

Abstract
We show that Trichostatin A (TSA)-induced partial histone hyperacetylation causes a unidirectional shift in the position of a previously defined binding domain for the centromere-specific histone H3 homologue CENP-A at a human neocentromere. The shift of ∼320 kb is fully reversible when TSA is removed, but is accompanied by an apparent reduction in the density of CENP-A per unit length of genomic DNA at the neocentromere. TSA treatment also instigates a reversible abolition of a previously defined major domain of differentially delayed replication timing that was originally established at the neocentromeric site. None of these changes has any measurable deleterious effects on mitosis or neocentromere function. The data suggest pliability of centromeric chromatin in response to epigenetic triggers, and the non-essential nature of the regions of delayed replication for centromere function. Reversibility of the CENP-A-binding position and the predominant region of delayed replication timing following removal of TSA suggest strong memory at the original site of neocentromeric chromatin formation.
Keywords: CENP-A/centromere/chromatin/neocentromere/replication time
Introduction
Centromeres govern the proper segregation of eukaryotic chromosomes during cell division (Choo, 1997a). Human neocentromeres are ectopic centromeres formed de novo from previously non-centromeric regions of the human genome (Choo, 1997b). They are functionally identical to normal centromeres in mitotic activity and their binding to essential centromeric and pericentromeric proteins (Aagaard et al., 2000; Saffery et al., 2000). Unlike normal human centromeres that contain megabase tracts of tandemly repetitive α-satellite DNA, most reported human neocentromeres originate from regions of euchromatin (Choo, 1997b, 2001; Warburton et al., 2000). The first and best studied example of a neocentromere comes from the 10q25 region of a rearranged mardel(10) chromosome (Voullaire et al., 1993; du Sart et al., 1997; Barry et al., 1999, 2000; Lo et al., 2001a). The absence of any detectable difference in DNA sequence between the precursor 10q25 genomic region and the neocentromere implies an epigenetic process of neocentromerization (Choo, 2000). Using chromatin immunoprecipitation and genomic DNA array (CIA) analysis, we have previously defined a binding domain of ∼330 kb for the centromere-specific histone H3 homologue CENP-A at the 10q25 neocentromere (Lo et al., 2001a). We also showed that neocentromerization is associated with significant delay in replication to either side of, but not coincident with, the CENP-A-binding region (Lo et al., 2001a; also see Figure 4).

Fig. 4. Replication timing at the 10q25 neocentromere region. (A) No TSA treatment (0; data taken from Lo et al., 2001a). (B) Cells treated for 17 h with 33 nM TSA (33). (C) Cells treated for 17 h with 33 nM TSA followed by culture without TSA for 3 days (33/0). Average percentages of S phase FISH signal doublets (inversely proportional to replication time) for each probe are plotted against the midpoint position of each probe (shown above the graph) within the 5 Mb 10q25 contig for 1f (normal chromosome 10; red circles) and 5f [mardel(10); blue squares]. Results from untreated cells (0; Lo et al., 2001a) are shown with open symbols; results from TSA-treated cells obtained in the present study are shown as filled symbols. The dotted horizontal line at 50% FISH doublets represents the approximate mid-S phase time-point. Minimal CENP-A-binding domains before and after TSA-treatment are designated A1 and A2 and shown as light and darker grey areas, respectively. R1 and R2 boxes denote the regions of differentially delayed replication on mardel(10) compared with normal chromosome 10 in the absence of TSA (Lo et al., 2001a). Results are the averages ±1 SD of four separate experiments. Experiments for the untreated cells reported previously (Lo et al., 2001a) and the TSA-treated cells presented here were performed, and the two data sets collected and analysed, simultaneously.
Centromere function has been reported to be intimately linked to a hypoacetylated state of the core histones (Choo, 2000; Pidoux and Allshire, 2000). In fission yeast, heterochromatin proteins mediate this histone modification at sites flanking, but not including, the CENP-A-associated chromatin (Saitoh et al., 1997; Goshima et al., 1999; Partridge et al., 2000; Nakayama et al., 2001). Histone hypoacetylation also appears to be a feature of at least some mammalian centromeres (Jeppesen et al., 1992; Jeppesen and Turner, 1993). Furthermore, specific inhibition of histone deacetylase activity using Trichostatin A (TSA) has been shown to compromise centromere integrity and result in mislocalization of heterochromatin proteins and missegregation of chromosomes in yeast and mammals (Ekwall et al., 1997; Taddei et al., 2001).
In mammals, replication timing is closely linked to chromatin structure and transcriptional competency (Bickmore and Craig, 1997; Zink et al., 2001). For some mammalian imprinted genes, the inactive allele is associated with lower levels of histone acetylation and, where studied, a later replication time (Bickmore and Carothers, 1995; Hu et al., 1998; Pedone et al., 1999; El Kharroubi et al., 2001). These observations, together with the demonstration that differences in replication timing between the active and inactive alleles of imprinted loci are abolished in the presence of TSA (Bickmore and Carothers, 1995), suggest that histone acetylation status has a direct influence on replication time. Although mammalian centromeres, including the 10q25 neocentromere and other regions of hypoacetylated heterochromatin, generally replicate during the second half of S phase (Camargo and Cervenka, 1982; Ten Hagen et al., 1990; O’Keefe et al., 1992; Lo et al., 2001a), nothing is known about the effect that disrupting histone acetylation status may have on centromere replication patterns.
Using the 10q25 neocentromere as a model system, we have investigated the relationship between core histone acetylation, replication timing and centromere protein integrity. Specifically, we studied the effects of partial inhibition of histone deacetylation using a low concentration of TSA that caused minimal disruption to cell division. The results indicate significant and definable changes to CENP-A binding and replication at the neocentromere, providing new insight into the epigenetic regulation and pliability of mammalian centromeres.
Results
The following studies were based on the use of two somatic cell hybrid lines generated previously (du Sart et al., 1997; Lo et al., 2001a): 1f, which contained a normal human chromosome 10; and 5f, which contained the mardel(10) chromosome.
Effects of TSA concentration on cell growth and chromosome segregation
TSA-induced histone deacetylation can result in differentiation and cell cycle arrest in addition to chromosome segregation defects (Yoshida et al., 1995). We wanted to investigate the plasticity of centromeres under TSA-induced conditions that maintained normal or near-normal centromere function. This required the determination of a concentration of TSA that would result in minimal disruption to cellular functions and chromosome segregation properties while bestowing a partial but significant increase in histone acetylation at the level of the cell, chromosome and centromere. Treatment with 30–33 nM TSA for 16–18 h had previously been shown to induce partial relief of transcriptional repression in frog oocytes (Jones et al., 1998) and to induce replication timing changes in human lymphocytes (Bickmore and Carothers, 1997). In addition, Taddei et al. (2001) showed that 75 and 120 nM TSA enabled progression through multiple cell divisions in mouse and human cell lines, in addition to inducing some disruption in centromere structure. We have therefore used a range of concentrations of TSA (33, 75, 100, 150 and 300 nM) to treat 5f cells for up to 6 days. Concentrations of 150 and 300 nM significantly reduced cell viability and caused cell growth rates to fall below zero within 24 h (data not shown), and were excluded from further analysis. Figure 1 shows a comparison of the effects of 0, 33, 75 and 100 nM TSA on cell growth, viability and level of chromosome missegregation. Some effect in the slowing of cell growth rate was observed at 33 nM TSA (raising the average doubling time from 23.8 to 32.9 h; P < 0.0001), with this slowing effect noticeably accentuated at 75 nM (50.0 h) and 100 nM (51.7 h) compared with untreated cells (Figure 1A; P values <0.01 and <0.05 for 75 and 100 nM, respectively). No significant drop in cell viability was observed under these three drug concentrations (Figure 1B). An examination of unsynchronized anaphase II and telophase cells for lagging and bridging chromosomes indicated that 33 nM was the only concentration that did not induce a significant rise in the level of missegregating chromosomes compared with untreated cells (P values >0.05, <0.05 and <0.05 for 33, 75 and 100 nM TSA, respectively; Figure 1C). Similarly, when cells were analysed for the number of micronuclei present, 33 nM TSA was once again the only concentration showing no significant increase over untreated cells (P values >0.05, <0.01 and <0.01 for 33, 75 and 100 nM TSA, respectively; Figure 1D). Further analysis of cells treated with 33 nM TSA has indicated that the incidences of polyploidy and apoptotic-like cells were also not significantly elevated compared with untreated cells (data not shown).

Fig. 1. Effects of TSA concentration on cell growth and chromosome segregation. (A) Doubling time was determined by maintaining cells in exponential phase (with splitting every 2 days) for a total of 6 days. Results were averaged from six separate experiments. (B) Viability was determined by Trypan Blue staining from a sample of cells during each of the culture splits in (A). (C) The level of missegregation was calculated by scoring 50 cells for the number of anaphase II/telophase cells exhibiting lagging or bridging chromosomes. Results averaged from four separate experiments were divided by those of untreated cells to give relative levels of missegregation. (D) The level of micronuclei was calculated by scoring 1000 cells in four separate experiments and dividing the value by that of the untreated cells.
Based on the above results showing that 33 nM TSA did not measurably disrupt general chromosome segregation behaviour (but elicited a significant elevation in histone acetylation level; see below), we decided to further investigate the effect of this TSA concentration on the mitotic stability of the mardel(10) chromosome specifically. A 10q25 neocentromere-specific bacterial artificial chromosome (BAC) probe (bA153G5; see Figure 3) was used in fluorescence in situ hybridization (FISH) experiments to detect the mardel(10) chromosome. The results indicated that neither short-term (17 h) nor extended (3 months) exposure to 33 nM TSA had any measurable effect on mardel(10) stability (Table I). Immuno-FISH analysis performed with antibodies against centromere proteins CENP-A (CREST 6) and CENP-C, and the BAC probe bA153G5, further demonstrated the presence of these proteins on all the 10q25 neocentromeres scored (Table I). Taken together, these results indicate that short- or long-term 33 nM TSA treatment has no measurable deleterious effects on the mitotic functions of chromosomes in general, and mardel(10) in particular.

Fig. 3. Effects of TSA on CENP-A distribution at the mardel(10) neocentromere. (A) No TSA treatment (0; data taken from Lo et al., 2001a). (B) Treatment with 33 nM TSA for 17 h (33). (C) Treatment with 33 nM TSA for 17 h followed by 3 days without TSA (33/0). Data were collected using a previously described CIA analysis procedure (Lo et al., 2001a,b). Percent differences between the normalized values of 5f [mardel(10)] and 1f (normal chromosome 10) are plotted against the midpoint position of each BAC within the 5 Mb 10q25 BAC contig described previously (Lo et al., 2001a). Each data point is the mean of two to five independent experiments. All data points are shown with error bars (±SE). The grey box shows the CENP-A binding region defined in (A) (Lo et al., 2001a). Experiments for the untreated cells reported previously (Lo et al., 2001a,b) and the TSA-treated cells presented here were performed, and the two data sets collected and analysed, simultaneously. The designations for several pertinent BACs are shown.
Table I. Effects of 33 nM TSA treatment on the mardel(10) chromosome.
| TSA concentration(nM)a | % mardel(10)(17 h)b | % mardel(10)(3 months)b | % mardel(10) withCENP-A (17 h)c | % mardel(10) withCENP-C (17 h)c |
|---|---|---|---|---|
| 0 | 94.0 ± 1.0 | 94.7 ± 3.1 | 100 | 100 |
| 33 | 99.0 ± 1.7 | 95.7 ± 0.6 | 100 | 100 |
| P values | >0.05 | >0.05 | N/A | N/A |
a5f cells treated with 33 nM TSA for 17 h or 3 months were compared with untreated cells grown for the same amount of time. Results represent averages ± SD and t-test P values for four separate experiments.
bFifty to 100 nuclei were scored for the presence of mardel(10) chromosome, identified by FISH with the neocentromere-specific BAC probe bA153G5 (Lo et al., 2001a) following 17 h or 3 months of culture with or without 33 nM TSA.
cOne hundred metaphase spreads from unfixed, cytospun cells cultured with or without 33 nM TSA were examined by immuno-FISH using anti-CENP-A CREST serum (Lo et al., 2001a) and anti-CENP-C (Saffery et al., 1999) antibodies, and the bA153G5 probe, for the presence or absence of CENP-A and CENP-C on mardel(10).
Effects of TSA concentration on histone acetylation
The 5f cell line was further treated with 0, 33, 150 or 300 nM TSA for 17 h and the levels of acetylated histones H3 and H4 determined using western blotting (Figure 2A and B). The results showed significant elevation of acetylated H3 and H4 at 33 nM TSA, peaking at ∼150 nM TSA. As the core CENP-A-associated centromeric chromatin is known to be depleted of histone H3 (Choo, 2001; Lo et al., 2001a), further investigations focused only on histone H4.
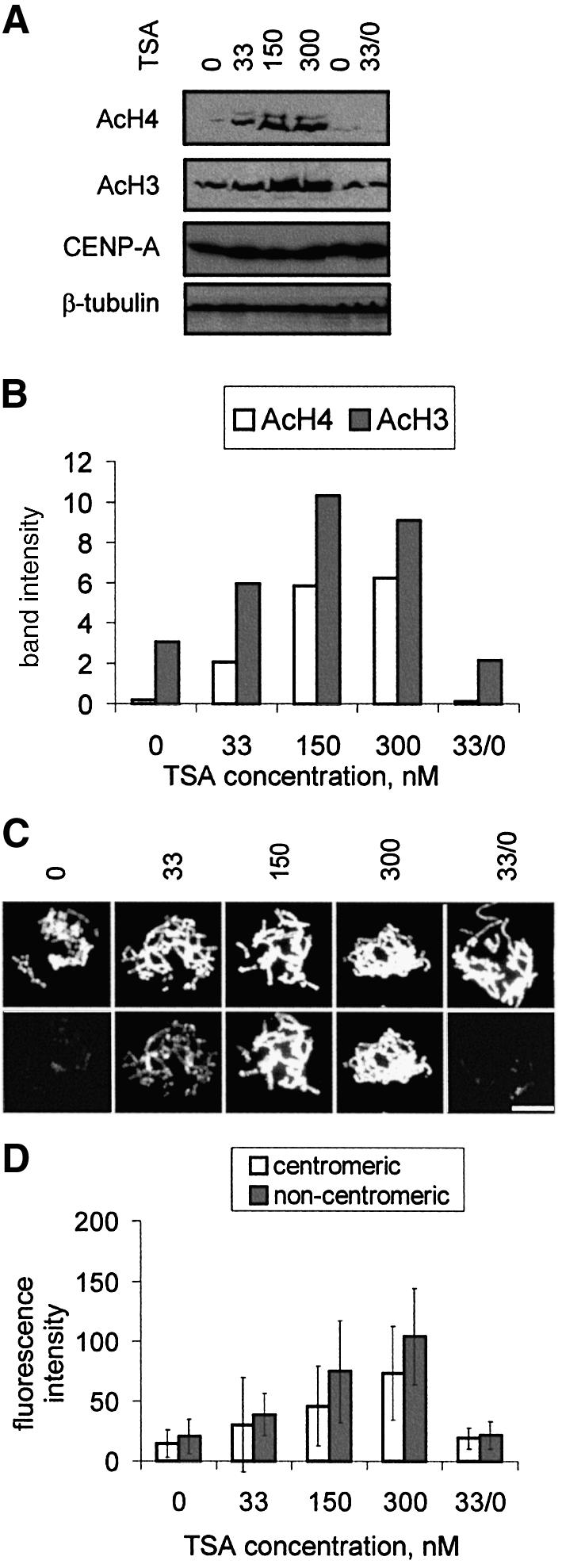
Fig. 2. Effects of TSA concentration on cellular levels of acetylated histones H4 (AcH4) and/or H3 (AcH3). (A and B) Western blotting. (A) 5f cells were cultured without TSA or with 33, 150 or 300 nM TSA for 17 h. In some experiments (33/0), cells were grown in the presence of 33 nM TSA for 17 h, washed, and grown in TSA-free medium for a further 3 days. Untreated controls (0) were grown for the same length of time. Total cell lysates from equivalent numbers of cells were run on denaturing SDS–PAGE and blotted with antibodies against acetylated histone H4 (AcH4), histone H3 (AcH3) or CENP-A. In all cases, equal gel loadings were confirmed using an anti-β-tubulin antibody. (B) Quantitation of band intensity in (A) for 0, 33, 150, 300 and 33/0 nM TSA for acetylated histones. (C and D) Immunofluorescence. (C) Metaphase chromosomes (DAPI stained in upper panels) from unfixed and cytospun 5f cells cultured as in (A) were stained with an antibody against acetylated histone H4 (lower panels). Bar, 10 µm. (D) Quantitation of fluorescence intensities in (C) at the centromeres and non-centromeric regions following TSA treatment. Signal intensities in (B) and (D) were given arbitrary units after correcting for background signals on the gels or slides.
Immunofluorescence using an antibody against acetyl ated histone H4 on unfixed 5f cells treated with different levels of TSA for 17 h indicated significant staining at 33 nM and maximal staining at 150 and 300 nM (Figure 2C), providing independent evidence that the elevation of cellular acetylated histone H4 detected by western blotting following TSA treatment was reflected at the chromosome level. Closer examination of the chromosomes indicated that some degree of banding was produced by the anti-acetylated H4 antibody without TSA, as has been described previously (Jeppesen et al., 1992; Jeppesen, 1997), and after each of the TSA treatments (data not shown). In addition to the visual observation of a gross elevation of acetylated H4 staining on chromosomes, we quantified the relative levels of antibody signals at the centromeres compared with surrounding regions. The results (Figure 2D) indicated that for all treatments, centromeric histone H4 acetylation was lower than those of surrounding non-centromeric chromatin (P = 0.001, 0.0003, 0.0002 and 00005 for 0, 33, 150 and 300 nM TSA, respectively), in agreement with previously published data (Jeppesen et al., 1992; Jeppesen and Turner, 1993; Johnson et al., 1998) and indicating that TSA affects centromeric histone acetylation to the same extent as on bulk chromatin. We also found a similar difference between the mardel(10) neocentromere and its surrounding non-centromeric chromatin (data not shown). It is noteworthy that since the CREST serum was used to counterstain the centromere, it is possible that this might have sterically hindered the binding of hyperacetylated histone H4 antibodies at the centromere, although to our knowledge no such phenomenon has been reported for such a standard dual-antibody immunofluorescence technique.
Since a key aspect of these studies involved addressing the question of whether the effects of TSA treatment at this concentration was reversible, we ascertained the histone H4 and/or H3 acetylation status in cells that were cultured for an additional 3 days in TSA-free media following the initial 17 h of incubation at 33 nM (33/0). We found a complete return to normal levels of acetylated histones by western blotting (Figure 2A and B) and immunofluorescence analysis (Figure 2C and D), demonstrating a full recovery of histone acetylation status from the TSA treatment, including a significant difference between centromeres and arms (P = 0.004), within this time span. We also found that a complete return to the normal level of hyperacetylated histone H4 could be achieved following 1 day of recovery in TSA-free culture media (see Supplementary data, available at The EMBO Journal Online), agreeing with the results shown in a previous similar study (Taddei et al., 2001). In addition, we have shown that the level of hyperacetylated histone H4 remained stable on chromosomes even after prolonged exposure to 33 nM TSA for 3 months (see Supplementary data).
Based on the above analyses, a level of 33 nM was chosen for the studies described below.
TSA alters CENP-A binding at the mardel(10) neocentromere
We have previously identified and delimited the CENP-A-binding domain at the mardel(10) neocentromere to a genomic region of ∼330 kb (Lo et al., 2001a). The method involved chromatin immunoprecipitation using a CENP-A-specific antibody on somatic cell hybrids containing either a normal chromosome 10 (1f) or a mardel(10) (5f). Immunoprecipitated chromatin was captured by protein A–Sepharose (bound fraction). DNA extracted from both input and bound fractions was randomly amplified by DOP (degenerate oligonucleotide-primed)-PCR for use as probes on an array of dot-blotted BACs mapped to the 10q25 sequence contig (Lo et al., 2001a). The difference between the ratios of enhancement (bound/input signal intensity) between 1f (normal chromosome 10) and 5f [mardel(10)] was calculated for each BAC, and the results plotted (Figure 3A; Lo et al., 2001a).
The above CIA analysis was repeated for 1f and 5f cells that had been exposed to 33 nM TSA for 17 h. Two significant effects on CENP-A binding were observed (Figure 3B). First, the treatment resulted in a shift in the position of the CENP-A binding domain toward the p′ side of the mardel(10) chromosome by ∼320 kb (measuring from the centres of the peaks). This shift, which is approximately the length of two average BACs, was accompanied by an apparent expansion of the CENP-A-binding domain from 330 to 480 kb. However, these domain sizes should be regarded as upper estimates, since the precise boundary for CENP-A binding within the two outermost BAC clones constituting the shifted (i.e. BACs bA87E14 and bA69K10) and pre-shift (i.e. BACs bA87P3 and bA87E14) peaks remains to be determined. Secondly, measurement of the area under the curves suggested that the magnitude of the CENP-A-binding domain was diminished by approximately one-third following TSA treatment, suggesting some loss of CENP-A at the neocentromere. This occurred without any measurable changes in the overall level of cellular CENP-A expression (Figure 2A).
We next studied the effects of TSA removal. A full return of the CENP-A-binding domain to both its original position and expanse was observed (Figure 3C). However, the level of CENP-A binding (area under the curve) appeared to be further diminished by ∼50% compared with the original value in untreated cells. We have also shown that the reversible return of the CENP-A-binding domain to its original position can occur within 1 day of the removal of TSA (data not shown), coincident with the return of the levels of chromosomal acetylated histones to baseline 1 day after the removal of TSA (see Supplementary data).
To estimate the density of CENP-A molecules per unit length of DNA, we divided the area under the curve for each treatment by the linear extent of the CENP-A binding domain defined above (i.e. area under BACs bA87P3, bA153G5 and bA87E14 for untreated cells, and BACs bA87E14, bA48L24 and bA69K10 for TSA-treated cells). This showed that the density of CENP-A dropped by approximately half after TSA treatment and remained at that level after removal of TSA. Thus the effect of TSA treatment is reversible in terms of the position and extent of the CENP-A-binding domain, but not in terms of the overall amount of CENP-A bound at the neocentromere.
Changes in replication timing following TSA treatment
We previously measured the replication timing of DNA sequences within the 10q25 region in 1f and 5f using a modified FISH assay (Lo et al., 2001a). In this assay, a stretch of genomic DNA corresponding to the probe region is judged to have replicated when it can be detected as a double spot, as distinct from a single spot prior to replication. Replication time is inferred from the relative number of S phase nuclei in an asynchronous cell population with single- or double-FISH signals. Results are expressed as percent FISH doublets (%D), which is directly proportional to the percentage of S phase remaining at the time the probe region completes replication (Bickmore and Carothers, 1995).
In the current study, 1f and 5f cells were cultured for 17 h in the presence of 33 nM TSA before replication times were determined for the different probe regions along the 5 Mb 10q25-BAC contig, using the above method. Results for the normal chromosome 10 (1f) indicated an overall change to a slightly earlier replication timing compared with untreated cells for the region tested (Figure 4A and B). For the mardel(10) (5f), changes were similar to those observed in 1f, except for the stretches of DNA within the differentially delayed replicating regions designated R1 and R2 in the previous study (Lo et al., 2001a). Regions within R1 and R2 showed a larger shift to an earlier replication time, making them no longer significantly different from those obtained with similarly treated 1f cells (Figure 4A and B). It was also noted that the regions involved in CENP-A binding either before or after TSA treatment (designated as domains A1 and A2, respectively) were among the regions of DNA showing the smallest changes in replication time.
We next investigated the effects of removal of TSA. As shown in Figure 4C, for a number of the probed regions, this shifted replication times to later in S phase so that these regions now replicated much later than before the addition of TSA. Noted exceptions were the probes that resided within the A1 and A2 CENP-A-binding domains, which again remained relatively unchanged. Furthermore, the replication times of 1f and 5f remained statistically identical throughout all the regions tested, including those within the R1 and R2 domains (Figure 4C). Of note, on the mardel(10) chromosome, the replication times within the R2 domain (but not the much smaller R1 domain) returned to values not significantly different from the replication times seen in untreated 5f cells. Interestingly, most of the assayed regions outside the R1/R2 and A1/A2 domains on both chromosomes did not return to their original replication times following TSA removal.
The above results indicated that transient, partial histone deacetylation has different effects on different chromosomal regions. For the 10q25 contig region on the mardel(10) chromosome, it induced a reversible shift to an early replication time for the larger of two domains of delayed replication but did not significantly affect the replication time of the CENP-A-binding region. In addition, many regions outside of both of these domains failed to return to their original replication times after addition and removal of TSA. For the equivalent regions on the normal chromosome 10, TSA treatment induced a change to a slightly earlier replication timing; however, after removal of TSA, replication occurred much later in S phase. Notable exceptions were the A1 and A2 domains corresponding to the CENP-A-binding regions of the neocentromeric chromosome, the replication times of which remained relatively unchanged both during TSA treatment and following TSA removal. In addition, the replication timing differences between the normal and neocentromeric 10q25 regions were abolished on TSA treatment, and the differences were not reinstated after TSA removal.
Discussion
We have demonstrated that partial inhibition of histone deacetylation has significant and definable effects on neocentromere structure that are not associated with any detectable deleterious effects on its function. Western blot analysis showed that 33 nM TSA significantly increases (although not to the full extent as with 150 or 300 nM) the cellular acetylated levels of histone H3 and H4, and that such an increase is completely reversible following 1–3 days of culture without TSA. Immunofluorescence on metaphase spreads shows that the levels of acetylated histone H4 on chromosomes respond to TSA in the same manner as total cellular acetylated histone H4. We confirm that centromeres have consistently lower levels of acetylated histone H4 than surrounding chromatin, which remains the case even after global hyperacetylation. This suggests that some ‘imprint’ remains at the centromere, marking it out from adjacent chromatin.
The FISH spot-counting assay allows the replication timing of individual stretches of genomic DNA to be determined at a resolution that is not affordable by other more conventional cytogenetic-based methods. Using this assay, it was found that partial inhibition of histone deacetylation resulted in significant changes in replication timing within the previously defined R1 and R2 regions of differentially delayed replication timing. The R2 domain, and to a much lesser extent the smaller R1 domain at the mardel(10) neocentromere, were shifted to a substantially earlier replication time, to the extent that these domains no longer showed significant differences in replication timing compared with the same regions in normal chromosome 10. This finding that TSA equalizes replication time at the neocentromeric and normal 10q25 regions agrees with that of a previous study demonstrating that 33 nM TSA caused an equalization of the replication timings of previously replication time-discordant imprinted alleles (Bickmore and Carothers, 1995). Furthermore, both studies have shown that the neocentromeric locus or inactive allele exhibited a larger shift in replication time than the normal chromosomal locus or active allele. These results suggest that the non-neocentromeric or non-imprinted loci may possess a chromatin ‘ground state’, and that neocentromere formation or gene silencing establishes a chromatin structure that happens to be particularly sensitive to TSA-induced histone hyperacetylation.
To our knowledge, the present study is the first to describe the combined effects that TSA treatment and its removal have on replication time. Our results indicate that the differentially delayed replication property originally established at the R2 domain during neocentromere formation is irreversibly abolished. This suggests that such a delayed replication property/domain is not essential for neocentromere function. The apparently non-essential nature of the R2 domain for neocentromere function is further supported by a comparison of the boundaries of the differentially delayed replication domains with those of the smallest mitotically stable minichromosome constructed from mardel(10) (NC-MiC5), which shows a near-complete absence of the R2 replication domain on the minichromosome (Saffery et al., 2001) (Figure 5).

Fig. 5. Summary of the distribution of different domains at the 10q25 neocentromere region. A1 designates the position of the minimal CENP-A-binding domain before TSA (Lo et al., 2001a) or following 3 days of recovery from 33 nM TSA treatment, whereas A2 denotes the position of the minimal CENP-A-binding domain after 17 h of 33 nM TSA treatment. Solid R1 and R2 boxes denote the regions of delayed replication on mardel(10) compared with normal chromosome 10 in the absence of TSA (Lo et al., 2001a). Open R1 and R2 boxes denote the same regions whose delayed replication characteristics are abolished following treatment with TSA, noting that the major domain R2 (but not R1) is reversible after removal of TSA. Regions of high AT (>62%) and LINE interspersed repeat contents and regions of depressed Alu repeats and predicted gene contents are shown (Lo et al., 2001a). NC-MiC5 represents a 0.65 Mb stable minichromosome previously constructed in our laboratory (Saffery et al., 2001).
Removal of TSA resulted in replication times within the R2 but not the R1 domain returning to values not significantly different from the values found at the neocentromere before TSA treatment. This observation implies that although histone acetylation status generally influences replication time, regions such as R2 at the mardel(10) neocentromere possess a stronger memory for replication timing than some other genomic regions. This may be due to ‘imprinting’ factors that are present within this region only, such as histone deacetylases or heterochromatin proteins. Regions of chromosomes exhibiting a different replication time than before the addition of TSA may therefore represent regions with a looser chromatin memory.
It is also of note that the A1/A2 CENP-A-binding domain and its equivalent region on the normal chromosome 10 seems to be more resistant than surrounding regions to TSA-induced changes in replication time. This raises the possibility that replication timing at certain genomic sites may be intrinsically resistant to the effects of TSA-induced histone hyperacetylation. Furthermore, the possibility that such genomic regions may constitute preferred sites for neocentromere formation in general is tantalizing, but additional data for other independent genomic sites would be necessary to provide support for this suggestion.
It has been proposed that centromeres may be defined as the last regions of chromosomal replication that occur in synchrony with the time of CENP-A expression (Csink and Henikoff, 1998). However, experiments in yeast, Drosophila and human cells have dispelled this theory by demonstrating that centromeres replicate at different times in different species and that CENP-A can be incorporated throughout the cell cycle (Shelby et al., 2000; Ahmad and Henikoff, 2001; Choo, 2001; Sullivan and Karpen, 2001). As human centromeres are known to replicate later in S phase, and the few cases of neocentromeres studied to date have all been found in AT-rich regions (Lo et al., 2001a,b; Satinover et al., 2001) that are usually late replicating in the human genome, it may be speculated that neo centromeres only form in regions that are already late replicating, as is the case with the A1 domain (Choo, 2001; Lo et al., 2001a; this study). In this regard, it is of interest that a recent study has shown that histone deacetylase 2, which has a role in nucleosomal remodelling, is preferentially associated with late replicating foci (Rountree et al., 2000).
Our data have shown for the first time that the position of a mammalian centromere can be experimentally shifted. Through a partial inhibition of histone deacetylation, we have demonstrated a lateral movement by ∼320 kb in the CENP-A-binding domain of the mardel(10) neocentromere and an apparent expansion in size from ∼330 to 480 kb. We have further shown that this shift is reversible and is not accompanied by any significant changes in the total cellular level of CENP-A expression when normal histone deacetylation status is restored. Several major inferences can be made from these observations. First, the TSA-induced position-shift is unidirectional, suggesting that CENP-A-redistribution is a directed rather than dispersed or random process. Secondly, the amount of CENP-A bound to the neocentromere was reduced by ∼30% after TSA treatment and by ∼50% of the original amount after removal of TSA. Taken together, these results show that the density of CENP-A per unit length of genomic DNA within its binding region was reduced after TSA treatment and remained at a reduced level after removal of TSA. This implies that the absolute amount of CENP-A present at the 10q25 neocentromere may show considerable latitude (by as much as a 50% reduction) without compromising neocentromere function. As the number of nucleosomal particles per unit length of genomic DNA is expected to remain constant, a corollary of our finding is a prediction of a significant increase in the level of ‘placeholder’ histone H3 in the 330 and 480 kb CENP-A-binding domains to compensate for the loss and/or ‘dilution’ of CENP-A. The deposition of ‘placeholder’ histone H3 has recently been proposed as a possible mechanism to counteract the temporary reduction of CENP-A molecules following centromeric DNA replication (Sullivan, 2001). Consistent with this model, we presented in our previous analysis evidence that CENP-A replaces histone H3 at the neocentromere (Lo et al., 2001a). In addition, a number of studies have shown that CENP-A homodimers replace histone H3 homodimers in centromeric nucleosomes both in vitro and in vivo (Glowczewski et al., 2000; Shelby et al., 2000; Yoda et al., 2000; Ahmad and Henikoff, 2001; reviewed in Smith, 2002). In addition, Blower et al. (2002) showed, using immunodetection on stretched chromatin and by co-immunoprecipitation, that CENP-A and histone H3 occupy alternate and separate domains at the centromere.
The ability to reversibly alter the position of a previously marked centromere domain provides direct support for the epigenetic hypothesis of centromere formation and propagation (Karpen and Allshire, 1997; Choo, 2000). Our data further point to changes in the threshold of core histone acetylation as a possible determinant in the epigenetic marking of a genomic site for CENP-A binding. There are at least two ways in which this may occur. The first is that elevated levels of histone acetylation lower the affinity of CENP-A for the normally CENP-A-binding chromatin at region A1. This could be achieved directly through a reduced affinity of acetylated histone H4 for CENP-A or by a perturbation of components that may participate in CENP-A loading (Sullivan, 2001). The second possibility is that deacetylation causes the loss or the lateral movement of a ‘boundary’ protein(s), such as one of a number of heterochromatin proteins, some of which are known to flank centromeric heterochromatin in fission yeast and Drosophila and to be mislocalized by histone hyperacetylation in fission yeast and humans (Ekwall et al., 1997; Partridge et al., 2000; Blower and Karpen, 2001; Taddei et al., 2001). We have investigated one of these proteins on the mardel(10) neocentromere by defining the binding region of the heterochromatin protein HP1α using the same CIA procedure as described in this study. We found that before TSA treatment HP1α binds a region ∼100 kb in size, located ∼1 Mb away on the p′ side of the CENP-A-binding domain, and that HP1 dissociates from chromatin following TSA treatment, as has been described previously (Ekwall et al., 1997; Taddei et al., 2001; data not shown). Thus the HP1-binding region neither forms an immediate boundary of the CENP-A-binding domain nor fully or substantially encompasses the domains of delayed replication, suggesting that HP1 is not the primary determinant defining the position of CENP-A binding or the establishment of the regions of delayed replication timing. The reversible nature of the TSA-induced CENP-A-binding position, together with the fact that the lateral shift in this position appears to be spatially confined, may also imply that certain genomic DNA characteristics may be more favourable for CENP-A binding under normal and TSA-induced conditions. A detailed analysis of the nucleotide sequences at the 10q25 region (Lo et al., 2001a; Figure 5) shows that the distribution of the A1 and A2 CENP-A-binding domains before and after TSA treatment are coincident with the peaks for AT-nucleotide and long interspersed nuclear element (LINE) contents, and the troughs for Alu repeats and predicted gene contents. The tight confinement of both the A1 and A2 domains within the high-AT peak suggests the possibility that high AT content may favour human neocentromere formation and that the drop-off in AT content at either side of the A1 and A2 regions may limit CENP-A to this region. Other studies have also shown that mammalian centromeres and neocentromeres contain AT-rich DNA (Choo, 1997b; Lo et al., 2001b; Satinover et al., 2001). Finally, in addition to the expected direct correlation between high AT content and low gene density (Craig and Bickmore, 1993), the coincidence of the A1/A2 domains with the predicted gene-poor region may reflect a preference for neocentromere formation in gene-poor regions, which through the possibility of gene silencing at the neocentromere, may select against gene-dense regions and lead to a clinical ascertainment bias.
We have demonstrated that human neocentromeres are a valuable model system for dissecting the structural and functional domains of complex mammalian centromeres. We have shown that suitable modulation of cellular histone acetylation levels yields unexpected outcomes, which have provided insight into the regulatory dynamics, including the pliability and ‘memory’ of a previously established centromeric chromatin state. This ability to modulate varying centromeric chromatin states may play important adaptive roles to meet the changing requirements of the cellular environment, such as through the cell cycle, during neocentromere formation or in genome evolution.
Materials and methods
Cell lines and 10q25 BAC contig
Somatic cell hybrids containing the normal chromosome 10 (1f) and the neocentromeric mardel(10) (5f) were established previously (du Sart et al., 1997). TSA was dissolved in ethanol at a stock concentration of 66 µg/ml for final concentrations of <100 nM, and 595 µg/ml for final concentrations of >100 nM. It is worth noting that we have found TSA to have a half-life in culture of ∼24 h (data not shown), necessitating daily medium changes. A 5 Mb BAC contig from the human 10q25 region covering the critical mardel(10) neocentromere domain has been described fully elsewhere (Lo et al., 2001a).
Cell growth and chromosome segregation assays
The 5f cell line was used for all cell growth and chromosome segregation assays as it contains the mardel(10) chromosome and has identical growth characteristics to the 1f cell line. For cell growth assays, cells were maintained in logarithmic phase by seeding at a concentration of 5 × 104/ml and splitting back to the same concentration after 2 days of logarithmic growth. At this stage, cells were counted and viability determined with Trypan Blue, and the process repeated for a total of 6 days. At least six separate cultures were assayed for each treatment. The average doubling (cell cycle) time was calculated as the total growth time (6 days = 144 h) divided by the log2 of the factor increase in cell number over this time. Information on cell morphology, micronuclei number and chromosome segregation were determined by growing cells overnight on microscope slides, fixing with 3:1 methanol:acetic acid and staining with Giemsa. For cell morphology and micronuclei determination, 1000 cells were scored from four independent experiments. For chromosome segregation, 50 mitotic cells were counted for each of four independent experiments. Missegregation was calculated by scoring a total of 50 anaphase II/telophase nuclei for the presence of bridging and lagging chromosomes relative to those of untreated cells.
Antisera and antibodies
Human anti-centromere serum CREST 6, rabbit anti-mouse CENP-A antibody and anti-CENP-C antibody (for immunofluorescence experiments) have been described elsewhere (du Sart et al., 1997; Saffery et al., 1999; Lo et al., 2001a). Rabbit anti-(tetra)acetylated histone H4 (penta) and anti-(di)acetylated histone H3 antibodies were obtained from Upstate Biotech (Waltham, MA). Mouse anti-β-tubulin antibody was obtained from Roche Molecular Biochemicals (Indianapolis, IN).
Western immunoblotting
Cells were analysed by conventional SDS–PAGE and western blotting techniques using 15% gels and 5 × 104 cells per lane. Immunodetection was performed using the enhanced chemiluminescence kit ECL-Plus (AP Biotech, Uppsala, Sweden). Secondary antibodies used included horseradish peroxidase (HRP)-conjugated sheep anti-rabbit IgG and HRP-conjugated goat anti-mouse IgG antisera, from Jackson Immunoresearch Laboratories (West Grove, PA).
Immunofluorescence
Immunofluorescence was performed essentially as described previously (Sullivan and Warburton, 1999). Antibodies/sera were diluted as follows: CREST 6, 300×; CENP-A and CENP-C, 50×; and hyperacetylated histones H3 and H4, 200×. Antibodies were incubated for 30 min at 37°C. Secondary antibodies used were goat anti-rabbit Texas Red (200×) and goat anti-human FITC (200×) (Jackson Immunoresearch Laboratories). Images were captured using IPLab software (Scanalytics, Fairfax, VA). For comparisons of signal intensity, all images were exposed for the same length of time. Fluorescence intensity of acetylated histone H4 signal was measured at single locations over the centromere (marked by the CREST 6 signal), or at non-centromeric chromosomal arm locations three to four pixels away on each side of the centromere, both corrected for nearby non-chromosomal background signals. For each cell line and all conditions, signals for 50–100 chromosomes were quantified and average values calculated.
CIA analysis
Chromatin immunoprecipitation was carried out using a CENP-A-specific antibody as previously described (Lo et al., 2001a,b).
Replication timing experiments
The procedure and DNA probes used for the analysis of replication timing were as described previously (Lo et al., 2001a,b). Briefly, 5-bromo-2′-deoxyuridine (BrdU) was added to cultures in log phase and incubated for 1.5 h prior to harvesting. FISH was carried out using standard techniques and digoxygenin-labelled probes were detected using sheep anti-dig-FITC (Roche; 1:50) followed by donkey anti-sheep-FITC (Jackson Immunoresearch Laboratories; 1:100). BrdU was detected using rat anti-BrdU (Harlan, IN; 1:10 in 0.5% w/v BSA/PBS). Slides were washed in 0.5% w/v BSA/PBS, 0.5% Tween 20 and incubated with anti-rat-AMCA (Jackson Immunoresearch Laboratories; 1:100). All spot-counting was performed blind. Two hundred BrdU-positive (S phase) nuclei were scored per hybridization experiment for the presence of single or double signals. Each probe was hybridized in four independent experiments.
Sequence analysis
Sequence analysis was performed as described previously (Lo et al., 2001a) except that to provide a direct comparison with the CIA results, only the BACs used in the CIA analysis were analysed for sequence content and the domains for each sequence feature were defined as for CENP-A.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
We thank A.Stafford, D.Irvine and H.Sumer for BAC DNA. A.W.I.L. was supported by a Melbourne International Research Scholarship and an International Postgraduate Research Scholarship. K.H.A.C. is a Senior Principal Research Fellow of NH&MRC of Australia. This work was supported by funding from NH&MRC to K.H.A.C.
References
- Aagaard L., Schmid,M., Warburton,P. and Jenuwein,T. (2000) Mitotic phosphorylation of SUV39H1, a novel component of active centromeres, coincides with transient accumulation at mammalian centromeres. J. Cell Sci., 113, 817–829. [DOI] [PubMed] [Google Scholar]
- Ahmad K. and Henikoff,S. (2001) Centromeres are specialized replication domains in heterochromatin. J. Cell Biol., 153, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry A.E., Howman,E.V., Cancilla,M.R., Saffery,R. and Choo,K.H.A. (1999) Sequence analysis of an 80 kb human neocentromere. Hum. Mol. Genet., 8, 217–227. [DOI] [PubMed] [Google Scholar]
- Barry A.E., Bateman,M., Howman,E.V., Cancilla,M.R., Tainton,K.M., Irvine,D.V., Saffery,R. and Choo,K.H.A. (2000) The 10q25 neocentromere and its inactive progenitor have identical primary nucleotide sequence: further evidence for epigenetic modification. Genome Res., 10, 832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickmore W.A. and Carothers,A.D. (1995) Factors affecting the timing and imprinting of replication on a mammalian chromosome. J. Cell Sci., 108, 2801–2809. [DOI] [PubMed] [Google Scholar]
- Bickmore W.A. and Craig,J.M. (1997) Chromosome Bands: Patterns in the Genome. R.G. Landes Company, Austin, TX.
- Blower M.D. and Karpen,G.H. (2001) The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. Nat. Cell Biol., 3, 730–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower M.D., Sullivan,B.A. and Karpen,G.H. (2002) Conserved organization of centromeric chromatin in flies and humans. Dev. Cell, 2, 319–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo M. and Cervenka,J. (1982) Patterns of DNA replication of human chromosomes. II. Replication map and replication model. Am. J. Hum. Genet., 34, 757–780. [PMC free article] [PubMed] [Google Scholar]
- Cameron E.E., Bachman,K.E., Myohanen,S., Herman,J.G. and Baylin,S.B. (1999) Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat. Genet., 21, 103–107. [DOI] [PubMed] [Google Scholar]
- Choo K.H.A. (1997a) Centromere DNA dynamics: latent centromeres and neocentromere formation. Am. J. Hum. Genet., 61, 1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo K.H.A. (1997b) The Centromere. Oxford University Press, Oxford, UK.
- Choo K.H.A. (2000) Centromerization. Trends Cell Biol., 10, 182–188. [DOI] [PubMed] [Google Scholar]
- Choo K.H.A. (2001) Domain organization at the centromere and neocentromere. Dev. Cell, 1, 165–177. [DOI] [PubMed] [Google Scholar]
- Craig J.M. and Bickmore,W.A. (1993) Chromosome bands—flavours to savour. BioEssays, 15, 349–354. [DOI] [PubMed] [Google Scholar]
- Csink A.K. and Henikoff,S. (1998) Something from nothing: the evolution and utility of satellite repeats. Trends Genet., 14, 200–204. [DOI] [PubMed] [Google Scholar]
- du Sart D. et al. (1997) A functional neo-centromere formed through activation of a latent human centromere and consisting of non-α-satellite DNA. Nat. Genet., 16, 144–153. [DOI] [PubMed] [Google Scholar]
- Eissenberg J.C. and Elgin,S.C. (2000) The HP1 protein family: getting a grip on chromatin. Curr. Opin. Genet. Dev., 10, 204–210. [DOI] [PubMed] [Google Scholar]
- Ekwall K., Olsson,T., Turner,B.M., Cranston,G. and Allshire,R.C. (1997) Transient inhibition of histone deacetylation alters the structural and functional imprint at fission yeast centromeres. Cell, 91, 1021–1032. [DOI] [PubMed] [Google Scholar]
- El Kharroubi A., Piras,G. and Stewart,C.L. (2001) DNA demethylation reactivates a subset of imprinted genes in uniparental mouse embryonic fibroblasts. J. Biol. Chem., 276, 8674–8680. [DOI] [PubMed] [Google Scholar]
- Glowczewski L., Yang,P., Kalashnikova,T., Santisteban,M.S. and Smith,M.M. (2000) Histone–histone interactions and centromere function. Mol. Cell. Biol., 20, 5700–5711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goshima G., Saitoh,H. and Yanagida,M. (1999) Proper metaphase spindle length is determined by centromere proteins Mis12 and Mis6 required for faithful chromosome segragation. Genes Dev., 13, 1664–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.F., Oruganti,H., Vu,T.H. and Hoffman,A.R. (1998) The role of histone acetylation in the allelic expression of the imprinted human insulin-like growth factor II gene. Biochem. Biophys. Res. Commun., 251, 403–408. [DOI] [PubMed] [Google Scholar]
- Jeppesen P. (1997) Histone acetylation: a possible mechanism for the inheritance of cell memory at mitosis. BioEssays, 19, 67–74. [DOI] [PubMed] [Google Scholar]
- Jeppesen P. and Turner,B.M. (1993) The inactive X chromosome in female mammals is distinguished by a lack of histone H4 acetylation, a cytogenetic marker for gene expression. Cell, 74, 281–289. [DOI] [PubMed] [Google Scholar]
- Jeppesen P., Mitchell,A., Turner,B. and Perry,P. (1992) Antibodies to defined histone epitopes reveal variations in chromatin conformation and underacetylation of centric heterochromatin in human metaphase chromosomes. Chromosoma, 101, 322–332. [DOI] [PubMed] [Google Scholar]
- Johnson C.A., O’Neill,L.P., Mitchell,A. and Turner,B.M. (1998) Distinctive patterns of histone H4 acetylation are associated with defined sequence elements within both heterochromatic and euchromatic regions of the human genome. Nucleic Acids Res., 26, 994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P.L., Veenstra,G.J., Wade,P.A., Vermaak,D., Kass,S.U., Landsberger,N., Strouboulis,J. and Wolffe,A.P. (1998) Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet., 19, 187–191. [DOI] [PubMed] [Google Scholar]
- Karpen G.H. and Allshire,R.C. (1997) The case for epigenetic effects on centromere identity and function. Trends Genet., 13, 489–496. [DOI] [PubMed] [Google Scholar]
- Lo A.W., Craig,J.M., Saffery,R., Kalitsis,P., Irvine,D.V., Earle,E., Magliano,D.J. and Choo,K.H.A. (2001a) A 330 kb CENP-A binding domain and altered replication timing at a human neocentromere. EMBO J., 20, 2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo A.W., Magliano,D.J., Sibson,M.C., Kalitsis,P., Craig,J.M. and Choo,K.H.A. (2001b) A novel chromatin immunoprecipitation and array (CIA) analysis identifies a 460-kb CENP-A-binding neocentromere DNA. Genome Res., 11, 448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J., Rice,J.C., Strahl,B.D., Allis,C.D. and Grewal,S.I.S. (2001) Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science, 292, 110–113. [DOI] [PubMed] [Google Scholar]
- O’Keefe R.T., Henderson,S.C. and Spector,D.L. (1992) Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific α-satellite DNA sequences. J. Cell Biol., 116, 1095–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partridge J.F., Borgstrom,B. and Allshire,R.C. (2000) Distinct protein interaction domains and protein spreading in a complex centromere. Genes Dev., 14, 783–791. [PMC free article] [PubMed] [Google Scholar]
- Pedone P.V., Pikaart,M.J., Cerrato,F., Vernucci,M., Ungaro,P., Bruni,C.B. and Riccio,A. (1999) Role of histone acetylation and DNA methylation in the maintenance of the imprinted expression of the H19 and Igf2 genes. FEBS Lett., 458, 45–50. [DOI] [PubMed] [Google Scholar]
- Pidoux A.L. and Allshire,R.C. (2000) Centromeres: getting a grip of chromosomes. Curr. Opin. Cell Biol., 12, 308–319. [DOI] [PubMed] [Google Scholar]
- Rice J.C. and Allis,C.D. (2001) Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr. Opin. Cell Biol., 13, 263–273. [DOI] [PubMed] [Google Scholar]
- Rountree M.R., Bachman,K.E. and Baylin,S.B. (2000) DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat. Genet., 25, 269–277. [DOI] [PubMed] [Google Scholar]
- Saffery R., Earle,E., Irvine,D.V., Kalitsis,P. and Choo,K.H.A. (1999) Conservation of centromere protein in vertebrates. Chromosome Res., 7, 261–265. [DOI] [PubMed] [Google Scholar]
- Saffery R., Irvine,D.V., Griffiths,B., Kalitsis,P., Wordeman,L. and Choo,K.H.A. (2000) Human centromeres and neocentromeres show identical distribution patterns of >20 functionally important kinetochore-associated proteins. Hum. Mol. Genet., 9, 175–185. [DOI] [PubMed] [Google Scholar]
- Saffery R. et al. (2001) Construction of neocentromere-based human minichromosomes by telomere-associated chromosomal truncation. Proc. Natl Acad. Sci. USA, 98, 5705–5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh S., Takahashi,K. and Yanagida,M. (1997) Mis6, a fission yeast inner centromere protein, acts during G1/S and forms specialized chromatin required for equal segregation. Cell, 90, 131–143. [DOI] [PubMed] [Google Scholar]
- Satinover D.L., Vance,G.H., Van Dyke,D.L. and Schwartz,S. (2001) Cytogenetic analysis and construction of a BAC contig across a common neocentromeric region from 9p. Chromosoma, 110, 275–283. [DOI] [PubMed] [Google Scholar]
- Shelby R.D., Monier,K. and Sullivan,K.F. (2000) Chromatin assembly at kinetochores is uncoupled from DNA replication. J. Cell Biol., 151, 1113–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.M. (2002) Centromeres and variant histones: what, where, when and why? Curr. Opin. Cell Biol., 14, 279–285. [DOI] [PubMed] [Google Scholar]
- Sullivan B.A. and Warburton,P.E. (1999) Studying progression of vertebrate chromosomes through mitosis by immunofluorescence and FISH. In Bickmore,W.A. (ed.), Chromosome Structural Analysis: A Practical Approach. Oxford University Press, Oxford, UK, pp. 81–101.
- Sullivan B. and Karpen,G. (2001) Centromere identity in Drosophila is not determined in vivo by replication timing. J. Cell Biol., 154, 683–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan K.F. (2001) A solid foundation: functional specialization of centromeric chromatin. Curr. Opin. Genet. Dev., 11, 182–188. [DOI] [PubMed] [Google Scholar]
- Taddei A., Maison,C., Roche,D. and Almouzni,G. (2001) Reversible disruption of pericentric heterochromatin and centromere function by inhibiting deacetylases. Nat. Cell Biol., 3, 114–120. [DOI] [PubMed] [Google Scholar]
- Ten Hagen K.G., Gilbert,D.M., Willard,H.F. and Cohen,S.N. (1990) Replication timing of DNA sequences associated with human centromeres and telomeres. Mol. Cell Biol., 10, 6348–6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voullaire L.E., Slater,H.R., Petrovic,V. and Choo,K.H.A. (1993) A functional marker centromere with no detectable α-satellite, satellite III, or CENP-B protein: activation of a latent centromere? Am. J. Hum. Genet., 52, 1153–1163. [PMC free article] [PubMed] [Google Scholar]
- Warburton P.E. et al. (2000) Molecular cytogenetic analysis of eight inversion duplications of human chromosome 13q that each contain a neocentromere. Am. J. Hum. Genet., 66, 1794–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoda K., Ando,S., Morishita,S., Houmura,K., Hashimoto,K., Takeyasu,K and Okazaki,T. (2000) Human centromere protein A (CENP-A) can replace histone H3 in nucleosome reconstitution in vitro. Proc. Natl Acad. Sci. USA, 97, 7266–7271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M., Horinouchi,S. and Beppu,T. (1995) Trichostatin A and trapoxin: novel chemical probes for the role of histone acetylation in chromatin structure and function. BioEssays, 17, 423–430. [DOI] [PubMed] [Google Scholar]
- Zink D., Bolzer,A., Mayr,C., Hofmann,W., Sadoni,N. and Uberla,K. (2001) Mammalian genome organization and its implications for the development of gene therapy. Gene Ther. Mol. Biol., 6, 1–24. [Google Scholar]