performed with Phyml using 100 replicates. The number of sequences and that of positions used in the construction of the different trees were, respectively: 67 and 1345 for Opisthokonta and Amoebozoa (figure 2), 190 and 1179 for the bikont diversity with the exception of alveolates, plants and excavates (figure 3) and 148 and 1099 for alveolates (figure 4). Alignments and detailed trees are available from the authors upon request. Sequences reported in this paper have been deposited in the GenBank database under accession numbers AY821916–AY821998.
Figure 2.
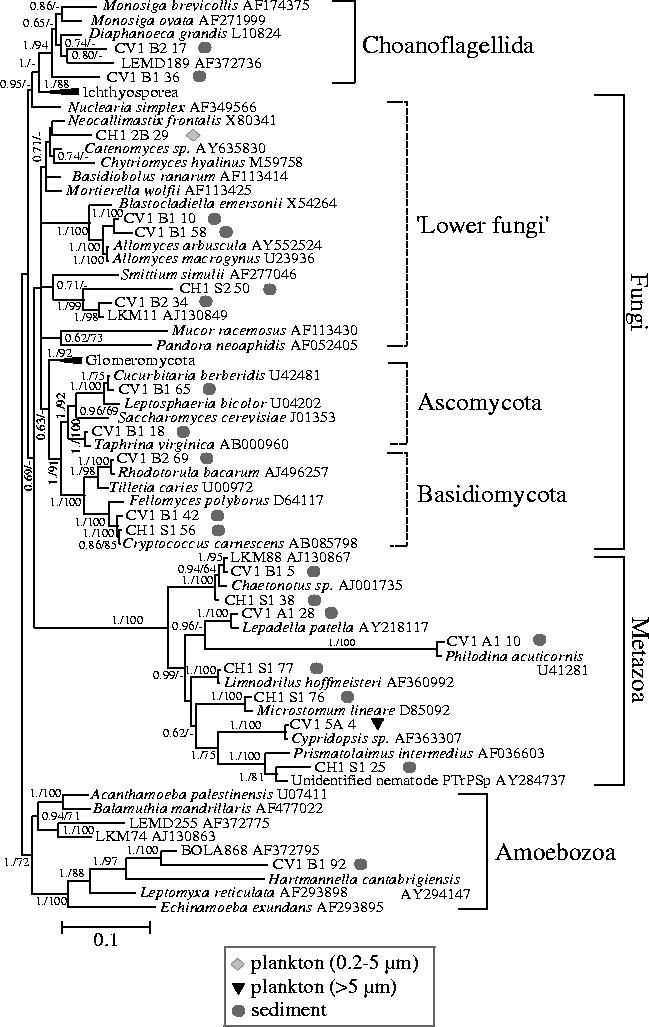
Bayesian phylogenetic tree of opisthokont and amoebozoan SSU rDNA sequences retrieved from sediment and plankton of a suboxic pond (CV1) and an oxic freshwater (CH1) shallow lake. Sixty-seven sequences and 1345 sites were used. For the chimerical phylotype LEMD255, only the amoebozoan portion (nucleotides 1–1379) was included in the analysis (see Berney et al. 2004). Sequences derived from this study are highlighted in bold. Geometric symbols indicate the type of environment in which each phylotype was identified. Posterior probabilities are given on the left at nodes. Bootstrap values obtained in maximum likelihood analyses are given at nodes (right) only when they are greater than 60%. The scale bar represents the number of substitutions per 100 positions per unit branch length.
Figure 3.
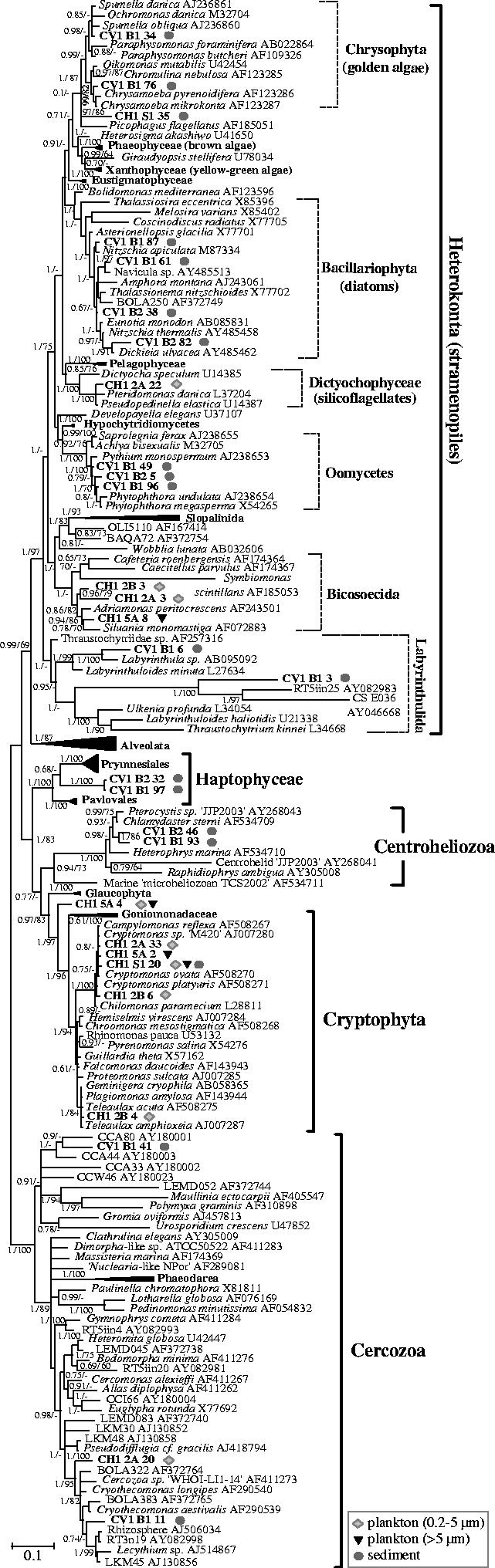
Bayesian phylogenetic tree of SSU rDNA sequences retrieved from sediment and plankton of a suboxic pond (CV1) and an oxic freshwater (CH1) shallow lake covering the diversity of heterokonts, haptophytes, centrohelid Heliozoa, cryptophytes and Cercozoa. One hundred and ninety sequences and 1179 sites were used. Sequences derived from this study are highlighted in bold. Geometric symbols indicate the type of environment in which each phylotype was identified. Posterior probabilities are given on the left at nodes. Bootstrap values obtained in maximum likelihood analyses are given at nodes (right) only when they are greater than 60%. The scale bar represents the number of substitutions per 100 positions per unit branch length.
Figure 4.
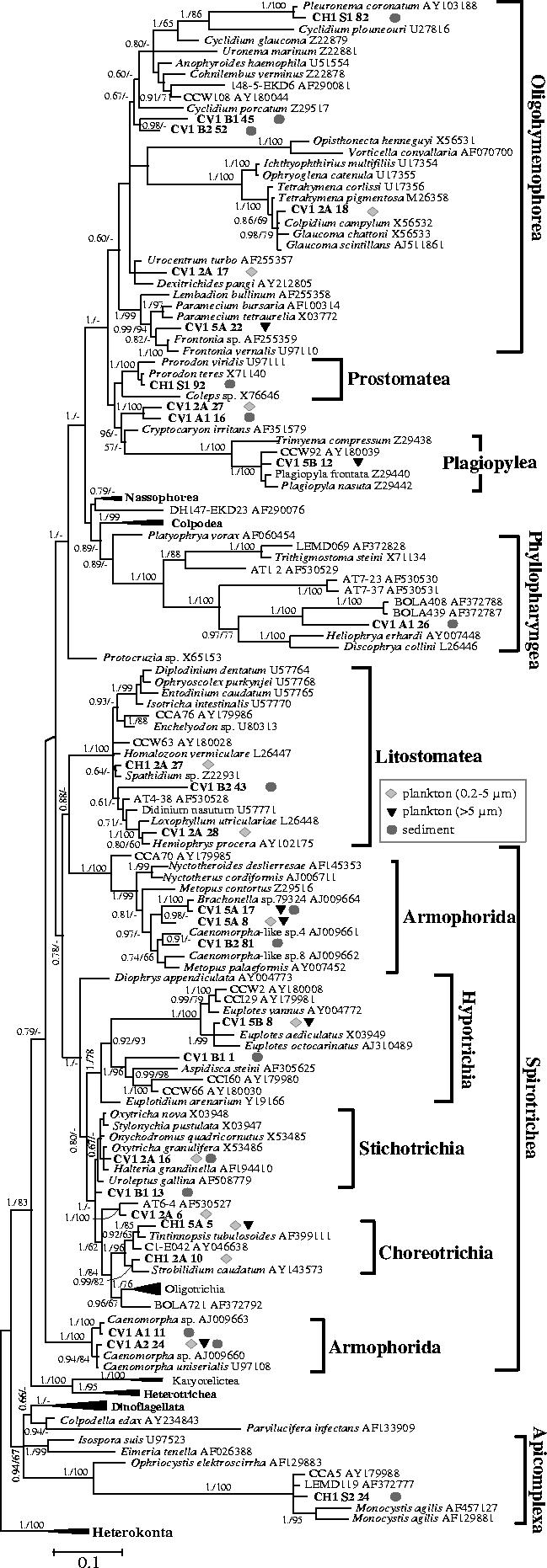
Bayesian phylogenetic tree of alveolate SSU rDNA sequences retrieved from sediment and plankton of a suboxic pond (CV1) and an oxic shallow lake (CH1). One hundred and forty eight sequences and 1099 sites were used. Sequences derived from this study are highlighted in bold. For the chimerical phylotypes AT6-4 and LEMD119 only the ciliate (nucleotides 332–1708) and gregarine (nucleotides 1–901) parts were included in the analysis (see Berney et al. 2004). For sequences retrieved in this work, geometric symbols indicate the type of environment in which each phylotype was identified. Posterior probabilities are given on the left at nodes. Bootstrap values obtained in maximum likelihood analyses are given at nodes (right) only when they are greater than 60%. The scale bar represents the number of substitutions per 100 positions per unit branch length.
3. Results and discussion
(a) Overall eukaryotic diversity in two, oxic and suboxic, freshwater systems
We carried out a molecular survey of the eukaryotic diversity in the sediment and plankton in two contrasting freshwater environments. One was a common permanent shallow lake in a temperate wooded region in France, while the other, also located in the same geographic area, was suboxic due to a very limited water circulation and a massive input of organic matter from tree leaves and the surrounding vegetation. The degradation of this organic material triggers a rapid oxygen consumption by heterotrophic oxygen-respiring microorganisms. Reduced gases, H2S and CH4, are abundantly degassed from the system when its waters are stirred, attesting to the presence of active sulphate-reducing bacteria and methanogenic Archaea which, being strict anaerobes, characterize oxygen-depleted environments. In both cases, we studied the diversity of eukaryotic SSU rDNA sequences retrieved from sediment and from two different planktonic fractions: that in the range 0.2–5 μm encompassing the picoplankton (lesser than or equal to 2 μm) and small nanoplankton (2–20 μm), and that greater than 5 μm encompassing larger planktonic organisms. Since small nanoplankton and, most especially, picoplankton in freshwater systems have been traditionally less studied, this type of analysis might reveal previously unrecognized eukaryotic lineages.
After DNA extraction and amplification of eukaryotic SSU rDNA with different primer combinations, 14 libraries were constructed. A total of 377 eukaryotic partial sequences (ca 750–1000 bp) were determined. Their taxonomic distribution was very wide, revealing the presence of members of 13 major eukaryotic phyla (figure 1). However, their partition in the plankton and sediment of the two freshwater systems was very unequal, revealing important differences in community composition. The most remarkable was the overwhelming presence of ciliate sequences in both the plankton and sediment of the suboxic pond (CV1). Ciliates were also present in sediment and water of the oxic lake (CH1) but, although relatively abundant, they did not dominate CH1 libraries. In turn, the most abundant clones in the oxic lake belonged to planktonic cryptophytes, which were absent from the suboxic pond libraries. Heterokonts (stramenopiles) were also remarkably abundant in sediment, but not in the plankton libraries from the suboxic system. However, they were present in plankton and sediment in the oxic lake in much lower proportions. Metazoan clones were similarly highly abundant in libraries obtained from sediment in both freshwater systems. Animals were not detected in plankton because they were probably filtered away during the pre-filtration step (greater than 150 μm). The high number of metazoan clones obtained resulted most likely from the bias derived from the fact that, being multicellular, they contributed large amounts of DNA. Nevertheless, animal sequences were relatively diverse (see below), implying that this bias is limited. The other eukaryotic group that was relatively abundant in our libraries corresponded to the fungi in the sediment of the suboxic system. The rest of eukaryotic groups were usually represented by one or a few sequences that were almost invariably retrieved from sediment libraries, either from the oxic or the suboxic pond. The only exception corresponded to two parabasalid sequences identified in the suboxic CV1 picoplanktonic fraction.
Figure 1.
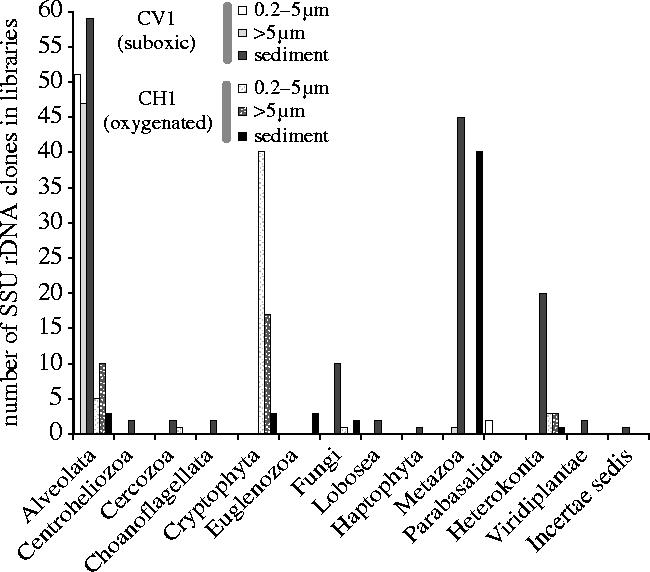
Taxonomic distribution of eukaryotic SSU rDNA phylotypes retrieved from sediment and two different planktonic size fractions in a suboxic (CV1) and a normal, oxygenated (CH1) freshwater system. For details see Electronic Appendix table S1.
The diversity of eukaryotic clones identified was not only very large among major eukaryotic phyla, but also the sequences belonging to each detected phylum were generally highly diverse. This was particularly striking for the fungi and heterokonts, with 77 and 64% of the detected sequences diverging by greater than 3% nucleotide identity, respectively (see below and Electronic Appendix—table S1). Once that we had had a first insight in the overall eukaryotic diversity of our samples, and from the total 377 eukaryotic partial sequences obtained, we identified 83 groups of sequences that shared more than 97% nucleotide identity, which we used as threshold to define phylotypes, and we selected one member of each to be fully sequenced. Detailed phylogenetic analyses were then carried out using different sets of sequences covering the whole eukaryotic diversity, and particularly that of the most represented phyla, in order to place our environmental phylotypes in trees.
(b) Opisthokonta and Amoebozoa
With the exception of one crustacean and one fungal sequence retrieved from plankton, all amoebozoan and opisthokont sequences were identified in sediment (figure 2 and Electronic Appendix—table S1). Only one amoebozoan phylotype, CV1-B1-92, was identified in our libraries that probably represents a true lobose amoeba. It was 88% identical to the environmental clone BOLA868, retrieved from marine sediment in a tidal flat (Dawson & Pace 2002), and very distantly related to Hartmannella cantabrigiensis. On the contrary, the fungal phylotypes were very diverse (figure 2), none of them being represented by more than two clones in our libraries. Notably, two nearly identical basidiomycete sequences (CV1-B1-42 and CH1-S1-56, 99% identity) were obtained from each one of the freshwater systems studied. They were almost identical, in turn, to Cryptococcus carnescens. The rest of basidiomycete and ascomycete phylotypes were found to be greater than 98% identical to known fungi as well. However, the remaining fungal phylotypes were in general much more divergent. They ascribed to the poorly resolved ‘lower fungi’ (figure 2), including phylotypes related to the ubiquitous water mold Allomyces spp. or to the environmental sequence LKM11, retrieved from an anaerobic digestor (van Hannen et al. 1999). The finding of such a level of divergence between our phylotypes and those from known species suggests that the diversity of this heterogeneous group of fungi (lower fungi) is still very poorly known despite the fact that these flagellated fungi occupy a pivotal phylogenetic position to understand fungal origin and evolution (Bowman et al. 1992; Cavalier-Smith & Chao 2003b).
Interestingly, we identified two quite divergent phylotypes (CV1-B2-17 and CV1-B1-36) related to choanoflagellates in the anoxic sediments of the suboxic pond. Choanoflagellates are phylogenetically among the closest relatives to animals (King & Carroll 2001; Snell et al. 2001). They are ubiquitous in aquatic habitats but, to our knowledge, they have never been described in suboxic or anoxic environments. The only putative exception could correspond to the phylotype LEMD189 from the sediment of lake Lemon, another divergent sequence apparently related to choanoflagellates but not robustly related to our sequences (Dawson & Pace 2002). Nevertheless, since our sequences are very divergent, they might also be morphologically distant from known choanoflagellates, and hence, difficult to identify as such. As mentioned, with the exception of one crustacean sequence (CV1-5A-4), metazoan phylotypes recovered from both freshwater sites were exclusively found in sediments (figure 2). In both cases, the most abundant sequences belonged to the gastrotrichs, represented by two almost identical phylotypes (CV1-B1-5 and CH1-S1-38) related to Chaetonotus spp. and to one environmental sequence (LKM88) retrieved from an anaerobic digestor (van Hannen et al. 1999). Chaetonotus is a genus of worm-like aquatic invertebrates frequent in the interstitial waters of fine sediments or on overlying detritus (Margulis & Schwartz 1998). Rotifers are widespread in freshwater habitats, and in fact, two rotifer phylotypes (CV1-A1-28, CV1-A1-10) were recovered from CV1 sediments. They were closely related to the typical benthic rotifers Lepadella patella and Philodina acuticornis, respectively. In addition, we retrieved two phylotypes closely related to the tubificid oligochaete Limnodrilus hoffmeisteri, a cosmopolitan and abundant bioturbator annelid often found in polluted areas (Margulis & Schwartz 1998), and to the turbellarian platyhelminth Microstomum lineare, and a third phylotype (CH1-S1-25) that was distantly related to several nematode species.
(c) Diverse bikont phyla
A large fraction of eukaryotic sequences in our libraries was distributed in a variety of bikont phyla including alveolates, cryptophytes, haptophytes, heterokonts (stramenopiles), Cercozoa, centrohelid Heliozoa, euglenids and parabasalids. Among the typical photosynthetic groups, the oxic freshwater lake was clearly dominated by cryptophyte sequences, while the suboxic pond showed a variety of photosynthetic heterokont lineages (figure 1). Cryptophytes were of relatively low diversity with two major lineages being detected, one closely related to Cryptomonas spp., and the other most similar to species of Teleaulax and other related marine genera (figure 3). Interestingly, most cryptophyte sequences were retrieved from the 0.2–5 μm fraction, where the most abundant phylotype was CH1-S1-20, almost identical to the freshwater Cryptomonas sp. M420 (Marin et al. 1998; Deane et al. 2002). This suggests that cryptophytes may be important contributors to the largely understudied freshwater eukaryotic picoplankton. In addition to bona fide cryptophytes, we retrieved a phylotype (CH1-5A-4), which branched robustly as sister group to the cryptophytes, including the plastid-lacking phagocytic Goniomonas spp. (figure 3 and Electronic Appendix—table S1). Its phylogenetic position is very interesting, and the identification and structural study of this lineage may shed light on the evolutionary history of cryptophytes. One appealing possibility would be that this phylotype belongs to kathablepharids, which are unclassified heterotrophic flagellates abundant in aquatic systems (Arndt et al. 2000) that have been proposed to be related to cryptophytes (Clay & Kugrens 1999; Cavalier-Smith 2004). Unfortunately, SSU rDNA sequences are not yet available for this group.
Cryptophytes were not detected in libraries from the suboxic pond, which suggests that they may be sensitive to oxygen-depleted environments, but a variety of phylotypes affiliated to typical heterokont photosynthetic groups were identified. Indeed, all likely photosynthetic heterokonts, diatoms and one chrysophyte lineage, were detected exclusively in the sediment fraction of the suboxic pool (figure 3 and Electronic Appendix—table S1). Four different phylotypes related to pennate diatom species were identified. Diatoms are mostly benthic, which explains their association with the sediment fraction. The chrysophyte phylotype CV1-B1-76 was highly similar to species of the photosynthetic genus Chrysamoeba, amoeboid organisms with filopodial extensions (Andersen et al. 1999). The other chrysophyte phylotype (CV1-B1-34) was most closely related to the colourless Spumella genus and to the photosynthetic genus Ochromonas and, therefore, its physiology cannot be deduced. No additional sequences belonging to chrysophytes were detected, although they represent abundant heterotrophic flagellates in freshwater (Arndt et al. 2000). Although many silicoflagellate species are photosynthetic, the only silicoflagellate sequence we detected, CH1-2A-22 from the oxic lake picoplankton, was closely related to the colourless genera Pteridomonas and Pseudopedinella, suggesting that it is a phagotrophic organism. As a matter of fact, a wide diversity of typical heterotrophic heterokont lineages was detected in both freshwater ponds. These comprised the saprophytic oomycetes (three phylotypes) and labyrinthulids (two phylotypes), which were retrieved from the anoxic and extremely organic matter-rich sediment of CV1, and the predatory bicosoecids (three phylotypes), detected in the plankton of the oxic lake (figure 3). Additionally, the phylotype CH1-S1-35 occupies an uncertain position in the heterokont tree. Within the labyrinthulids, our clone CV1-B1-3, from anoxic sediment, formed a very robust monophyletic cluster with two very distantly related environmental clones from hydrothermal sediment of the Guaymas basin (CS_E036) and from the extremely acidic river Tinto (RT5iin25) (Amaral Zettler et al. 2002; Edgcomb et al. 2002). Not only are the environmental sequences forming this group very divergent among themselves, but the group as a whole is very distant from known species of labyrinthulids.
An interesting cluster was formed by two phylotypes closely related to each other that were robustly placed in phylogenetic analyses either within the haptophytes (figure 3) or at their base (not shown). Together with Pavlovales and Prymnesiophyta, this cluster would form a third lineage of haptophytes. Although known haptophytes are photosynthetic, the degree of divergence of this third cluster does not allow any safe assumption about its actual lifestyle to be made. Haptophytes have been essentially described in oceanic waters, with only sparse records from freshwater (Green et al. 1990; Edvardsen et al. 2000). The discovery of a third, divergent, group indicates that the diversity and ecological distribution of this phylum may be much broader than previously thought.
Another group detected in the sediment of the suboxic pond was that of centrohelid Heliozoa, characteristic predators frequent in freshwater. We detected two phylotypes (CV1-B2-46 and CV1-B1-93) that were related to each other forming an independent lineage within this phylum. In this same sediment, two cercozoan lineages were identified. CV1-B1-11 was related to Cryothecomonas and Lecythium spp. and to a number of environmental sequences including several from anaerobic environments (BOLA383, LKM45; van Hannen et al. 1999; Dawson & Pace 2002). CV1-B1-41 formed a cluster with a group of environmental sequences coming from anoxic sediment in a salt marsh (Stoeck & Epstein 2003). In addition, the cercozoan clone CH1-2A-20 was retrieved from the picoplankton of the oxic lake, which was related, although more distantly, to the same group of sequences that the clone CV1-B1-11 (figure 3). Cercozoa comprises a vast variety of eukaryotic lineages with very different lifestyles, although predators dominate, which frequently display reticulate filose pseudopodia (Cavalier-Smith & Chao 2003a). Recent molecular surveys indicate that this group is even more diverse than classical studies suggested, with at least nine novel groups at the taxonomic order level or above (Bass & Cavalier-Smith 2004).
Three euglenid sequences were identified in the sediment of the oxic lake. Although euglenids comprise various photosynthetic members, our phylotypes are most closely related to Petalomonas spp., which are colourless heterotrophs either phagotrophic or osmotrophic (Electronic Appendix—figure S1; Fleedale & Vickerman 2000). One tree sequence, CV1-B1-85, was identified in the sediment, possibly from a natural contaminant integrating the partially decomposed vegetal matter in the sediment (Electronic Appendix—figure S1). Curiously, despite the fact that the sediment contained a large amount of plant debris, it was the only green plant sequence retrieved, suggesting that the organic matter forming the sediment is degraded very rapidly. One parabasalid phylotype was detected in the plankton of the suboxic system, CV1-2B-35, most likely corresponding to a parasite (Electronic Appendix—figure S1).
(d) Ciliates
By far, the most abundant and diverse group detected in our libraries corresponded to the alveolates and, most particularly, the ciliates (figures 1 and 4). Only one sequence (CH1-S2-24) belonging to one Apicomplexa affiliated to the parasitic gregarines, was identified. The rest of the alveolate sequences were widely distributed in different ciliate groups (figure 4). A total of 21 distinct phylotypes were detected in the suboxic system, where ciliates appeared to be dominant, while five different phylotypes were retrieved from the oxic lake. This corroborated previous observations by light microscopy revealing a remarkable diversity of ciliate morphologies (not shown), especially in the suboxic system. Ciliates are major consumers of bacteria and other protists and, although they are fundamentally aerobic protists, several ciliate groups have independently adapted to anoxic environments and are among the most usual eukaryotes in anaerobic communities (Fenchel & Finlay 1995). Known groups of anaerobic ciliates comprise the order Armophorida, including the families Caenomorphidae (Caenomorpha) and Metopidae (Metopus, Brachonella), and the order Plagiopylida, including the families Plagiopylidae (Plagiopyla), Trimyemidae (Trimyema) and Sonderiidae (Sonderia; Lynn & Small 2000). Phylotypes belonging to nearly all of them have been retrieved from the suboxic pond (figure 4 and Electronic Appendix—table S1). One of the two phylotypes closely related to Caenomorpha uniserialis, CV1-A2-24, was the most abundantly retrieved from both CV1 plankton and sediment libraries. CV1-5A-17 was almost identical (99.5%) to Brachonella sp., CV1-5A-8 and CV1-B2-81 were distantly related to other sequences of Brachonella and Metopus spp., and CV1-5B-12 grouped with sequences of anaerobic Plagiopyla spp. and an environmental clone from a suboxic salt marsh (CCW92; Stoeck & Epstein 2003). In addition to phylotypes related to characteristic anaerobic ciliates, many sequences affiliated to other ciliate groups, including oligohymenophorean, prostomatean, phyllopharyngean, litostomatean, and the spirotrichean subclasses Hypotrichia, Stichotrichia, and Choreotrichia (figure 4).
In total, we detected ciliate phylotypes ascribing to 6 out of the ten major ciliate classes defined (Lynn & Small 2000). In many cases, our sequences were very closely related to known species or genera of ciliates. However, in other cases, we identified sequences that, although clearly affiliating to a particular class or subclass, are very distant to sequenced species and most likely belong to genera or groupings of higher taxonomic ranks for which no sequences were available. For instance, CV1-B1-45 and CV1-B2-52 forming a cluster within the Oligohymenophorea, CV1-B2-43 within the Litostomatea, or CV1-B1-1 within the Hypotrichia. Furthermore, in the case of the cluster formed by CV1-2A-27 and CV1-A1-16, it was not even possible to ascribe it to a particular class of ciliates. Its position in phylogenetic analyses was unstable despite the fact that both sequences were slow-evolving. In addition, an extreme example may be constituted by the highly divergent, very fast-evolving sequence CV1-A2-16, which appeared to branch within ciliates in several, but not all, phylogenetic analyses, and was not related to any known ciliate group (Electronic Appendix—figure S1).
(e) How extensive is our knowledge of protist diversity?
Our observations appear to confirm a trend profiled in previous molecular eukaryotic diversity surveys carried out in different biotopes. Many phylotypes very distant from known species or genera were detected, even in environments that have been traditionally studied more such as freshwater systems (Berney et al. 2004 and this work). It is possible that a number of these phylotypes correspond to described species or groups for which the SSU rDNA sequence is not yet available (Baldauf 2003). This may be the case even for ciliates, which have been studied for several centuries and are one of the protist groups for which both, morphological and SSU rDNA records are better sampled. Nevertheless, ciliate SSU rDNA sequences for representatives of all ciliate classes and virtually all subclasses are available in databases (Lynn & Small 2000). In this study, we detected not only divergent ciliate sequences within classes or subclasses, but also sequences that were difficult to classify within the known subclasses and even classes. For instance, the environmental clones BOLA721 (Dawson & Pace 2002), AT6-4 (López-García et al. 2003) and CV1-2A-6 (this work) might define new spirotrichean subclasses. The case of the Phyllopharyngea is particularly remarkable, since most of the environmental sequences branching within this group are extremely distant from known species (figure 4), and the clusters defined by AT1-2, AT7-23 and AT7-37, and BOLA408, BOLA439 and CV1-A1-26 (Dawson & Pace 2002; López-García et al. 2003 and this work) could represent new subclasses as well. Furthermore, as mentioned above, CV1-2A-27 and CV1-2A-1 did not clearly affiliate to any ciliate class, and this was also the case for other environmental sequences such as DH147–EKD23 (López-García et al. 2001). Recent works have shown that morphologically well defined species may have highly divergent SSU rDNA sequences that do not branch within the expected ciliate classes (Johnson et al. 2004). This might be also the case for several of our divergent sequences and, consequently, our results have to be considered cautiously until their eventual validation by morphological identification. Remarkably, a relatively high frequency of divergent sequences is seen even when saturation has not been reached in the few molecular surveys carried out to date, including this study (not shown). Moreover, the criteria that we applied to define a phylotype may underestimate the actual diversity. We arbitrarily used a threshold of greater than 97% identity to define a phylotype in this work, but cells with greater than 97% identity at the level of their SSU rDNA may very well belong to different species.
Altogether, these observations suggest that an important fraction of the eukaryotic diversity is still awaiting discovery, even for very well known groups such as the ciliates. This is in sharp contrast with the idea of Finlay, Fenchel and co-workers (Finlay 1998, 2002, 2004; Finlay et al. 1998; Finlay & Fenchel 1999; Finlay & Esteban 2001) that nearly all free-living ciliate species have already been discovered, particularly in freshwater environments. On the contrary, even having taken into account the limitation imposed by the lack of sequenced SSU rDNAs from all described species, our data tend to support the opposite view, i.e. only a limited fraction of ciliate diversity has been sampled, in agreement with Foissner (1999). According to him, the number of free-living ciliate species may exceed by at least one order of magnitude that of those already described. The slow increase in new species descriptions would be due to the decreasing number of ciliate taxonomists rather than to an actual low species number, and to the fact that most environments remain underexplored, including the soil (Foissner 1999). The latter appears confirmed by the identification of many novel, divergent ciliate sequences coming from the deep-sea and other unstudied habitats (López-García et al. 2001; Dawson & Pace 2002; Edgcomb et al. 2002; López-García et al. 2003) but, nevertheless, even in the well studied freshwater habitats, the frequency of new divergent sequences seems remarkably high. An additional problem for ciliate taxonomists is the occurrence of cryptic species, which are morphologically indistinguishable but genetically different. Cryptic species have been observed in ciliates (Nanney et al. 1998) and other protozoan groups (de Vargas et al. 1999; Saez et al. 2003). It is true that the opposite case is known to occur as well, since dimorphic species have been identified in some protist groups, such as the cryptophytes (Hoef-Emden & Melkonian 2003). However, molecular surveys are independent of morphological observations, and therefore, overcome this type of bias.
What is observed for ciliates appears to be also the case for other, less-studied, eukaryotic phyla. Although Patterson suggested that most heterotrophic flagellates were already known (Patterson et al. 2000), similar to what Finlay & Fenchel (1999) stated for ciliates, recent molecular surveys suggest that general protist diversity is very poorly known both in freshwater and other environments. Thus, the global diversity of Cercozoa (Bass & Cavalier-Smith 2004), bodonids (López-García et al. 2003; von der Heyden et al. 2004) and heterotrophic heterokonts (Massana et al. 2004) appears to be huge. Altogether, this points to the existence of a large hidden eukaryotic diversity whose actual extent will be very difficult to evaluate until more comprehensive molecular surveys combined with classical studies in different environments become available.
Note added in proof
The SSU rDNA sequences from two kathablepharid species have recently been published by Okamoto and Inouye (2005, Protist 156, 163–179). Phylogenetic analysis indicates that those sequences form a monophyletic group with our clone CH1-5A-4, confirming the suspicion mentioned above.
Acknowledgments
This work was supported by an ATIP grant of the French Centre National de la Recherche Scientifique, section ‘Dynamique de la biodiversité’.
Supplementary Material
References
- Amaral Zettler L.A, Gomez F, Zettler E, Keenan B.G, Amils R, Sogin M.L. Microbiology: eukaryotic diversity in Spain's River of Fire. Nature. 2002;417:137. doi: 10.1038/417137a. [DOI] [PubMed] [Google Scholar]
- Andersen R.A, Van de Peer Y, Potter D, Sexton J.P, Kawachi M, LaJeunesse T. Phylogenetic analysis of the SSU rRNA from members of the Chrysophyceae. Protist. 1999;150:71–84. doi: 10.1016/s1434-4610(99)70010-6. [DOI] [PubMed] [Google Scholar]
- Arndt H, Dietrich D, Auer B, Cleven E.-J, Grafenhan T, Weitere M, Mylnikov A.P. Functional diversity of heterotrophic flagellates in aquatic ecosystems. In: Leadbeater B.S.C, Green J.C, editors. The flagellates: unity, diversity and evolution. Taylor & Francis; London: 2000. pp. 240–268. [Google Scholar]
- Baldauf S.L. The deep roots of eukaryotes. Science. 2003;300:1703–1706. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]
- Bass D, Cavalier-Smith T. Phylum-specific environmental DNA analysis reveals remarkably high global biodiversity of Cercozoa (Protozoa) Int. J. Syst. Evol. Microbiol. 2004;54:2393–2404. doi: 10.1099/ijs.0.63229-0. [DOI] [PubMed] [Google Scholar]
- Berney C, Fahrni J, Pawlowski J. How many novel eukaryotic ‘kingdoms’? Pitfalls and limitations of environmental DNA surveys. BMC Biol. 2004;2:13. doi: 10.1186/1741-7007-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman B.H, Taylor J.W, Brownlee A.G, Lee J, Lu S.D, White T.J. Molecular evolution of the fungi: relationship of the Basidiomycetes, Ascomycetes, and Chytridiomycetes. Mol. Biol. Evol. 1992;9:285–296. doi: 10.1093/oxfordjournals.molbev.a040720. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. Only six kingdoms of life. Proc. R. Soc. B. 2004;271:1251–1262. doi: 10.1098/rspb.2004.2705. 10.1098/rspb.2004.2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T, Chao E.E. Phylogeny and classification of phylum Cercozoa (Protozoa) Protist. 2003a;154:341–358. doi: 10.1078/143446103322454112. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T, Chao E.E. Phylogeny of Choanozoa, Apusozoa, and other Protozoa and early eukaryote megaevolution. J. Mol. Evol. 2003b;56:540–563. doi: 10.1007/s00239-002-2424-z. [DOI] [PubMed] [Google Scholar]
- Clay B, Kugrens P. Systematics of the enigmatic kathablepharids, including EM characterization of the type species, Kathablepharis phoenikoston, and new observations on K. remigera comb. nov. Protist. 1999;150:43–59. doi: 10.1016/S1434-4610(99)70008-8. [DOI] [PubMed] [Google Scholar]
- Cole J.R, et al. The Ribosomal Database Project (RDP-II): previewing a new autoaligner that allows regular updates and the new prokaryotic taxonomy. Nucleic Acids Res. 2003;31:442–423. doi: 10.1093/nar/gkg039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson S.C, Pace N.R. Novel kingdom-level eukaryotic diversity in anoxic environments. Proc. Natl Acad. Sci. USA. 2002;99:8324–8329. doi: 10.1073/pnas.062169599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane J.A, Strachan I.M, Saunders G.W, Hill D.R.A, McFadden G.I. Cryptomonad evolution: nuclear 18S rDNA phylogeny versus cell morphology and pigmentation. J. Phycol. 2002;38:1236–1244. [Google Scholar]
- de Vargas C, Norris R, Zaninetti L, Gibb S.W, Pawlowski J. Molecular evidence of cryptic speciation in planktonic foraminifers and their relation to oceanic provinces. Proc. Natl Acad. Sci. USA. 1999;96:2864–2868. doi: 10.1073/pnas.96.6.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diez B, Pedros-Alio C, Massana R. Study of genetic diversity of eukaryotic picoplankton in different oceanic regions by small-subunit rRNA gene cloning and sequencing. Appl. Environ. Microbiol. 2001;67:2932–2941. doi: 10.1128/AEM.67.7.2932-2941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgcomb V.P, Kysela D.T, Teske A, de Vera Gomez A, Sogin M.L. Benthic eukaryotic diversity in the Guaymas Basin hydrothermal vent environment. Proc. Natl Acad. Sci. USA. 2002;99:7658–7662. doi: 10.1073/pnas.062186399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edvardsen B, Eikrem W, Green J.C, Andersen R.A, Moon-van der Staay S.Y, Medlin L.K. Phylogenetic reconstructions of the Haptophyta inferred from 18S ribosomal DNA sequences and available morphological data. Phycologia. 2000;39:19–35. [Google Scholar]
- Fenchel T, Finlay B.J. Oxford series in ecology and evolution. Oxford University Press; 1995. Ecology and evolution in anoxic worlds. [Google Scholar]
- Finlay B.J. The global diversity of Protozoa and other small species. Int. J. Parasitol. 1998;28:29–48. doi: 10.1016/s0020-7519(97)00167-7. [DOI] [PubMed] [Google Scholar]
- Finlay B.J. Global dispersal of free-living microbial eukaryote species. Science. 2002;296:1061–1063. doi: 10.1126/science.1070710. [DOI] [PubMed] [Google Scholar]
- Finlay B.J. Protist taxonomy: an ecological perspective. Phil. Trans. R. Soc. B. 2004;359:599–610. doi: 10.1098/rstb.2003.1450. 10.1098/rstb.2003.1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay B.J, Esteban G.F. Exploring Leeuwenhoek's legacy: the abundance and diversity of protozoa. Int. Microbiol. 2001;4:125–133. doi: 10.1007/s10123-001-0027-y. [DOI] [PubMed] [Google Scholar]
- Finlay B.J, Fenchel T. Divergent perspectives on protist species richness. Protist. 1999;150:229–233. doi: 10.1016/S1434-4610(99)70025-8. [DOI] [PubMed] [Google Scholar]
- Finlay B.J, Esteban G.F, Fenchel T. Protozoan diversity: converging estimates of the global number of free-living ciliate species. Protist. 1998;149:29–37. doi: 10.1016/S1434-4610(98)70007-0. [DOI] [PubMed] [Google Scholar]
- Fleedale G, Vickerman K. Phylum Euglenozoa. In: Lee J.J, Leedale G.F, Bradbury P, editors. An illustrated guide to the Protozoa. vol. 2. Allen Press; Lawrence, KS: 2000. pp. 1135–1185. [Google Scholar]
- Foissner W. Protist diversity: estimates of the near-imponderable. Protist. 1999;150:363–368. doi: 10.1016/S1434-4610(99)70037-4. [DOI] [PubMed] [Google Scholar]
- Green J.C, Perch-Nielsen K, Westbroek P. Phylum Prymnesiophyta. In: Margulis L, Corliss J.O, Melkonian M, Chapman D.J, editors. Handbook of Protoctista. Jones and Bartlett Publishers; Boston: 1990. pp. 293–317. [Google Scholar]
- Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- Hoef-Emden K, Melkonian M. Revision of the genus Cryptomonas (Cryptophyceae): a combination of molecular phylogeny and morphology provides insights into a long-hidden dimorphism. Protist. 2003;154:371–409. doi: 10.1078/143446103322454130. [DOI] [PubMed] [Google Scholar]
- Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–2319. doi: 10.1093/bioinformatics/bth226. [DOI] [PubMed] [Google Scholar]
- Johnson M.D, Tengs T, Oldach D.W, Delwiche C.F, Stoecker D.K. Highly divergent SSU rRNA genes found in the marine ciliates Myrionecta rubra and Mesodinium pulex. Protist. 2004;155:347–359. doi: 10.1078/1434461041844222. [DOI] [PubMed] [Google Scholar]
- King N, Carroll S.B. A receptor tyrosine kinase from choanoflagellates: molecular insights into early animal evolution. Proc. Natl Acad. Sci. USA. 2001;98:15 032–15 037. doi: 10.1073/pnas.261477698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-García P, Rodríguez-Valera F, Pedrós-Alió C, Moreira D. Unexpected diversity of small eukaryotes in deep-sea Antarctic plankton. Nature. 2001;409:603–607. doi: 10.1038/35054537. [DOI] [PubMed] [Google Scholar]
- López-García P, Philippe H, Gail F, Moreira D. Autochthonous eukaryotic diversity in hydrothermal sediment and experimental microcolonizers at the Mid-Atlantic Ridge. Proc. Natl Acad. Sci. USA. 2003;100:697–702. doi: 10.1073/pnas.0235779100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn D.H, Small E.B. Phylum Ciliophora. In: Lee J.J, Leedale G.F, Bradbury P, editors. An illustrated guide to the Protozoa. vol. 1. Allen Press; Lawrence, KS: 2000. pp. 371–656. [Google Scholar]
- Margulis L, Schwartz K.V. 3rd edn. W. H. Freeman; New York: 1998. Five kingdoms: an illustrated guide to the phyla of life on Earth. 520pp. [Google Scholar]
- Marin B, Klingberg M, Melkonian M. Phylogenetic relationships among the cryptophyta: analyses of nuclear-encoded SSU rRNA sequences support the monophyly of extant plastid-containing lineages. Protist. 1998;149:265–276. doi: 10.1016/S1434-4610(98)70033-1. [DOI] [PubMed] [Google Scholar]
- Massana R, Castresana J, Balague V.V, Guillou L, Romari K, Groisillier A, Valentin K, Pedros-Alio C. Phylogenetic and ecological analysis of novel marine stramenopiles. Appl. Environ. Microbiol. 2004;70:3528–3534. doi: 10.1128/AEM.70.6.3528-3534.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon-van der Staay S.Y, De Wachter R, Vaulot D. Oceanic 18S rDNA sequences from picoplankton reveal unsuspected eukaryotic diversity. Nature. 2001;409:607–610. doi: 10.1038/35054541. [DOI] [PubMed] [Google Scholar]
- Moreira D, López-García P. The molecular ecology of microbial eukaryotes unveils a hidden world. Trends Microbiol. 2002;10:31–38. doi: 10.1016/s0966-842x(01)02257-0. [DOI] [PubMed] [Google Scholar]
- Nanney D.L, Park C, Preparata R, Simon E.M. Comparison of sequence differences in a variable 23S rRNA domain among sets of cryptic species of ciliated protozoa. J. Eukaryot. Microbiol. 1998;45:91–100. doi: 10.1111/j.1550-7408.1998.tb05075.x. [DOI] [PubMed] [Google Scholar]
- Patterson D.J, Vors N, Simpson A.G.B, O'Kelly C. Residual free-living and predatory heterotrophic flagellates. In: Lee J.J, Leedale G.F, Bradbury P, editors. An illustrated guide to the Protozoa. vol. 2. Allen Press; Lawrence, KS: 2000. pp. 1302–1328. [Google Scholar]
- Romari K, Vaulot D. Composition and temporal variability of picoeukaryote communities at a coastal site of the English Channel from 18S rDNA sequences. Limnol. Oceanogr. 2004;49:784–798. [Google Scholar]
- Ronquist F, Huelsenbeck J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003;19:1572–1574. doi: 10.1093/bioinformatics/btg180. [DOI] [PubMed] [Google Scholar]
- Saez A.G, Probert I, Geisen M, Quinn P, Young J.R, Medlin L.K. Pseudo-cryptic speciation in coccolithophores. Proc. Natl Acad. Sci. USA. 2003;100:7163–7168. doi: 10.1073/pnas.1132069100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell E.A, Furlong R.F, Holland P.W. Hsp70 sequences indicate that choanoflagellates are closely related to animals. Curr. Biol. 2001;11:967–970. doi: 10.1016/s0960-9822(01)00275-5. [DOI] [PubMed] [Google Scholar]
- Stoeck T, Epstein S. Novel eukaryotic lineages inferred from small-subunit rRNA analyses of oxygen-depleted marine environments. Appl. Environ. Microbiol. 2003;69:2657–2663. doi: 10.1128/AEM.69.5.2657-2663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeck T, Taylor G.T, Epstein S.S. Novel eukaryotes from the permanently anoxic Cariaco Basin (Caribbean Sea) Appl. Environ. Microbiol. 2003;69:5656–5663. doi: 10.1128/AEM.69.9.5656-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J.D, Gibson T.J, Plewniak F, Jeanmougin F, Higgins D.G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hannen E.J, Mooij W, van Agterveld M.P, Gons H.J, Laanbroek H.J. Detritus-dependent development of the microbial community in an experimental system: qualitative analysis by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 1999;65:2478–2484. doi: 10.1128/aem.65.6.2478-2484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Heyden S, Chao E.E, Vickerman K, Cavalier-Smith T. Ribosomal RNA phylogeny of bodonid and diplonemid flagellates and the evolution of Euglenozoa. J. Eukaryot. Microbiol. 2004;51:402–416. doi: 10.1111/j.1550-7408.2004.tb00387.x. [DOI] [PubMed] [Google Scholar]
- Weisse T. The significance of inter- and intraspecific variation in bacterivorous and herbivorous protists. Antonie Van Leeuwenhoek. 2002;81:327–341. doi: 10.1023/a:1020547517255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.