

Abstract
The hypothesis that RNA interference constrains L1 mobility seems inherently reasonable: L1 mobility can be dangerous and L1 RNA, the presumed target of RNAi, serves as a critical retrotransposition intermediate. Despite its plausibility, proof for this hypothesis has been difficult to obtain. Studies attempting to link the L1 retrotransposition frequency to alterations in RNAi activity have been hampered by the long times required to measure retrotransposition frequency, the pleiotropic and toxic effects of altering RNAi over similar time periods, and the possibility that other cellular machinery may contribute to the regulation of L1s. Another problem is that the commonly used L1 reporter cassette may serve as a substrate for RNAi. Here we review the L1-RNAi hypothesis and describe a genetic assay with a modified reporter cassette that detects approximately 4 times more L1 insertions than the conventional retrotransposition assay.
RNAi SILENCING OF TRANSPOSABLE ELEMENTS
RNAi is an evolutionarily conserved process of sequence-specific posttranscriptional gene silencing (reviewed in [1]). Double-stranded RNA (dsRNA) is cleaved by the ribonuclease DICER into small interfering RNA species (siRNAs). SiRNA molecules, in turn, target complementary RNA sequences for destruction (reviewed in [2]). RNAi is postulated to play a role in the silencing of transposable elements and viruses that produce dsRNA [3, 4]. One line of evidence linking RNAi to repressed transposition comes from the nematode, C elegans [5, 6]. Tc1 elements, a class of DNA transposons, mobilize in somatic cells, but are silenced in the germ line of C elegans. A number of mutant C elegans strains that have lost this silencing have also lost the ability to execute RNAi (though there were also RNAi mutants that lacked this transposon mobilization phenotype) [5]. The identification of specific genes, which when mutated show activation of germline transposition, indicates that an active transposon-silencing process exists in the germline [5, 6]. Another line of evidence linking RNAi (or a mechanism similar to RNAi) to the regulation of transposable elements involves the I-factor in Drosophila. Mobilization of the I-factor (an L1-like non-LTR retrotransposon) is regulated at least in part by a homology-dependent silencing mechanism in the female germline [7, 8]. This silencing mechanism has been linked to a series of molecules that are implicated in the RNAi pathway, including the Argonaute protein PIWI [9, 10].
By analogy, perhaps a sequence-dependent process of mobile element silencing, such as RNAi, is used to regulate L1 mobility. As with the above-mentioned examples, the regulation of L1 mobility may be particularly relevant in the germline and in embryos. Mobility in the germline or in embryos could result in inheritance of the new insertion. These sites are also where L1s are believed to be most active [11–14]. Other mechanisms for recognizing and responding to dsRNA, such as RNase L and PKR-mediated responses, can cause apoptosis. While apoptosis seems like a reasonable strategy for dealing with a wayward somatic cell, in the germline or early embryo, apoptosis could be detrimental to the fitness of the organism [14, 15]. Here we explore the thesis that the mobility of human L1s is regulated by RNAi.
L1 RETROTRANSPOSITION: HAZARDS AND CONSTRAINTS
The human genome contains roughly half a million long interspersed elements (L1s) that collectively account for 17% of its mass [16]. Most new L1 insertions are “dead on arrival” due to 5′ truncation and nearly all but perhaps 60–100 L1 sequences in the human genome are inactive due to truncation, inversion, or mutation [17].
As discussed elsewhere in this issue, retrotransposition can be hazardous because L1s can insert into genes, alter gene expression, shuffle exons, transduce 3′ flanking sequences, mobilize Alu elements, and their replicative mobilization adds significant DNA mass to the genome [18–24]. L1 insertions and recombination events involving genomic L1 and Alu insertions have been reported in a number of genetic disorders (reviewed in [25]). Although it is possible that some functions of L1 are beneficial to mammals (a most interesting recent demonstration involves the potential role of L1s as diversity generators in the CNS, [26]), most germline L1 insertions are likely to be neutral or negatively selected. Negative selection of L1s is suggested by the higher frequencies of full-length human L1 insertions on the sex chromosomes than the autosomes (the former not being as able as the latter to undergo purifying selection) and by the dominance and limited periods of activity of single L1 subfamilies in some primate lineages [27, 28].
L1 mobility in mammals appears to be actively constrained. An indirect line of evidence for this constraint is that different cell types exhibit different rates of retrotransposition, ranging from 30% or higher in some transformed cell lines to fewer than one per million cells. In the mouse, the rate of germline retrotransposition events using an L1-EGFP transgene is approximately one event in 100 offspring [11, 13]. Analysis of L1 transcription, protein production and retrotransposition, reveals different levels of L1 activity in different cell types, with highest levels of activity noted in germ cells, embryonal cells, and recently neuronal cells [11–13, 26, 29, 30]. The factor(s) that assist L1 mobilization in some cell lines, but not others, are not known.
L1 RNA IS A LOGICAL TARGET FOR LIMITING L1 MOBILITY
RNA is a logical target for cellular machinery to protect against unwanted L1 proliferation. L1 RNA is required and may be rate-limiting for retrotransposition. In cell-culture-based assays with tagged human L1 elements, it has been shown that a decrease in L1 mRNA leads to a decrease in L1 retrotransposition frequencies, lending support to the idea that L1 activity can be limited by regulating L1 transcript abundance [31, 32]. L1 RNA is critical for retrotransposition because it encodes the necessary ORF1 and ORF2 proteins, which act preferentially upon the RNA that encoded them [33, 34]. This effect, termed cis preference, may allow active L1s a greater proliferative advantage than retroelements that mobilize in trans because trans-mobilization can result in the expansion of mutated rather than active elements. RNAi may be able to counter this potential advantage of cis preference by using nonfunctional L1 RNAs to inhibit functional L1s. On the other hand, the high copy number of L1 insertions in mammals may have been selected for L1s that are inefficiently regulated by RNAi. If RNAi silences L1s, it does not do so with perfect efficiency since L1 transcripts are detected, and some L1s can still mobilize in the human genome.
How (or even if) L1 RNA is recognized by cellular machinery is unknown. If RNAi limits human L1 retrotransposition, the most obvious possibility is that RNAi posttranscriptionally targets L1 mRNA. The presumed trigger for RNAi is double-stranded RNA (dsRNA), although other forms of sequence-specific recognition or unusual RNA secondary structure are possible. DsRNA has been documented to be the target of RNAi-induced transposon silencing in other species, most notably the Tc1 DNA transposon in C elegans [5, 6]. Read-through transcription of dispersed Tc1 copies can form dsRNA as a result of “snap-back” of their terminal inverted repeats (TIRs), which are complementary in sequence. Human L1 retrotransposons are not flanked by complementary TIRs, however there is considerable nucleotide sequence similarity between active L1s [35]. This high level of sequence similarity amongst active human L1 elements might allow only a few L1 dsRNA molecules to silence many genomic L1s. Sense and antisense L1 transcripts have been documented in human teratocarcinoma cells [36]. There are two reports suggesting the presence of long L1 dsRNA [37, 38], although thus far an unequivocal demonstration of Dicer-derived L1 siRNAs or miRNAs from mammalian cells has remained elusive [39–41].
There are several ways in which L1 dsRNA could be formed (see Figure 1). First, antisense L1 RNA could arise as a read-through transcript from a heterologous promoter element (Figure 1(a)). If sense and antisense transcripts originating from different loci could form dsRNA, even inactive copies of L1 could contribute to the loss of L1 mobility (Figure 1(b)). As the genomic burden of L1 copies increases, the level of L1 repression might also increase. On the other hand, highly efficient silencing of all L1 copies in trans could be problematic since L1s may influence the human transcriptome significantly (reviewed in [20]). An alternative is to selectively target L1 dsRNA that arises in cis. In this connection, the L1 5′UTR has antisense promoter activity at positions 400–600, providing an additional source of antisense L1 RNA (Figure 1(c)) [42, 43]. RNAi targeting the L1 5′UTR would be expected to selectively restrain full-length (and therefore more likely to be active) elements. Finally, L1 dsRNA can originate from the transcription of inverted repeats (Figure 1(d)). A high copy number of L1 sequences in mammalian genomes increases the likelihood of generating inverted repeats either by genome rearrangements or by insertions of L1 elements into or near themselves [44]. We conducted a simple search for long inverted repeats (> 200 bp) in the human genome, and a preliminary analysis of several chromosomes indicates that the whole genome contains tens of perfect inverted repeats of L1 sequences. Transcription of such inverted repeats results in an efficient dsRNA formation because this dsRNA folding is a first-order reaction. Although the focus here is on dsRNA, there may be other forms of L1 RNA that are recognized. L1 RNA species are heterogeneous due to variable 5′ truncation, premature polyadenylation, and inversion [45, 46].
Figure 1.
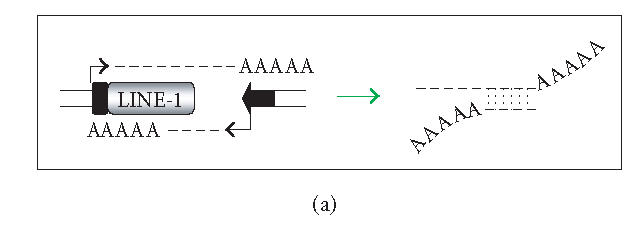
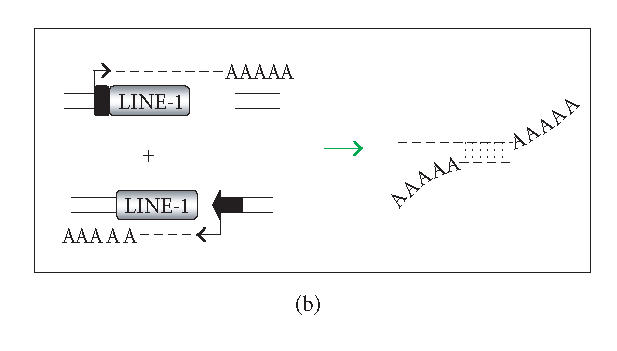
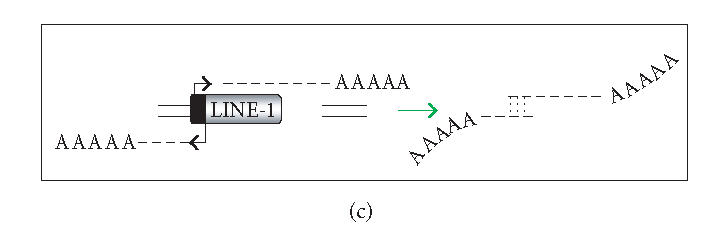
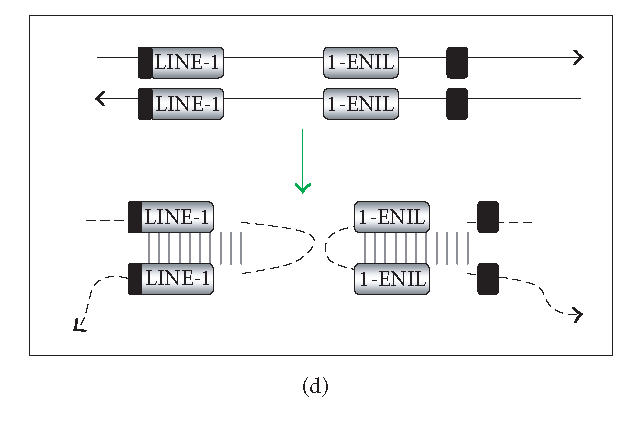
Generation of L1 dsRNA. L1 dsRNA could arise from different transcripts (shown in Figures 1(a), 1(b), and 1(c)) or from the same transcript (shown in Figure 1(d)). (a) An L1 transcript originates from the internal 5′UTR promoter, producing a sense-strand RNA. Neighboring the same element, an antisense-oriented heterologous promoter produces a read-through transcript that includes antisense L1 RNA. (b) An L1 is transcribed producing sense RNA and another L1 insertion, elsewhere in the genome, is transcribed off of a heterologous promoter yielding an antisense RNA. (c) An L1 is transcribed off of its 5′UTR producing a sense transcript, while antisense promoter activity of the 5′UTR produces an antisense transcript. (d) If an L1 inserts near another L1 sequence in the genome, it may be possible to create a hairpin. The figure shows two full-length L1 sequences facing each other, although it should be noted that hairpins could also form between truncated L1 copies that face each other, as long as there is a transcript that extends between the copies. DNA strands are shown with solid lines and RNA with dashed lines. In the scenario depicted, transcripts off of either DNA strand extending through the two L1 sequences will give rise to self-complementary regions: the forward facing L1 and the reverse complementary sequence of L1 on the same RNA strand can base pair, forming hairpins (dashed vertical lines) with stretches of dsRNA.
HOW MIGHT RNAi SILENCE L1s?
The most obvious possibility is that RNAi limits L1 retrotransposition by decreasing the amount of L1 RNA. In this case, disruption of RNAi should increase L1 RNA levels and result in an increased retrotransposition frequency. The relationship between RNAi and repression of the LTR retrotransposons MuERV-L and intracisternal A-particle (IAP) was recently investigated in early mouse embryos [47]. Knocking down DICER (with siRNA or dsRNA) resulted in a 50% increase in the abundance of MuERV-L and IAP transcripts [47]. Recently, conditional dicer knock-out ES cells were shown to exhibit slightly increased levels of IAP and L1 transcripts compared to dicer wild-type cells [48]. In further support of this theory, L1 retrotransposons can form dsRNA that is cleaved into siRNAs by DICER in cultured cells [49, 50]. This analysis reveals that L1s can serve as targets for RNAi, but does not address whether they do so in nature.
Another possibility is that one or more components of the RNAi machinery acts by silencing L1 insertions in chromatin via methylation of L1 DNA [47]. Methylation has been proposed as a genomic defense against transposable elements and may function in an RNAi-dependent or independent manner to limit L1 transcription [53, 54]. Methylation of the L1 5′UTR has been demonstrated in different cell types [55, 56]. Treatment of 3T3 cells with 5-azacytidine, a pyrimidine analog that inhibits DNA methyltransferase, increases L1 transcript abundance [57]. In mice, inactivation of methylases can result in mobilization of retrotransposons including IAP elements and L1s [58, 59]. On the other hand, methylation was not observed in response to stable dsRNA expression in murine oocytes [47]. Consistent with the latter observation, a recent analysis in human cancer cells suggests that RNAi-mediated transcriptional silencing can arise independently of methylation [60].
Another nonmutually exclusive possibility is that RNAi participates in altering chromatin accessibility. Heterochromatic silencing and histone methylation have been tied to the RNAi pathway in S pombe [61, 62]. Moreover, in the filamentous fungus, Neurospora crassa, repression of the L1-like retrotransposon Tad is dependent upon the Argonaute protein QDE2 and DICER [63]. DNA and histone methylation have also been implicated in transposon control in Arabidopsis [64]. In certain yeast and plant species, heterochromatin formation may be directed by siRNAs in an Argonaute complex with similarities to the RNA-induced silencing complex, suggesting that the processes of PTGS and transcriptional gene silencing are intertwined [63]. Although plant L1-like elements differ from mammalian L1 elements, a similar means of mammalian L1-associated chromatin silencing may be at work.
It is possible that RNAi acts upon L1s using all of these pathways: degradation of L1 RNA (which limits the production of new insertions), modification of L1 DNA sequences and chromatin silencing (which should limit the activity of new or existing functional L1s). The containment of L1s in regions of silenced chromatin provides protection by suppressing their transcription, mobility, and recombinational activity [65–67].
ESTABLISHING A FUNCTIONAL LINK BETWEEN RNAi AND L1 RETROTRANSPOSITION
Currently, the only direct evidence linking RNAi to the repression of L1 elements in mammals is a slightly increased level of L1 transcripts in dicer deficient mouse ES cells [48]. Current efforts to explore L1 regulation by RNAi in mammals are focused on three areas: (i) demonstration of siRNAs derived from native L1 elements; (ii) determining whether L1 dsRNA is assembled in cis (from the same L1) or if sense and antisense transcripts originating from two different elements (assembly in trans) can also trigger RNAi; and (iii) perturbing components of the RNAi pathway and seeing if there are corresponding alterations in the L1 retrotransposition frequency. Exploration of the first area is under active investigation and is discussed in detail elsewhere in this issue. Concerning the second area, it has been assumed that dsRNA formation in trans is minimal because xenogeneic L1s (eg, a human element in a mouse cell) do not appear to be more active than syngeneic L1s (a human element in a human cell) [32, 68]. However, the permissiveness for L1 retrotransposition in these different cell types is not controlled for. It is intriguing that L1 elements that have genetically modified RNA sequences, but identical protein coding sequences, can be far more active for retrotransposition [32, 46]. While there are other potential reasons for this (such as decreased premature polyadenylation and alterations in RNA structure), it will be interesting to see if some of the enhanced activity of synthetic L1s is due to different levels of RNAi. The third area of investigation attempts to establish a mechanistic link between RNAi and L1 retrotransposition.
Analyzing L1 retrotransposition in cells with altered RNAi activity is challenging. The first obstacle is to monitor the mobilization of an active L1 in a sea of L1 sequences in the genome. To get around this “needle-in-a-haystack” problem, L1 elements were tagged with antisense marker cassettes interrupted in the sense direction by an intron [52]. These tagged elements could then be monitored for retrotransposition by scoring for expression of the marker (which could only occur after a cycle of transcription, processing, reverse transcription, and integration into a transcriptionally permissive region of the genome, see Figure 2(a)). Because the L1 retrotransposition construct contains sense and antisense promoters, it may induce RNAi regardless of whether the L1 element induces RNAi naturally.
Figure 2.


(a) The standard L1 reporter construct contains opposing promoters. The standard L1 reporter construct used in our laboratory (L1-EGFP) consists of the CMV promoter, the human L1RP element, and the antisense EGFP gene cloned into the L1 3′UTR followed by the SV40 late poly-A sequence in pCEP4 (construct described in more detail in [51]). When L1-EGFP retrotransposes, a full-length L1 RNA is transcribed, the intron interrupting EGFP is spliced out, and the processed RNA is reverse transcribed, and a cDNA copy is inserted into the genome. If the insertion is of sufficient length and enters the genome in a transcriptionally permissive region, retrotransposition can be detected phenotypically by screening for EGFP expression. Retrotransposition can also be assayed genetically by performing PCR with primers that flank the EGFP intron. Because the EGFP marker is driven off of an antisense-oriented promoter relative to the L1, the potential exists for creating dsRNA. L1 and EGFP transcripts are given by dashed horizontal lines, promoters are denoted with black arrows, and blue arrows indicate intron-flanking primers used to distinguish new insertions from the parental L1. (b) Loss of an antisense promoter increases L1 retrotransposition in a cultured cell assay. 143B osteosarcoma cells were transfected with one of the following constructs as shown in Figure 2(a): L1-EGFP (the same wild-type L1 retrotransposition construct shown in Figure 2(a)), L1-EGFP-DelP (identical to L1-EGFP except that the CMV promoter driving EGFP was deleted), L1-EGFP-Stuffer (identical to L1-EGFP except that the CMV promoter driving EGFP was replaced with a piece of DNA lacking promoter activity or polyadenylation signals), or L1-EGFP-RIC (retrotransposition incompetent due to two missense mutations (marked with a red X over the L1 coding sequence) derived from the JM111 L1 mutant [52]). Boxes indicate coding sequences except for the yellow box in the L1-EGFP-Stuffer construct that denotes the stuffer sequence. Arrows denote the promoters and the black line separating the EGFP cassette denotes the intron. Cells were selected in hygromycin for two weeks and individual clones were picked and expanded. PCR using primers that flank the intron/exon splice site in EGFP (as described in [11]) was used to monitor individual clones of antibiotic-resistant cells for L1 retrotransposition (loss of the intron in EGFP). The percentages of clones that had the spliced EGFP are shown in Figure 2(b). The number of clones surveyed for each genotype is given to the right of each of the bars. None of the retrotransposition incompetent L1 transfectants had a spliced EGFP product.
To circumvent the potential problem of having bidirectional transcription in the L1-EGFP construct, we created a series of EGFP-tagged L1 elements that lacked antisense promoter activity (Figure 2(b)). Using a genetic assay to monitor retrotransposition, our preliminary data reveal a 2-fold increase in retrotransposition when constructs lacking the antisense promoter in the EGFP marker were used, compared to the conventional L1-EGFP construct (Figure 2(b)). To control for length effects (better detection of retrotransposition events due to a shorter marker cassette), we created a construct with a “stuffer fragment” in place of the EGFP promoter. This L1-stuffer construct also exhibited increased retrotransposition compared to the standard L1-EGFP construct, indicating that the basis for the increased retrotransposition frequency was due to the absence of promoter activity rather than being due to differences in marker length. Because the retrotransposition frequency in this assay approaches 90%, we may be underestimating the true retrotransposition frequency (there may be more than one insertion per clone). The basis for the increased retrotransposition activity of constructs lacking the antisense promoter driving the EGFP marker cassette is unresolved. Perhaps these constructs will be helpful in future studies that attempt to link RNAi to the regulation of L1 retrotransposition.
ALTERNATIVE SILENCING PATHWAYS MEDIATED BY DsRNA
DsRNA can induce several different pathways in mammals. One of them is RNA editing, a process in which adenosines are converted to inosine in nuclear dsRNA by the enzyme adenosine deaminase (ADAR). Editing of dsRNA can occur in a site-selective or promiscuous fashion. The latter results in the generation of a series of variably mutated RNA species. DsRNA longer than 50 bp in which > 20% A-to-I editing has occurred is referred to as hyperedited [69]. Based largely on work with polyoma virus, hyperedited RNA may be retained and/or sequestered in the nucleus [69].
L1 RNA can serve as a substrate for RNA editing [70, 71]. However, the effects of RNA editing on L1 activity are unknown. Since RNA editing affects dsRNA without targeting homologous copies of single-stranded RNA, editing may have a smaller impact on L1 retrotransposition than RNAi. If L1 RNA editing is similar to Alu editing, most RNA duplexes would be formed intramolecularly due to base pairing between two oppositely oriented Alus residing in the same RNA molecule [72]. Such duplexes would be expected to have imperfect base pairing between neighboring oppositely oriented L1 elements and could promote editing rather than RNAi. RNA editing may further help L1 to evade RNAi because hyperedited L1 dsRNA would be probably processed less efficiently into siRNAs and such siRNAs would not base pair as well with their targets. This idea is consistent with the observation that RNAi is antagonized by hyperediting [73] and that the phenotype of ADAR mutants can be rescued by mutations in RNAi [74].
In addition to siRNA and RNA editing, longer L1 dsRNA molecules can induce additional cellular responses [75]. Longer dsRNA molecules can be recognized by the dsRNA-dependent protein kinase PKR, which, when activated, results in interferon-mediated activation of the Jak-Stat pathway and cellular upregulation of interferon-regulated genes [76]. This mechanism of cytokine defense is an innate immune response that likely arose to combat viruses, which frequently produce dsRNA. Activation of PKR by dsRNA results in its autophosphorylation and subsequent phosphorylation of the eukaryotic initiation factor 2α (eIF2α), causing general inhibition of cellular protein synthesis [76]. Another pathway of dsRNA regulation involves RNaseL, a potent riboendonuclease. RNaseL can be indirectly triggered by dsRNA through an increase in 2′–5′ oligoadenylates. 2′–5′ oligoadenylates are produced from ATP by 2′–5′ oligoadenylate synthetases, which are activated by dsRNA [77]. In addition to the nucleolytic properties of RNaseL, the enzyme also upregulates type I interferon genes by sequestering NFκB transcription factors [77].
Certain cell types, for example those of myeloid origin, constitutively express receptors that recognize dsRNA [78]. The toll-like receptor (TLR)3 recognizes and binds to dsRNA [79]. TLR3 is expressed on the cell surface as well as in intracellular vesicles [80]. Thus, dsRNA can be recognized by TLR3 internally, as an intermediate in viral replication, or externally, as dsRNA leaks from dying cells [78]. Recognition of dsRNA by TLR3 initiates the binding of NFκB and IRF-3 transcription factors to the promoters of type I interferon genes leading to their upregulation, which can eventually cause cell death via apoptosis [80].
Mammalian oocytes, embryos, and embryonic stem cells do not induce a dsRNA-mediated interferon response, but utilize the RNAi pathway to respond to long dsRNA [14, 15, 81]. In contrast, somatic cells might be more likely to use an interferon pathway when confronted with long dsRNA species [82]. The rationale for using different dsRNA recognition pathways in progenitor cells versus somatic cells is that embryos may not be able to afford the luxury of shutting down individual cells if trouble arises. In contrast, adult mammalian cells can apoptose with little to no effect on the organism as a whole. It may be that the secondary products of L1 dsRNA dicer-mediated endonucleolytic cleavage activate the PKR-interferon pathway in adult differentiated cells, which induces cell death.
CONCLUSION
L1 retrotransposons have shaped the mammalian genome and contribute significantly to its mass, yet their mobility appears to be actively constrained. Along with other cellular defense mechanisms, RNAi may participate in cell-type-specific, multifaceted defense against L1 mobility that includes RNA destruction, DNA methylation, and heterochromatin formation. The development of new genetic models of RNAi deficiency in mammals, coupled with a genetic assay for monitoring L1 retrotransposition events, may help to advance our understanding of how L1 mobility is regulated.
ACKNOWLEDGMENTS
We thank Richard Schultz, Greg Hannon, and members of the Luning Prak Lab for helpful discussions and we gratefully acknowledge Sarah Fox and Janet Sallit for skilled technical assistance. This work was supported by NIH R01 CA108812 to Eline T Luning Prak and training grant support to Shane R Horman (T32 CA 09140).
References
- 1.Tomari Y, Zamore PD. Perspective: machines for RNAi. Genes & Development. 2005;19(5):517–529. doi: 10.1101/gad.1284105. [DOI] [PubMed] [Google Scholar]
- 2.Carmell MA, Hannon GJ. RNase III enzymes and the initiation of gene silencing. Nature Structural & Molecular Biology. 2004;11(3):214–218. doi: 10.1038/nsmb729. [DOI] [PubMed] [Google Scholar]
- 3.Plasterk RHA. RNA silencing: the genome's immune system. Science. 2002;296(5571):1263–1265. doi: 10.1126/science.1072148. [DOI] [PubMed] [Google Scholar]
- 4.Bagasra O, Prilliman KR. RNA interference: the molecular immune system. Journal of Molecular Histology. 2004;35(6):545–553. doi: 10.1007/s10735-004-2192-8. [DOI] [PubMed] [Google Scholar]
- 5.Sijen T, Plasterk RHA. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature. 2003;426(6964):310–314. doi: 10.1038/nature02107. [DOI] [PubMed] [Google Scholar]
- 6.Vastenhouw NL, Plasterk RHA. RNAi protects the Caenorhabditis elegans germline against transposition. Trends in Genetics. 2004;20(7):314–319. doi: 10.1016/j.tig.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 7.Jensen S, Gassama M-P, Heidmann T. Taming of transposable elements by homology-dependent gene silencing. Nature Genetics. 1999;21(2):209–212. doi: 10.1038/5997. [DOI] [PubMed] [Google Scholar]
- 8.Jensen S, Gassama M-P, Heidmann T. Cosuppression of I transposon activity in Drosophila by I-containing sense and antisense transgenes. Genetics. 1999;153(4):1767–1774. doi: 10.1093/genetics/153.4.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aravin AA, Naumova NM, Tulin AV, Vagin VV, Rozovsky YM, Gvozdev VA. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Current Biology. 2001;11(13):1017–1027. doi: 10.1016/s0960-9822(01)00299-8. [DOI] [PubMed] [Google Scholar]
- 10.Kalmykova AI, Klenov MS, Gvozdev VA. Argonaute protein PIWI controls mobilization of retrotransposons in the Drosophila male germline. Nucleic Acids Research. 2005;33(6):2052–2059. doi: 10.1093/nar/gki323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prak ETL, Dodson AW, Farkash EA, Kazazian HH Jr. Tracking an embryonic L1 retrotransposition event. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(4):1832–1837. doi: 10.1073/pnas.0337627100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Branciforte D, Martin SL. Developmental and cell type specificity of LINE-1 expression in mouse testis: implications for transposition. Molecular and Cellular Biology. 1994;14(4):2584–2592. doi: 10.1128/mcb.14.4.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ostertag EM, DeBerardinis RJ, Goodier JL, et al. A mouse model of human L1 retrotransposition. Nature Genetics. 2002;32(4):655–660. doi: 10.1038/ng1022. [DOI] [PubMed] [Google Scholar]
- 14.Svoboda P. Long dsRNA and silent genes strike back: RNAi in mouse oocytes and early embryos. Cytogenetic and Genome Research. 2004;105(2–4):422–434. doi: 10.1159/000078215. [DOI] [PubMed] [Google Scholar]
- 15.Stein P, Zeng F, Pan H, Schultz RM. Absence of non-specific effects of RNA interference triggered by long double-stranded RNA in mouse oocytes. Developmental Biology. 2005;286(2):464–471. doi: 10.1016/j.ydbio.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 16.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 17.Brouha B, Schustak J, Badge RM, et al. Hot L1s account for the bulk of retrotransposition in the human population. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luning Prak ET, Kazazian HH Jr. Mobile elements and the human genome. Nature Reviews. Genetics. 2000;1(2):134–144. doi: 10.1038/35038572. [DOI] [PubMed] [Google Scholar]
- 19.Han JS, Szak ST, Boeke JD. Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature. 2004;429(6989):268–274. doi: 10.1038/nature02536. [DOI] [PubMed] [Google Scholar]
- 20.Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? BioEssays. 2005;27(8):775–784. doi: 10.1002/bies.20257. [DOI] [PubMed] [Google Scholar]
- 21.Moran JV, DeBerardinis RJ, Kazazian HH., Jr Exon shuffling by L1 retrotransposition. Science. 1999;283(5407):1530–1534. doi: 10.1126/science.283.5407.1530. [DOI] [PubMed] [Google Scholar]
- 22.Pickeral OK, Makaowski W, Boguski MS, Boeke JD. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Research. 2000;10(4):411–415. doi: 10.1101/gr.10.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodier JL, Ostertag EM, Kazazian HH., Jr Transduction of 3′-flanking sequences is common in L1 retrotransposition. Human Molecular Genetics. 2000;9(4):653–657. doi: 10.1093/hmg/9.4.653. [DOI] [PubMed] [Google Scholar]
- 24.Dewannieux M, Esnault C, Heidmann T. LINE-mediated retrotransposition of marked Alu sequences. Nature Genetics. 2003;35(1):41–48. doi: 10.1038/ng1223. [DOI] [PubMed] [Google Scholar]
- 25.Kazazian HH., Jr Mobile elements and disease. Current Opinion in Genetics & Development. 1998;8(3):343–350. doi: 10.1016/s0959-437x(98)80092-0. [DOI] [PubMed] [Google Scholar]
- 26.Muotri AR, Chu VT, Marchetto MC, Deng W, Moran JV, Gage FH. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435(7044):903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 27.Boissinot S, Entezam A, Furano AV. Selection against deleterious LINE-1-containing loci in the human lineage. Molecular Biology and Evolution. 2001;18(6):926–935. doi: 10.1093/oxfordjournals.molbev.a003893. [DOI] [PubMed] [Google Scholar]
- 28.Boissinot S, Roos C, Furano AV. Different rates of LINE-1 (L1) retrotransposon amplification and evolution in New World monkeys. Journal of Molecular Evolution. 2004;58(1):122–130. doi: 10.1007/s00239-003-2539-x. [DOI] [PubMed] [Google Scholar]
- 29.Goto T, Jones GM, Lolatgis N, Pera MF, Trounson AO, Monk M. Identification and characterisation of known and novel transcripts expressed during the final stages of human oocyte maturation. Molecular Reproduction and Development. 2002;62(1):13–28. doi: 10.1002/mrd.10118. [DOI] [PubMed] [Google Scholar]
- 30.Ergun S, Buschmann C, Heukeshoven J, et al. Cell type-specific expression of LINE-1 open reading frames 1 and 2 in fetal and adult human tissues. The Journal of Biological Chemistry. 2004;279(26):27753–27763. doi: 10.1074/jbc.M312985200. [DOI] [PubMed] [Google Scholar]
- 31.Yang N, Zhang L, Zhang Y, Kazazian HH., Jr An important role for RUNX3 in human L1 transcription and retrotransposition. Nucleic Acids Research. 2003;31(16):4929–4940. doi: 10.1093/nar/gkg663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han JS, Boeke JD. A highly active synthetic mammalian retrotransposon. Nature. 2004;429(6989):314–318. doi: 10.1038/nature02535. [DOI] [PubMed] [Google Scholar]
- 33.Wei W, Gilbert N, Ooi SL, et al. Human L1 retrotransposition: cis preference versus trans complementation. Molecular and Cellular Biology. 2001;21(4):1429–1439. doi: 10.1128/MCB.21.4.1429-1439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nature Genetics. 2000;24(4):363–367. doi: 10.1038/74184. [DOI] [PubMed] [Google Scholar]
- 35.Boissinot S, Chevret P, Furano AV. L1 (LINE-1) retrotransposon evolution and amplification in recent human history. Molecular Biology and Evolution. 2000;17(6):915–928. doi: 10.1093/oxfordjournals.molbev.a026372. [DOI] [PubMed] [Google Scholar]
- 36.Skowronski J, Fanning TG, Singer MF. Unit-length line-1 transcripts in human teratocarcinoma cells. Molecular and Cellular Biology. 1988;8(4):1385–1397. doi: 10.1128/mcb.8.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kramerov DA, Bukrinsky MI, Ryskov AP. DNA sequences homologous to long double-stranded RNA. Transcription of intracisternal A-particle genes and major long repeat of the mouse genome. Biochimica et Biophysica Acta. 1985;826(1):20–29. doi: 10.1016/s0167-4781(85)80004-x. [DOI] [PubMed] [Google Scholar]
- 38.Martens JH, O'Sullivan RJ, Braunschweig U, et al. The profile of repeat-associated histone lysine methylation states in the mouse epigenome. The EMBO Journal. 2005;24(4):800–812. doi: 10.1038/sj.emboj.7600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Houbaviy HB, Murray MF, Sharp PA. Embryonic stem cell-specific MicroRNAs. Developmental Cell. 2003;5(2):351–358. doi: 10.1016/s1534-5807(03)00227-2. [DOI] [PubMed] [Google Scholar]
- 40.Suh MR, Lee Y, Kim JY, et al. Human embryonic stem cells express a unique set of microRNAs. Developmental Biology. 2004;270(2):488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 41.Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294(5543):853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- 42.Speek M. Antisense promoter of human L1 retrotransposon drives transcription of adjacent cellular genes. Molecular and Cellular Biology. 2001;21(6):1973–1985. doi: 10.1128/MCB.21.6.1973-1985.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nigumann P, Redik K, Matlik K, Speek M. Many human genes are transcribed from the antisense promoter of L1 retrotransposon. Genomics. 2002;79(5):628–634. doi: 10.1006/geno.2002.6758. [DOI] [PubMed] [Google Scholar]
- 44.Szak ST, Pickeral OK, Makalowski W, Boguski MS, Landsman D, Boeke JD. Molecular archeology of L1 insertions in the human genome. Genome Biology. 2002;3(10):research0052. doi: 10.1186/gb-2002-3-10-research0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ostertag EM, Kazazian HH Jr. Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Research. 2001;11(12):2059–2065. doi: 10.1101/gr.205701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perepelitsa-Belancio V, Deininger P. RNA truncation by premature polyadenylation attenuates human mobile element activity. Nature Genetics. 2003;35(4):363–366. doi: 10.1038/ng1269. [DOI] [PubMed] [Google Scholar]
- 47.Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Developmental Biology. 2004;269(1):276–285. doi: 10.1016/j.ydbio.2004.01.028. [DOI] [PubMed] [Google Scholar]
- 48.Kanellopoulou C, Muljo SA, Kung AL, et al. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes & Development. 2005;19(4):489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ. A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic Acids Research. 2005;33(3):846–856. doi: 10.1093/nar/gki223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang N, Zhang L, Kazazian HH., Jr L1 retrotransposon-mediated stable gene silencing. Nucleic Acids Research. 2005;33(6):e57. doi: 10.1093/nar/gni056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH Jr. Determination of L1 retrotransposition kinetics in cultured cells. Nucleic Acids Research. 2000;28(6):1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH Jr. High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87(5):917–927. doi: 10.1016/s0092-8674(00)81998-4. [DOI] [PubMed] [Google Scholar]
- 53.Bestor TH. The host defence function of genomic methylation patterns. Novartis Foundation Symposium. 1998;214:187–195. doi: 10.1002/9780470515501.ch11. discussion 195–199, 228–232. [DOI] [PubMed] [Google Scholar]
- 54.Bestor TH. Cytosine methylation mediates sexual conflict. Trends in Genetics. 2003;19(4):185–190. doi: 10.1016/S0168-9525(03)00049-0. [DOI] [PubMed] [Google Scholar]
- 55.Woodcock DM, Lawler CB, Linsenmeyer ME, Doherty JP, Warren WD. Asymmetric methylation in the hypermethylated CpG promoter region of the human L1 retrotransposon. The Journal of Biological Chemistry. 1997;272(12):7810–7816. doi: 10.1074/jbc.272.12.7810. [DOI] [PubMed] [Google Scholar]
- 56.Burden AF, Manley NC, Clark AD, Gartler SM, Laird CD, Hansen RS. Hemimethylation and non-CpG methylation levels in a promoter region of human LINE-1 (L1) repeated elements. The Journal of Biological Chemistry. 2005;280(15):14413–14419. doi: 10.1074/jbc.M413836200. [DOI] [PubMed] [Google Scholar]
- 57.Tchenio T, Segal-Bendirdjian E, Heidmann T. Generation of processed pseudogenes in murine cells. The EMBO Journal. 1993;12(4):1487–1497. doi: 10.1002/j.1460-2075.1993.tb05792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Walsh CP, Chaillet JR, Bestor TH. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nature Genetics. 1998;20(2):116–117. doi: 10.1038/2413. [DOI] [PubMed] [Google Scholar]
- 59.Bourc'his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431(7004):96–99. doi: 10.1038/nature02886. [DOI] [PubMed] [Google Scholar]
- 60.Ting AH, Schuebel KE, Herman JG, Baylin SB. Short double-stranded RNA induces transcriptional gene silencing in human cancer cells in the absence of DNA methylation. Nature Genetics. 2005;37(8):906–910. doi: 10.1038/ng1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Verdel A, Jia S, Gerber S, et al. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303(5658):672–676. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nature Genetics. 2005;37(8):809–819. doi: 10.1038/ng1602. [DOI] [PubMed] [Google Scholar]
- 63.Nolan T, Braccini L, Azzalin G, De Toni A, Macino G, Cogoni C. The post-transcriptional gene silencing machinery functions independently of DNA methylation to repress a LINE1-like retrotransposon in Neurospora crassa. Nucleic Acids Research. 2005;33(5):1564–1573. doi: 10.1093/nar/gki300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lippman Z, Gendrel AV, Black M, et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;430(6998):471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- 65.Birchler JA, Bhadra MP, Bhadra U. Making noise about silence: repression of repeated genes in animals. Current Opinion in Genetics & Development. 2000;10(2):211–216. doi: 10.1016/s0959-437x(00)00065-4. [DOI] [PubMed] [Google Scholar]
- 66.Bender J. DNA methylation and epigenetics. Annual Review of Plant Biology. 2004;55:41–68. doi: 10.1146/annurev.arplant.55.031903.141641. [DOI] [PubMed] [Google Scholar]
- 67.Lippman Z, May B, Yordan C, Singer T, Martienssen R. Distinct mechanisms determine transposon inheritance and methylation via small interfering RNA and histone modification. PLoS Biology. 2003;1(3):E67. doi: 10.1371/journal.pbio.0000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Naas TP, DeBerardinis RJ, Moran JV, et al. An actively retrotransposing, novel subfamily of mouse L1 elements. The EMBO Journal. 1998;17(2):590–597. doi: 10.1093/emboj/17.2.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DeCerbo J, Carmichael GG. Retention and repression: fates of hyperedited RNAs in the nucleus. Current Opinion in Cell Biology. 2005;17(3):302–308. doi: 10.1016/j.ceb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 70.Blow M, Futreal PA, Wooster R, Stratton MR. A survey of RNA editing in human brain. Genome Research. 2004;14(12):2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Levanon EY, Eisenberg E, Yelin R, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nature Biotechnology. 2004;22(8):1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 72.Athanasiadis A, Rich A, Maas S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biology. 2004;2(12):e391. doi: 10.1371/journal.pbio.0020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scadden AD, Smith CW. RNAi is antagonized by A → I hyper-editing. EMBO Reports. 2001;2(12):1107–1111. doi: 10.1093/embo-reports/kve244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tonkin LA, Bass BL. Mutations in RNAi rescue aberrant chemotaxis of ADAR mutants. Science. 2003;302(5651):1725. doi: 10.1126/science.1091340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Billy E, Brondani V, Zhang H, Müller U, Filipowicz W. Specific interference with gene expression induced by long, double-stranded RNA in mouse embryonal teratocarcinoma cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(25):14428–14433. doi: 10.1073/pnas.261562698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sledz CA, Holko M, de Veer MJ, Silverman RH, Williams BR. Activation of the interferon system by short-interfering RNAs. Nature Cell Biology. 2003;5(9):834–839. doi: 10.1038/ncb1038. [DOI] [PubMed] [Google Scholar]
- 77.Castelli JC, Hassel BA, Maran A, et al. The role of 2′−5′ oligoadenylate-activated ribonuclease L in apoptosis. Cell Death and Differentiation. 1998;5(4):313–320. doi: 10.1038/sj.cdd.4400352. [DOI] [PubMed] [Google Scholar]
- 78.Guillot L, Le Goffic R, Bloch S, et al. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded RNA and influenza A virus. The Journal of Biological Chemistry. 2005;280(7):5571–5580. doi: 10.1074/jbc.M410592200. [DOI] [PubMed] [Google Scholar]
- 79.Matsumoto M, Kikkawa S, Kohase M, Miyake K, Seya T. Establishment of a monoclonal antibody against human Toll-like receptor 3 that blocks double-stranded RNA-mediated signaling. Biochemical and Biophysical Research Communications. 2002;293(5):1364–1369. doi: 10.1016/S0006-291X(02)00380-7. [DOI] [PubMed] [Google Scholar]
- 80.Matsumoto M, Funami K, Oshiumi H, Seya T. Toll-like receptor 3: a link between toll-like receptor, interferon and viruses. Microbiology and Immunology. 2004;48(3):147–154. doi: 10.1111/j.1348-0421.2004.tb03500.x. [DOI] [PubMed] [Google Scholar]
- 81.Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saunders LR, Barber GN. The dsRNA binding protein family: critical roles, diverse cellular functions. The FASEB Journal. 2003;17(9):961–983. doi: 10.1096/fj.02-0958rev. [DOI] [PubMed] [Google Scholar]