

Abstract
Alzheimer's disease is the principal cause of dementia throughout the world and the fourth cause of death in developed economies.This brain disorder is characterized by the formation of brain protein aggregates, namely, the paired helical filaments and senile plaques. Oxidative stress during life, neuroinflamamtion, and alterations in neuron-glia interaction patterns have been also involved in the etiopathogenesis of this disease. In recent years, cumulative evidence has been gained on the involvement of alteration in neuronal lipoproteins activity, as well as on the role of cholesterol and other lipids in the pathogenesis of this neurodegenerative disorder. In this review, we analyze the links between changes in cholesterol homeostasis, and the changes of lipids of major importance for neuronal activity and Alheimer's disease. The investigation on the fine molecular mechanisms underlying the lipids influence in the etiopathogenesis of Alzheimer's disease may shed light into its treatment and medical management.
INTRODUCTION
Alzheimer's disease (AD), one of the major types of dementia in the elderly, is characterized by the formation of protein aggregates in the brain, namely paired helical filaments composed of hyperphosphorylated tau and senile plaques of the Aβ amyloid [1]. AD is a multifactorial disease, where four main factors appear to be involved in its pathogenesis, and influence tau pathology: (i) the action of Aβ1−42, Aβ1−40, and oligomers of these peptides [2]; (ii) oxidative stress molecules [3–5]; (iii) proinflammatory cytokines produced by activated glial cells [6]; and (iv) overproduction of NO by glial cells [7]. However, more recently a relevant link between changes in cholesterol homeostasis and AD has been evidenced [8–10].
The different factors triggering the degeneration of neurons modify various signalling pathways. Their actions appear to be mediated by the activation of the protein kinase systems cdk5/p35 and GSK3β, with the consequent hyperphosphorylation on tau [11]. The mechanisms involving the sequence of events after the neuronal insult by these molecules have been analyzed using as biological models either cell lines, primary cultured hippocampal cells, or transgenic mice models, such as Tg2576 [12] which expresses the Swedish mutation of the amyloid precursor protein APP, and other transgenic models of tau protein [13]. Aβ peptides and their oligomers induce alterations in the signalling cascades via activation of glial cells, or directly in neurons. Oxidative stress appears to be an early event in AD pathogenesis. The equilibrium between phosphatases and protein kinase activities is altered, and tau hyperphosphorylations occur as a consequence of deregulation in the cdk5 and MAP kinases signalling cascades [3]. These studies are consistent with some clinical findings in which cognitive decline in mild cognitive impairment (MCI) and AD patients, analyzed by neuropsychological tests, correlated with the levels of hyperphosphorylated tau markers in the cerebrospinal fluid [14, 15]. Moreover, these studies are relevant to the elucidation of the mechanisms involved in the etiopathogenesis of AD, and provide clues toward novel diagnostic approaches for this disease.
Lee et al [16] have provided evidence that points to alterations in rafts physiology in amyloid processing, a phenomenon, which appears to be modified by changes in cholesterol content in “lipid rafts,” suggesting a direct link between cholesterol and AD. On the other hand, tau protein has also been found in rafts, and tau modifications by the Src kinase Fyn have been reported [16]. Fyn is present in rafts, microdomains composed of cholesterol, and sphingolipids that participate in signal transduction systems. In addition, tau phosphorylation by Src kinases such as Fyn seems to be overactive in AD.
ETIOPATHOGENESIS OF ALZHEIMER'S DISEASE
Neuropathologically, AD has been characterized by extensive degeneration of cholinergic projection neurons of the basal forebrain nucleus basalis (NB), the presence of extracellular neuritic plaques mainly constituted by amyloid β-peptide (Aβ), intracellular deposits of neurofibrillary tangles formed by paired helical filaments (PHFs) containing the hyperphosphorylated tau protein, microglial-mediated inflammatory reaction, and neuronal death [1, 17, 18]. Even though, the pathogenic process leading to AD development has not been clearly defined; molecular and genetic factors are involved. In this review we emphasize the involvement of lipids components among the biochemical risk factors for AD.
Genetic factors and the familial AD
Among the genetic causes of AD, mutations and polymorphisms stand out in at least four genes. Mutations are associated with early-onset familial AD (EOAD) that account for 3% of all cases of AD and usually occur between the age of 30 and 60. These are different than the sporadic AD cases (more than 97%), even though genetic succeptibility and other molecular risk factors appear to be also involved. Alzheimer disease type 1 (AD1) is linked to mutations in the amyloid precursor gene (APP) [19]; while AD3 is caused by a mutation in the gene of presenilin-1 (PSEN1), located in chromosome 14 that encodes for a 7-transmembrane domain protein [20]; whereas AD4 accounts for mutations in the gene of presenilin-2 (PSEN2) on chromosome 1 [21] that encodes a similar 7-transmembrane domain protein. On the other hand, the late-onset of familial AD after age 65 is correlated with mutations AD2, related to the APOE4 allele on chromosome 19 [22], while AD7 and AD8 correspond to mutations that have been mapped to chromosomes 10p, 13p, and 20p, respectively [23, 24]. Mitochondrial DNA polymorphisms are also considered as a genetic risk factor [25, 26]. Furthermore, there is an association between a polymorphism in alpha-2 macroglobulin with low density lipoprotein-related protein-1 (LRP1), which is the receptor for A2M; and with APOE and APP [27, 28]. These studies suggest the possibility that all these proteins, A2M, LRP1, APOE, and APP, may participate in a common neuropathogenic pathway contributing to AD-related neurodegeneration.
Important links between the main biochemical events in AD
One of the hallmarks of AD is the observation of neurofibrillary tangles (NFT), intracellular filamentous aggregates of the microtubule-associated protein tau. Physiologic functions of tau stem from its ability to stabilize microtubules during axonal transport and its capability to help in the neurite growth [29]. Tau is regulated by phosphorylation. In a hyperphosphorylated status, tau detaches from the microtubules and, consequently, the microtubules fall apart and tau tends to aggregate in paired helical filaments (PHF), thus inducing breaks in the microtubular tracks and neuronal death [30]. Specific sites appear to be preferentially phosphorylated early in patients with AD, as, for example, the KXGS motifs targeted by the enzyme MARK, a serine-threonine kinase important for maintaining a polar network of microtubules, and, thus, cell polarity in neurons [31]. In addition to the intracellular NFT, the extracellular lesions are the amyloid plaques (or senile plaques) produced by the accumulation of amyloid (Aβ) beta peptides. Once cell-bound beta-amyloid precursor protein (APP) is cleaved by the β-secretase (BACE), it generates a soluble ectodomain sAPPβ and a C-terminal fragment CTFb, that is subsequently cleaved by γ-secretase originating neurotoxic soluble Aβ peptides that aggregate in oligomers to form these fibrillar structures [32]. APP interacts with multiple components of the nervous system mediating functions that include neuronal trophism, cell adhesion, neuronal migration, neurite outgrowth, cell-cell signalling, synapse formation, and plasticity. The active movement of APP within neurons contributes to transcription in the nucleus and apoptosis in the cytoplasm. Another cleavage of APP at the ε-site results in a fragment that can be stabilized by interaction with the factor Fe65. APP, Fe65, and Tip60 form a transcriptionally active complex that participates in gene transcription, thus making APP a gene regulator. However, a regulatory mechanism should exist to modulate APP levels since an excess of APP may lead to APP oligomerization, caspase activation, and neuronal apoptosis [33].
Oxidative damage and mitochondrial DNA alterations are involved in the neurodegeneration associated with AD. In AD, brain mitochondrial DNA point mutations have been found to appear specific to this condition. Some of these were associated with defects in oxidative phosphorylation. Additionally, the incidence of mitochondrial DNA mutations has been found to increase by 50% in AD patients [34, 35]. The mechanism that links mitochondrial alterations with neurodegeneration, as well as aging, is related to the oxidative and molecular damages that are being inflicted over time. Also, the oxygen-reactive species (ORS) scavenging mechanisms deteriorate with age and can be associated to functional deficits [36].
Even though, the early molecular events that occur in AD are not clear, the synapse loss is considered to be one of the morphological correlates related to the impairment of cognitive function observed in mid to late stages of AD [37]. It has been postulated that synaptic dysfunction precedes this synapse loss in AD [32]. In this regard, changes in the levels of proteins involved in synaptic vesicle biogenesis and/or recycling have been reported, like SNAP-25, syntaxin, and synaptotagmin in AD [38]. A second example is the critical reduction in the levels of dynamin 1 observed in AD brains, an essential protein in synaptic vesicle recycling [39]. This reduction is attributed to Aβ since it has been shown that Aβ decrease the dynamin 1 levels involving calpain-mediated proteolysis and down-regulation of dynamin-1 gene expression [40].
General risk factors and the changes in lipids as a risk factor
Epidemiologic studies have evidenced several risk factors for AD. Age represents one of the stronger risk factors for AD [41]. The prevalence of AD doubles every 5 years after the age of 60, increasing from a prevalence of 2% among those 60- to 64-year-old to up to 50% of those aged 85 years and older [42]. Studies have shown that AD is more common among women than men by a ratio of 1.2 to 1.5 [43]. Another important risk factor is the presence of the apolipoprotein e-4 (APOE epsilon-4) allele [44]. Of its three forms, ε-2, -3, and 4, only the ε-4 allele increases the likelihood of developing AD. The lifetime risk of AD for an individual without the ε-4 allele is approximately 9%; the lifetime risk of AD for an individual carrying at least 1 e-4 allele is 29%. While representing a substantial risk of AD, the ε4/ε4 genotype is not sufficiently specific or sensitive to allow its use as a diagnostic test. Moreover, ε-4 allele appears to increase the risk of AD more in white and Asian populations than in black and Hispanic populations [45, 46]. Additionally, hypertension [47, 48], heart disease [49], obesity at midlife [50], smoking [51], elevated plasma homocysteine levels [52], diabetes [53, 54], as well as hypercholesterolemia [55, 56] are also considered as risk factors for AD.
In relation to hypercholesterolemia, several reports have shown that elevated serum cholesterol levels and elevated levels of Aß are linked with AD risk [57, 58]. Additionally a cluster of polymorphisms in cholesterol-related genes such as APOE, SOAT1, APOE 5′-untranslated region, OLR1, CYP46A1, LPL, LIPA, and APOA4 has been shown to correlate with levels of the brain cholesterol catabolite 24S-hydroxycholesterol in the cerebrospinal fluid conferring significant susceptibility to AD [59]. Studies using statins, lipid-lowering agents that inhibit HMG-CoA reductase: the key enzyme of the endogenous cholesterol synthesis, indicate that statins also can affect γ-secretase activity, thereby decreasing the breakdown of APP and reducing the risk of AD [60, 61]. Consequently, the proteolytic activity of γ-secretase is stimulated by neutral glycosphingolipids (cerebrosides), anionic glycerophospholipids, and sterols (cholesterol), showing the involvement of lipids and rafts in the modulation of BACE activity [62]. Furthermore statins exhibiting anti-inflammatory actions [63, 64] have been able to down-regulate the Aβ-mediated inflammatory response independent of cholesterol reduction [65]. These anti-inflammatory effects involve the functional inactivation of members of the Rho subfamily of small G-proteins, which regulate the actin-based cytoskeleton and participate in proinflammatory signalling pathways inducing cytokines and chemokines. This inactivation occurs through a mechanism that blocks the isoprenylation, a lipid modification of Rho-family members that facilitates specific interactions with cytoplasmic regulators, cellular membranes, and effectors [66, 67]. Recent findings demonstrate that the anti-inflammatory action of statins depends on the disruption of Rho family functions, as a consequence of reduction of isoprenoid cellular levels, preventing Rho family members from interacting with RhoGDI, resulting in increased levels of GTP-loaded G-proteins and reducing the Rac translocation to the plasma membrane [68]. In this context, Rac1 signalling as well as the enzyme BACE1 have been postulated as targets for developing novel therapies for AD [69]. These observations underlie a mechanism for the statin-mediated reduction in AD risk that involves down-regulation of neuronal APP processing and Aβ production and attenuation of microglia-mediated inflammation.
REGULATION OF TAU PROTEIN AND ITS POST-TRANSLATIONAL MODIFICATIONS
Since modified tau variants have been found in membrane domains, their interactions with lipids and the involvement of these molecules in AD are analyzed below. Tau is produced from a single gene in the chromosome 17 by alternative splicing, mainly during neuronal development [74]. The splicing products are 6 isoforms that differ in molecular weight according to the number of N-terminal repetitions and the number of microtubule binding sequences at their C-terminal domains [75]. The expression of tau isoforms also differs in central and peripheral nervous system and depends on the developmental stage. In rat brain tau starts its expression at the embryonic day 13, only the shortest isoform. From the postnatal day 8, this fetal tau isoform decreases, and the new isoforms, called “adult forms,” start their expression. In the adult peripheral nervous system, there is another kind of tau, the high molecular weight isoform (110 kDa), which contains two additional expressed exons. In general, tau is a highly soluble protein, with a majority of hydrophilic residues; these characteristics mean that tau is a protein that has a natively unfolded structure, that confers resistance to heat and acid treatments.
Tau can be regulated post-translationally by phosphorylations, ubiquitinations, and O-glycosylations. This last consists of the addition of an O-linked N-acetylglucosamine (O-GlcNAc) on a Ser or Thr residues in the proximity of a Pro. Tau phosphorylation appears to be especially relevant considering that anomalous phosphorylations are involved in AD. The equilibrium between the protein kinases and phosphatases regulates the existence of these phosphorylations (Table 1).
Table 1.
Summary of the different protein kinases implicated in Tau phosphorylation, the exact residues that are modified by them and the phosphatases that participate in the dephosphorylation process of each residue. Phosphatases are (1) PP1, (2) PP2A, and (3) PP2B. See [70–73].
| Kinase | Phosphatase | ||||||||||||||||||
| 2,3 | 2 | 2,3 | 1,2,3 | 1,2,3 | 2,3 | 3 | 3 | 1,3 | 3 | 1,2,3 | 1,2,3 | 3 | |||||||
| PKA | — | — | — | — | — | — | — | — | S214 | — | T234 | S262 | T293 | S324 | S356 | — | S404 | S409 | S416 |
| PKB | — | — | — | — | — | — | — | T212 | S214 | — | — | — | — | — | — | — | — | — | — |
| PKC | — | T123 | — | — | — | — | — | — | — | — | — | — | — | — | — | S396 | S404 | — | S416 |
| CaMKII | — | — | — | — | — | — | — | — | — | — | — | S262 | — | — | S356 | — | — | — | S416 |
| p110mapk | — | — | — | — | — | — | — | — | — | — | — | S262 | — | — | — | — | — | — | — |
| JNK | — | — | T175 | T181 | — | S202 | T205 | T212 | — | — | — | S262 | — | — | — | S396 | S404 | — | — |
| p38 | S46 | — | T175 | T181 | — | S202 | T205 | T212 | — | — | — | — | — | — | S356 | S396 | S404 | — | — |
| SAPK3 | — | — | — | T181 | — | — | — | — | — | — | — | S262 | — | — | S356 | — | — | — | — |
| ERK2 | S46 | — | T175 | T181 | — | S202 | T205 | T212 | — | — | — | — | — | — | — | S396 | S404 | — | — |
| GSK3β | — | — | T175 | T181 | S199 | — | — | T212 | — | T231 | — | — | — | — | — | S396 | S404 | — | — |
| Cdk5 | — | — | — | T181 | — | S202 | T205 | T212 | T231 | — | — | — | — | — | S396 | S404 | — | — | |
| Cdk2 | S46 | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — | — |
Tau phosphorylations at Ser/Thr residues
Two different Serine-Threonine kinases have been described. (i) Proline-directed protein kinases that recognize consensus sequences that are followed by prolines. This group involves glycogen synthase kinase 3 (GSK3), cdk2 and cdk5; mitogen-activated protein kinases MAPK family, Erk1 and Erk2; and the stress-activated protein kinases (SAPK), JNK and p38 [11, 76]. These enzymes affect tau self-aggregation and phosphorylations occurring in the N-terminal and in the C-terminal tau moieties. (ii) Nonproline-directed protein kinases that recognize consensus sequences that are not followed by prolines. These include protein kinase C (PKC), protein kinase A (PKA), Ca+2/calmodulin-dependent kinase II (CaM kinase II), Ser 262 kinase/p100mapk, and microtubule affinity regulating kinase (MARK). These phosphorylations mainly occur at the tubulin-binding region of tau [77, 78].
Nonproline-directed serine-threonine phosphorylations of tau affect the microtubule affinity. Ser262/Ser356 sites, in the repeat domain of tau, are critical for tau binding to microtubules; therefore, when tau is phosphorylated in these residues, its affinity for microtubules diminishes. Tau phosphorylation status at these Ser residues plays a key role in the extension of cell processes [79]. Since tau stabilizes microtubules when it is bound to them, and neurites extension process needs the elongation of microtubules, then phosphorylations in Ser262 and Ser356 render microtubules less stable. Proline-directed serine-threonine phosphorylation of tau comprises almost 80% of total tau phosphorylation; it has a weak effect on microtubule binding and is regulated during neuronal development. This phosphorylation is enhanced in AD. It has been observed [78, 80] that the decrease in Ser/Thr-Pro phosphorylations favors the extension of processes, that is, when these sites are dephosphorylated. Phosphorylations are done by proline-directed protein kinases and mainly occur on the C-terminal region, thus modifying tau affinity for microtubules [78–80]. An imbalance in kinases triggers tau hyperphosphorylation, producing the paired helical filaments (PHFs), the basic components of neurofibrillary tangles (NFTs) [81].
Phosphorylations at Tyr residues
Tau can associate cell plasma membrane by its N-terminal region [82] without altering its microtubules binding. Tau can be phosphorylated by Fyn, a tyrosine kinase from the nonreceptor Src family kinases [83]. Fyn participates in the signal transduction system related to integrins having responses such as the Ras and Rho activations and the consequent actin reordering [84]. Bhaskar et al [85] have shown that Src also phosphorylate tau in Tyr 18 in vitro. In addition, Derkinderen et al [86] demonstrated that Abl phosphorylates tau in Tyr 394; both phosphorylations are present in PHFs. Tyr 18 is conserved in all tau isoforms and the sequence around this residue (GTYG) is close to the binding sequence of Fyn to its target (ETYG). Among the 7 PXXP motifs of tau, one has the major affinity to Fyn SH3 domain [84]. Moreover, Fyn has more affinity for 3R tau than for 4R tau, however, the low affinity for 4R tau is considerably increased by phosphorylations present in AD-like Ser 199/Ser202 and Ser 396/Ser 404, epitopes recognized by AT8 and PHF1 antibodies [85]. Since tau 3R plays an important role in neuronal development, its affinity with Fyn has a major function in this process. Meanwhile, tau 4R is mainly present in adult brain, and the increase in its affinity for Fyn may have an important effect in AD etiopthogenesis (Figure 1).
Figure 1.
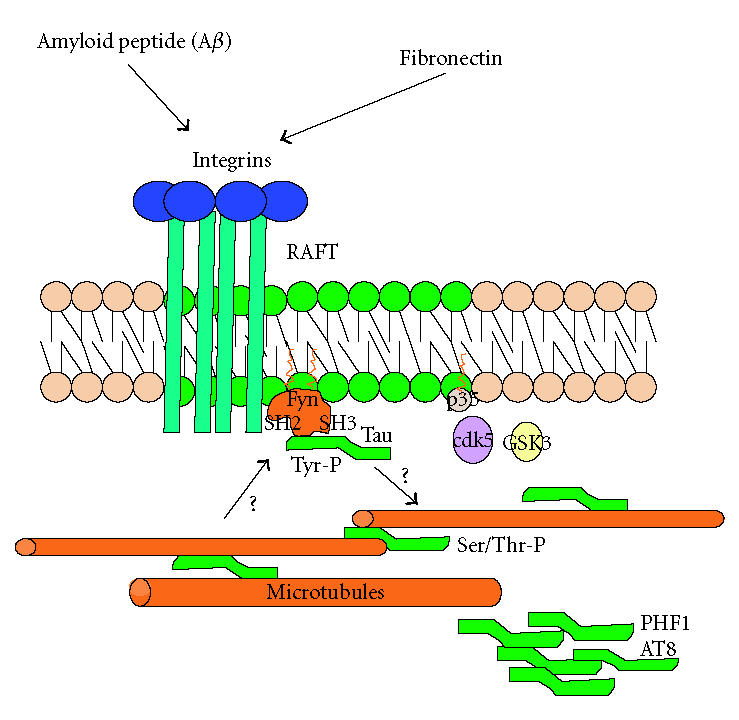
TAU PROTEIN IN PLASMA MEMBRANE
In the context of the involvement of lipids in AD, membrane tau appears to be relevant. The C-terminal tau domain is related to the binding of microtubules and actin filaments, participates in the stabilization of the cytoskeleton, and is a target for phosphorylations in Ser/Thr residues that regulate microtubule binding and the aggregation of tau in neurological diseases. The N-terminal moiety, is a projection of the protein and does not participate in the stabilization of microtubules and extension of neurites, as shown in deletion experiments [87, 88]. Brandt and Lee [87] showed that the projection domain is present in the plasma membrane of transfected PC12 cells with tau and its N-terminal domain. In nonneuronal models such as rat fibroblast (RAT-1), tau was not enriched in the periphery of the cell, indicating that its membrane localization requires the presence of neural-specific factors [87].
The association of tau to the plasma membrane is determined by its phosphorylation patterns [82]. Tau-1 antigens were detected in the membrane (tau dephosphorylated in Ser 199). When tau phosphorylation increases, it dissociates from membranes. Thus, tau phosphorylation status regulates its capability to bind to microtubules. Then, the pathological hyperphosphorylation of tau could lead migration of the subpopulation of tau associated to the plasma membrane to the cytosol, triggering its binding to microtubules or its aggregation. This is consistent with the lesser amount of tau phosphorylated at PHF1 and AT8 epitopes in the plasma membranes in AD [82].
It has been proposed that tau binds the plasma membrane in association with the membrane cortex, that is, the actin cytoskeleton present in the periphery of the cell [82]. Tau binds actin by the same amino acids used for the microtubule binding [88], and is regulated by the phosphorylation status. The accumulation of tau in the somatodendritic compartment produced in AD could be triggered by a loss of tau-cortical interaction as a result of the phosphorylation in PHF-like epitopes.
The phosphorylation pattern of tau is affected by phospholipids [89]: incubation of tau with phosphatidyl serine (PS), and MAP kinases showed that the phosphorylation of tau in PHF epitopes decreased, increasing the Tau1 reactivity. Phosphatidyl choline (PC) showed the same results, but phosphatidyl inositol (PI) did not. PS also reduced the cosedimentation of tau with microtubules in experiments of tau-tubulin association and enhanced the tau proteolysis by calpain. The lipid components of the plasma membrane are able by this way to alter tau function, making the tau subpopulation present in this compartment, different from the cytosolic tau.
In rafts, the exoplasmic leaflet is enriched with sphingomyelin and glycosphingolipids, the cytoplasmic leaflet is enriched with glycerolipids (phosphatidyl serine and phosphatidyl ethanolamine). Cholesterol is present in both leaflets. Some proteins such as those with GPI anchors are also present in rafts, leaving the GPI in the exoplasmic leaflet; also present in rafts are proteins with transmembrane domains, like the influenza virus proteins and haemaglutinin (HA) [90] and the palmitoylated such as Src family kinases. The hydrogen-bonding properties of glycosphingolipids with themselves and with the GPI anchor the GPI-linked proteins to stabilize the complexes formed in the microdomains. In addition, cholesterol has a planar shape that favors its organization in the membrane, also giving stabilization. The lipids can move in and out of rafts individually, making this system highly dynamic.
In oligodendrocytes, the myelination process is led in great part by Fyn. Fyn knockout mice lack correct myelination. Fyn interacts with tau, and tau interacts with microtubules. Both are present in the rafts in the oligodendrocytes (see [91]), forming a complex as evidenced by tau-Fyn coimmunoprecipitation in raft and nonraft fractions. It was also observed that tubulin is capable of binding SH2 and SH3 domains of Fyn, whereas tau only binds the SH3 domain. When the cells were transfected with the N-terminal tau moiety containing the PXXP motif that interacts with Fyn, the number and length of processes diminished because tau N-terminal competes for the Fyn binding. Klein et al [91] proposed a model in which Fyn interacts with tau in rafts, and in turn tau interacts with microtubules allowing the extension process of oligodendrocytes when these cells contact neighbor neurons.
POTENTIAL ROLE OF LIPIDS AND CHOLESTEROL IN AD
Membrane lipids are essential for biological functions ranging from processes involving trafficking of molecules and signalling transduction. Lipids and cholesterol are transported through the blood stream by lipid-protein particles, lipoproteins that can be classified in different subsets according ot their density: high density (HDL), medium density (IDL), low density (LDL), and very low density (VLDL). LDL has an elevated fat proportion and participates in lipids transported from blood stream to peripheral tissues. HDL has a major protein fraction with respect to lipids, and takes part in reverse cholesterol transport from peripheral tissues to liver where conjugation and excretion occur. Cholesterol is transported in the plasma predominantly as cholesteryl-esters associated with lipoproteins. Cholesterol derived from diet is transported from the intestines to the liver within chylomicrons. Cholesterol can be synthesized de novo by the liver using acetyl-CoA as precursor molecule. In this biochemical pathway, the enzyme HMG-CoA reductase has a main role in the pharmacological intervention to diminish the endogenous cholesterol synthesis. Finally, cholesterol is excreted in the bile as free cholesterol or as bile salts following conversion to bile acids in the liver [92].
Brain cholesterol is mainly synthesized locally within the CNS. It is estimated that during CNS development, neurons synthesize most of the cholesterol needed for their growth and synaptogenesis. Later, when neurons are mature, they reduce their endogenous cholesterol's synthesis and become more dependent on cholesterol synthesized and secreted by the astrocytes [93]. Cholesterol in the CNS is turned over in a proportion of 0.7% of the total amount every day. Even though, it is not a great fraction, it is a relatively high amount of cholesterol considering that the CNS accounts for 2.1% of body weight and contains 23% of the total sterol in the whole body. The brain is therefore the most cholesterol-rich human organ. In addition, cholesterol accounts for 20–25% of the total lipids in neurons plasma membranes. Thus, neurons require a continuous supply of new cholesterol to maintain its constant concentration in the plasma membranes. Eukaryotic cells incorporate cholesterol through at least three mechanisms: (1) de novo synthesis within the cell from acetyl-CoA, which is the most important mechanism for neurons, (2) uptake of unesterified or esterified cholesterol from the external environment using the LDL receptors (LDLR), or (3) the Niemann-Pick C1-like protein (NPC1L1) (90).
LDLR bind particles that contain either apoE or ApoB-100 (remnants of chylomicrons, very low density lipoproteins (VLDL), and LDL) [94]. These particles are then processed through the clathrin-coated pit pathway to late endosomes and lysosomes. Then, the hydrolysis of the cholesteryl esters takes place and cholesterol becomes part of the metabolically active pool within the cell. At this final stage, cholesterol performs a variety of functions: it can be transferred to the plasma membrane, metabolized to other products, or act as a regulator of cell sterol metabolism [94]. Two other proteins; Niemann-Pick type C1 and C2 (NPC1 and NPC2), are also required to move the unesterified cholesterol to the metabolically active pool. However, their roles remain uncertain so far [92]. Current evidence indicates that cells with dysfunctional NPC1 or NPC2 accumulate unesterified cholesterol in late endosomes, which reflects a failure of cholesterol to exit efficiently this compartment and travel to the plasma membrane and endoplasmic reticulum [95]. This failure of cholesterol trafficking can be explained, in part, by its genetic basis, which involves mutations of either of two functionally related genes, NPC1 and NPC2, accounting, respectively, for 95% and 5% of the cases [95].
Cholesterol cannot pass directly through the blood-brain barrier (BBB). Firstly, it has to be oxidized into 27-hydroxycholesterol and/or 24S-hydroxycholesterol in the peripheral organs or in the CNS, respectively. As 24S-hydroxycholesterol is synthesized in the brain and spinal cord, concentrations of this late metabolite are higher in the CNS than in any other tissue. Some authors have even suggested that 24S-hydroxycholesterol peripheral levels can be used as a marker for AD [96]. In fact, during the early stages of AD, 24S-hydroxycholesterol levels are high in CSF and in peripheral circulation, even though the physiological explanation for this fact and its implications for AD are still unknown [96, 97]. The transport of excess cholesterol out of cells is mediated by ABCA1, a membrane protein that facilitates the formation of APOE-cholesterol-phospholipid (ApoE-Chol-PL) complex and requires the hydrolysis of ATP for its activity. Upon secretion of the APOE-Chol-PL complex into the extracellular environment, the complex may bind to LDL receptor-related protein (LDLRP) to be taken up by neurons or form HDL-like particles for transport to the systemic circulation [93].
Cholesterol influences the activity of the enzymes involved in the metabolism of the amyloid precursor protein and in the production of Aβ [58]. In some animal studies, dietary cholesterol accelerated Aβ deposition in the brain, whereas cholesterol-lowering drugs lowered it [58–60]. Also, in other in vivo studies a lower-cholesterol environment resulted in an increased production of soluble amyloid precursor protein [98]. The mechanism by which cholesterol affects Aβ production and metabolism is not fully understood. It has been suggested that possible mechanisms related to changes in plasma membrane stiffness and fluidity could explain the influence of cholesterol on enzymes like BACE 1. The high cholesterol content in lipid rafts, where these enzymes are located, could facilitate the clustering of α and β secretases with their substrates into an optimum configuration, thereby promoting the undesirable pathogenetic cleavage of amyloid precursor protein [58, 69]. This suggests that the use of statins might decrease the levels of cholesterol in neurons thereby altering the organization of lipids rafts. However, Abad-Rodriguez et al have demonstrated that some of the most commonly used statins are poor penetrators of the blood-brain barrier [99], so their benefits in the prevention of AD development might derive from anti-inflammatory or antioxidant properties rather than a direct effect on cholesterol concentration in the plasma membrane of neurons. In neuronal and glial membranes, cholesterol is distributed asymmetrically among the two membrane leaflets. In normal physiological conditions, the plasma membrane cytofacial leaflet contains more than 85% of the membrane cholesterol in the synaptic plasma membrane, whereas the cholesterol content of the exofacial leaflet is low [93]. Some statins may raise this ratio by lowering the cholesterol content of the exofacial leaflet [100]. However, these effects of statins in the brain vary depending on the lipophilicity of each molecule. This suggests that, rather than reducing brain cholesterol, a feasible explanation for the effect of statins might be that they alter the cholesterol balance in the plasma membranes of brain neurons. However, the significance of these effects on the human brain is still unknown.
In vivo experiments have shown that there are differences between the effects of simvastatin, a lipophilic statin; and pravastatin, a hydrophilic statin, when given as a high-dose short-term treatment [70]. Simvastatin reduced the levels of lathosterol, a cholesterol precursor, but did not affect the formation of 24(S)-hydroxycholesterol. The HMG-CoA reductase and ABCA1 mRNA expression in the brain was significantly upregulated in animals treated with simvastatin compared to those treated with pravastatin or with placebo. These findings suggest that cholesterol synthesis is significantly affected by short-term treatment with high doses of lipophilic simvastatin, while whole brain cholesterol turnover is not disturbed [70].
Altogether, there is still no evidence for the net transfer of sterol from the systemic circulation into the brain. Details about the implications of cholesterol blood levels in the development of AD are therefore an open field for research. Niemann-Pick type-C disease (NPC) is a juvenile, fatal autosomal recessive neurovisceral lipid storage disorder. NPC is associated with a progressive neurodegeneration, thereby resulting in dementia caused by dysfunction of the neuronal network [101]. Hallmarks of NPC are ballooned neurons and massive neuronal loss with massive intracellular cholesterol accumulations both in human and in murine NPC brain [102].
Although NPC differs in major respects from AD, intriguing parallels exist in the cellular pathology of these two diseases, including neurofibrillary tangle formation, prominent lysosome system dysfunction, and influences of the apolipoprotein E4 genotype. In addition, accumulation of cleaved APP and Aβ peptides within endosomes has been observed in NPC [95]. In vivo experiments using murine models of NPC have demonstrated that tau protein is phosphorylated at epitopes considered to represent early stages of AD [102]. This strengthens the concept that an alteration in cholesterol metabolism could play a pivotal role in early stages of AD.
CHOLESTEROL IN LIPID MEMBRANE MICRODOMAINS (RAFTS) AS AN APPROACH TO STUDY ALZHEIMER'S DISEASE
Several protein systems use lipid rafts as a platform for signalling. “Lipid raft” is basically a definition given to those membrane domains rich in cholesterol and glycosphingolipids isolated from cells or cell membranes preparations through detergent and nondetergent methods [71]. Names like cholesterol enriched membranes (CEMs), glycosphingolipid enriched membranes (GEMs), detergent-insoluble, glycosphingolipid-enriched membranes (DIGs), and detergent-resistant membranes (DRMs) have been given to these rigid plasma membrane fragments based on their preferential lipid composition and/or nonionic detergent resistance properties [103]. Rafts can be invaginated or not depending on whether they are composed of a protein called caveolin. This protein interacts with the membrane making a hairpin loop and raising caveolae; these are considered as specialized forms of rafts implicated in cell transduction.
During the last ten years, numerous data obtained from a wide variety of biophysical and biological techniques have helped to explain the composition and function of lipid rafts. However, there are still many unanswered questions, especially in the area of cholesterol/lipids and cholesterol/proteins biochemical interactions and differential segregation. In the light of currently available scientific evidence, it seems clear that lipid rafts composition strongly depends on the “isolation” method used to obtain them [72, 104]. A plausible hypothesis to explain the differences occurring in cholesterol, glycosphingolipids, and proteins composition of lipid rafts, based on different isolation methods, has been recently described. In this hypothesis, scientific evidence is presented and discussed in order to support the existence of three different models of raft structure based on the utilization of different raft isolation protocols. The first two predict that all rafts isolated by the same method will have similar composition and the third predicts that rafts isolated by the same method could be heterogeneous in composition because the domains themselves are heterogeneous [105]. Conversely, Rajendran et al showed that, in lymphocytes, raft proteins like lck, lyn, and LAT were released from lipid rafts by treatment with methyl-β-cyclodextrin; whereas flotilins markers remained in detergent resistant membranes, suggesting that some rafts require less cholesterol than others to maintain their integrity, or that some rafts retain their cholesterol more effectively than the others by cholesterol sequestering agents [73].
It seems, therefore, of pivotal importance to focus on the raft isolation methods when discussing cholesterol rafts and their possible role in any pathophysiological event. Altogether, despite the heterogeneous composition found in lipid rafts, it is widely accepted that they are membrane domains rich in cholesterol and glycosphingolipids [106, 107]. Lipid composition is approximately 1 : 1 : 1, phosphoglycerolipids: sphingolipids (including sphingomielin (SM) and glycosphingolipids (GSL)): cholesterol. In rafts isolated using detergent protocols, the phospholipid population is enriched in saturated acyl chains relative to the average for whole cell phospholipids [108]. Conversely, little consistent evidence has been provided regarding the mechanism underlying cholesterol selective migration, residence, and dynamics in these membrane lipid rafts.
Ipsen et al have proposed a lipid membrane spatial distribution theory that partially helped to explain the clustering of lipids into the cholesterol rich domains; they proposed that cholesterol and phospholipids form a liquid-ordered (Lo) phase characterized by a high level of molecular order in the lipid packaging [109]. According to this hypothesis, the Lo phase would coexist with the liquid-disordered phase (Ld) or liquid-crystalline phase, characterized by a high degree of disorder and very high lipid mobility, and with the gel phase, in which lipid molecules are virtually immobile. Lipids and proteins that “prefer” liquid-ordered phase (including both glycosphingolipids and glycosylphosphatidylinositol-anchored proteins) would segregate into the Lo-phase domains and thus into lipid rafts. However, this hypothesis appears to be controversial since, according to Brown [110], it does not have enough support. Importantly, a significantly lower fluidity was found in acyl chain of caveolae/raft domains when purified without the use of detergents [103].
In spite of its limitations, giant unilamellar vesicles (GUVs), an artificial membrane system, have provided a very useful tool to elucidate the biophysical mechanisms of raft assembly, especially allowing for the study of cholesterol and lipids dynamics in a noninvasive manner [111]. Application of FCS combined with confocal scanning microscopy allows for the determination of single molecule diffusion, chemical kinetics, and conformational equilibrium. Thus, it has become a valuable insight into lipid/cholesterol interactions and lipid dynamic organization during lipid rafts assembly [111, 112]. By applying these techniques, Kahaya et al [112–114] have established that both dioleoyl-phosphatidylcholine (DOPC) and DLPC, unsaturated phospholipids, gradually decrease their mobility as a function of cholesterol concentration [113]. When compared to DOPC, bigger changes in mobility were exhibited by DLPC, meaning that cholesterol would interact more intimately with the saturated phospholipids. They found that cholesterol interacts more strongly with SM than phosphatidylcholine (PC), implying that the latter interaction may have a role in the stiffness of SM membranes with respect to PC bilayers. In addition, in ternary mixtures, a weaker tendency to form extensive domains was observed for DOPC/dipalmitoylphosphatidylcholine (DPPC)/cholesterol as compared with DOPC/DSPC/cholesterol. In both cases, this tendency was much weaker than that for the DOPC/SM/cholesterol mixture. This reflects the weaker DPPC/cholesterol with respect to SM/cholesterol interactions. Altogether, it seems that cholesterol clearly interacts more strongly with sphingolipids than with phospholipids even for those that are unsaturated [114]. Interestingly, it has been found that ceramides, generated from SM by the action of SMase, displace membrane cholesterol due to a competition for associating with lipid rafts [115]. Other GUVs/FCS studies published by Kahya in 2003 [112] proved that the lipophilic fluorescent probe DiI-C18 was excluded from SM enriched regions, while the raft marker GM1 was present. Furthermore, cholesterol was shown to promote lipid segregation in DOPC-enriched liquid disordered and SM-enriched liquid ordered phases. Lipids mobility in SM-enriched regions significantly increased by increasing the cholesterol concentration.
Another feasible model to explain the generation of lipid rafts and the differential cholesterol distribution was proposed by Anderson and Jacobson and is based on protein/lipid interaction [116]. In this model, also named “the shell hypothesis,” raft proteins like GPI-anchored proteins and selected transmembrane and peripheral proteins would be able to adopt a “shell” formation via interactions with cell membrane lipids. Caveolins, the proteolipid MAL, flotilins, and stomatin have been reported to interact with the bilayer and have an intimate contact with membrane lipids forming the so-called “lipid shells,” which are thought to be thermodynamically stable and diffusible unit that coalesce based on protein-protein interactions. As a result, a larger functional unit called “lipid raft” would be created. However, it remains unknown which of the raft proteins are capable of interacting with lipids in a way to form these lipids shells.
Pankov et al [117] have hypothesized that integrins, important protein components of the cholesterol-enriched domains, may have a fundamental role in cholesterol levels and in the stability of lipid rafts. They demonstrated that integrin presence in fibroblast plasma membrane increased lipid rafts cholesterol and sphingomyelin content as well as the nonraft constituent phosphatidylethanolamine. Even though, they do not provide details about the mechanism underlying this fact, a likely explanation based on phosphatidylethanolamine thermodynamic incompatibility with cholesterol was proposed.
Rafts and caveolae contain several proteins implicated in signal transduction pathways [118, 119]. Thus, it is believed that rafts are implicated directly in transduction events; possibly allowing a group of proteins to participate in a certain pathway and isolating others, avoiding the crosstalk of different pathways. They could make the encounter of interacting proteins easier. Rafts are implicated in the integrin signalling and also in the processing of amyloid precursor protein (APP) to Aβ [120]. The secretases implicated in the cleavage of APP are present in rafts, and the processing in Aβ [121] is dependent on the cholesterol amount in the cell. It has been proposed that if the cell has more cholesterol, then it has more rafts, allowing the APP cleavage and the posterior accumulation of this peptide at the exterior of the cell [120].
AMYLOID PEPTIDE, APP, AND THEIR LINKS WITH RAFTS AND MEMBRANE LIPIDS
By using silver [122] and Congo-red staining [123] it was possible to see in the tissue affected with Alzheimer's disease the lesions produced in the cortical regions of the brain. The neurons are surrounded by a characteristic large, dark, and circular inclusion called Alzheimer's plaque. This plaque is made of amyloid deposits composed of a peptide of 4 kDa in a β-sheet conformation; this was called β-amyloid [124]. Later, it was discovered that the peptide was part of a 79 kDa protein with 695 amino acids recognized as an amyloid precursor protein (APP). Hydrophobic analysis of this protein revealed membrane-spanning segment, a C-terminal cytoplasmic domain, and an N-terminal extracellular domain; it is characterized as a ubiquitously expressed type-1 membrane glycoprotein [125]. APP undergoes alternative splicing originating three varieties depending on the amount of residues: APP695, APP741, APP751, and APP770. The two longer isoforms contain the exon 7, which encodes a serine protease inhibitor domain. This kind of inhibitors promotes the outgrowth of neurites providing a possible nonpathological function for this protein. The most common isoform present in the body is APP751, but in the brain, the most abundant is APP695. APP is widely expressed in cell surface, especially on neurons, but also in astrocytes, microglia, endothelial cells, smooth muscles, and all peripheral cells.
APP is processed by three enzymes called α, β, and γ secretases. The β-secretase is a member of the ADAM family of metalloproteases and the γ-secretase is a membrane-bound aspartyl protease, also called BACE. These secretases cleave the ectodomain of APP in different sites, resulting in two fragments named after the involved secretase: APPsβ and APPsα. After these cleavages, the α-secretase processes the transmembrane domain of the APP C-terminal fragments (α-CTF and β-CTF) producing two smaller fragments called p3 (for the α previous clip) and Aβ (for the β-clip). The first fragment dissolves easily within the brain, but the second does not dissolve and therefore it accumulates in the brain, forming the senile plaques. The Aβ fragments can have 40 or 42 residues (Aβ1−40 or Aβ1−42). The Aβ1−42 is neurotoxic and more easily aggregates in plaques. Since the fragment produced by the previous cleavage of the γ-secretase does not produce Aβ or plaques, it is called “nonamyloidogenic pathway” and is neuroprotective. This is the major way of APP metabolism in most cells. Therefore, the stimulation of easier γ-secretase decreases the Aβ formation and protects from AD. There are several reagents that can stimulate this secretase, such as estrogen, testosterone, various neurotransmitters, growth factors, and the activation of protein kinase-C (PKC) by phorbol esters. β- and γ-secretases are considered as participants in the amyloidogenic pathway, especially γ-secretase because it is associated with presenilins. Two highly conserved residues in the transmembrane domains 6 and 7 of presenilins seem to be necessary for the normal γ-secretase activity. Since the presenilin genes PSEN1 and PSEN2 participate in familial AD, one possible treatment for the prevention of AD would be the control of γ-secretase activity. Both β- and -secretases are present in lipid rafts and these lipid microdomains containing cholesterol have been implicated in AD [126]. These membrane domains could function as a microenvironment, where APP is processed, but while the generation of Aβ seems to be dependent on rafts and what occurs there, the processing of the APP by the γ-secretase occurs outside the lipid rafts [120]. The levels of Aβ are not dependent on the cholesterol amount in the cell in vivo [121], due to cholesterol having no effect over γ-secretase activity, but the increment in the cholesterol at the membrane can increase the lipid rafts formation and thus allow the APP processing by BACE1 (γ-secretase) [126].
OXIDIZED LIPIDS AND CHOLESTEROL IN AD
Neurodegenerative disorders like Alzheimer's disease encompass oxidative modifications of lipids and cholesterol detected in serum and cerebrospinal fluid (CSF) of Alzheimer's disease patients (AD). The major protein modification by trans-4-hydroxy-2-nonenal (HNE) is the adduction to lysines, which was observed in postmortem brains of AD [127, 128]. The neurotoxic effect of HNE, a potent prooxidant, was determined in a cellular culture model. Acrolein is a more reactive product of the metal-catalyzed oxidation of polyunsaturated fatty acids [129] detected at increased level in the brain of AD and becomes neurotoxic to hippocampal neurons by a calcium-dependent mechanism [130].
Efforts have been made to find reliable biological markers for the initial phase in AD, even before the first clinical evidence for the disease. In this context, the detection of oxidative markers such as 4-HNE and 24S-hydroxycholesterol in plasma and urine samples can help in the early diagnosis of AD, thus facilitating the treatment approaches.
An elevated level of HNE, produced by the oxidation of arachidonic acid, was detected in the plasma of AD [131, 132] malondialdehyde, an end product of lipid peroxidation, and was increased in the plasma of AD patients versus age-matched and nutritionally evaluated control subjects [133]. Nevertheless, malondialdehyde failed as a predictive AD marker in several other studies [134, 135]. Elevated levels of 24S-hydroxycholesterol, produced via an enzymatic oxidation of brain cholesterol by CYP46, were detected in the plasma of AD compared with control age-matched volunteers [136, 137]. Concomitantly, treatment with statins lowered the levels of LDL-cholesterol and 24S-hydroxycholesterol in plasma [138], and it has been suggested that the oxysterol may be an important marker of AD risk instead of total cholesterol. In summary, 24S-hydroxycholesterol and HNE are good candidates to be a marker of AD.
A key role in AD pathology is now assumed to be vascular in origin due to hypoperfusion of microvasculature that induces hypoxia in brain tissue, events that are associated with oxidative stress [139]. Moreover, postmortem AD brains show atherosclerotic hypoperfused microvessels lesions that are closely related to oxidative stress markers and amyloid plaques. This scenario is completed by a recent work that has shown an induction of iNOS with a consequent peroxynitrite production in astrocyte cell culture treated with LDL from AD patients [140], which is in agreement with an association among lipids/cholesterol, oxidative stress production and pathology of AD.
An interesting connection between cholesterol and amyloid-β-peptide (Aβ1−42) has been demonstrated. Aβ : Cu2+ complexes oxidize cholesterol selectively at the C-3 hydroxyl group, catalytically producing 4-cholesten-3-one and therefore mimicking the activity of cholesterol oxidase [141]. Moreover, it was demonstrated that amyloid peptide precursor (APP) protein promotes cholesterol oxidation, yielding 7β-hydroxycholesterol, a proapoptotic oxysterol, but Aβ1−42 was shown to be 200 times more potent as prooxidant [142, 143].
Sulfatides are a class of sulfated galactocerebrosides that mediate diverse biological processes including cell growth regulation, protein trafficking, signal transduction, adhesion, neuronal plasticity, and cell morphogenesis which has been characterized as potential AD marker. A decrease in sulfatide concentration in very mild dementia patients has been found [144] but a correlation with Alzheimer's disease should be done.
Interestingly, knockout mice for ApoE have between 61–114% more sulfatides and mice over-expressing ApoE4 exhibit 60% less sulfatides than the “wild type” counterparts. However, no modifications in the contents of phospholipids, sphingolipids, and cholesterol were detected, suggesting a novel role for ApoE in the brain in mediating sulfatides metabolism [145].
EXPERIMENTAL MODELS FOR AD AND THE STUDY OF THE ROLES OF LIPIDS
Studies revealed that an elevated serum cholesterol level is a risk factor for AD [146]. Based on this epidemiological data, a wide variety of in vitro and in vivo experimental models has been used to elucidate potential mechanisms underlying cholesterol/AD relationships. The involvement of cholesterol in modulating BACE activity was assessed, and it was observed that cholesterol stimulates the proteolytic activity of purified BACE in 100 nm unilamellar vesicles [147]. George et al reported that a diet which induced hypercholesterolemia increased APP intracellular domain and reduced soluble Aβ in the transgenic mouse Tg2576, an AD mouse model that overexpresses human amyloid β-protein precursor [148].
Even though statins have pleiotropic effects, they have been widely used to study potential specific links between AD and cholesterol in a variety of AD experimental models [149]. Gender differences observed in the risk for AD development in humans led Park et al to assess the effect of lovastatin in male and female Tg2576 mice separately. Results showed a reduction of cholesterol levels in both sexes, but Aβ1−40 peptide levels were increased in female mice only. As well, no changes were observed in the amounts of full-length, α-secretase processed amyloid precursor protein (APP) or presenilin 1 (PS1) in either sex [150].
Li et al (see [151]) reported that in the transgenic mice B6Tg2576, an AD mice model that also develops atherosclerosis, there is a positive relationship between the presence of aortic atherosclerotic lesions and cerebral β-amyloidosis. Atherogenic diets and spatial learning impairment were also positively correlated in this model. Cordle and Landreth demonstrated that statins reduce neuronal Aβ production and inhibit the microgial mediated inflammation by means of a reduction of isoprenyl intermediates in the cholesterol biosynthetic pathway [152].
Recently, experiments performed with microglial cultures demonstrated that statins blocks Aβ stimulated phagocytosis through inhibition of Rac1. In addition, these experiments paradoxically demonstrated that statins mediate the inactivation of G-proteins by increasing GTP loading of Rac and RhoA and by disrupting the interaction of Rac with its negative regulator RhoGDI [153].
MAIN CURRENT PHARMACOLOGICAL APPROACHES FOR AD
To understand the therapeutic approaches for AD based on the changes in lipids metabolism, it is worth to review the current pharmacological treatments for this disease. The cholinergic hypothesis of AD is based on the evidence that cholinergic brain deficits lead to alterations in attention and memory in animal models, healthy elderly, and cognitively impaired patients. Cholinesterase inhibitors have been widely accepted by clinical and basic researchers as a pivotal part of AD treatment. Furthermore, there is strong evidence supporting the improvement in cognitive and global function with the use of rivastigmine, donepezil, and galantamine [154]. However, a recent critical analysis of the scientific evidence has seriously questioned the benefits of these drugs in AD [153]. Several studies have established that in patients who are cognitively impaired, donepezil may delay the onset of AD [155]. Rivastigmine, which also inhibits butyrylcholinesterase, has shown benefits in mild to moderate AD in clinical trials, but side effects at high doses have led to the suspension of this therapy. A study designed to assess the effects of rivastigmine on the cognitive functioning of 92 patients suffering from dementia with Lewy bodies indicated that rivastigmine produces a significant benefit over placebo on tests of attention, working memory, and episodic secondary memory [156]. Galantamine, a competitive reversible inhibitor of acetylcholesterase and allosteric modulator of nicotinic receptors, has shown no great cognition improvements in mild to moderate AD, thus galantamine does not seem to be significantly better than donepezil [157].
In addition to its role as an excitatory neurotransmitter, glutamate can also be neurotoxic in excessive levels leading to disproportionate depolarization, influx of calcium into neurons, and cell death. Due to its role in memory impairment and dementia, glutamate has been considered a potential aggravating factor in AD [159]. On the other hand, three randomized clinical trials have shown that memantine, a glutamate antagonist causes just mild improvements in the cognitive performance of severe AD patients, though side effects were not very significant. Use of memantine has been shown to slow the decline of the treated patients, rather than cause an actual improvement. Therefore, there seems to be no reason to expect promising results from memantine or better efficacy than cholinesterase inhibitors [160].
As we previously mentioned, the key event leading to cognitive impairment in a variety of AD is the formation of the peptide Aβ1−42, which clusters into amyloid plaques in brains of patients with AD. β-secretase cleavage followed by γ-secretase proteolysis are both necessary to cleave APP into the toxic Aß1−42 [1]. No sufficiently successful therapeutic strategies based on the inhibition of both β and γ secretases have been developed so far. γ-secretase inhibitors are already being tested in clinical trials, while β-secretase inhibitors are still in preclinical development. Other approaches include interfering with the deposition of the Aβ1−42 into plaques and enhancing its clearance, but none of these approaches has gained enough scientific and clinical evidence to support a feasible therapeutic intervention [161, 162].
It is widely accepted that hyperphosphorylated tau is the main component of neurofibrillary tangles (NFTs), a broadly known marker for AD neurodegeneration. Indeed, several reports point out that tau aggregation requires an anomalous previous hyperphosphorylation. Both the prolonged activity of glycogen synthase kinase-3 (GSK3β) as well as cdk5/p35 system deregulation are thought to be pivotal events in tau hyperphosphorylation [11, 12]. Therapeutic drugs like valproic acid and lithium, which are approved for other neurological and psychiatric disorders, have the ability to inhibit GSK-3β. However, according to clinical evidences, both the efficacy and tolerability are limited. Thus, current scientific evidence is still too weak to support a feasible pharmacological therapeutic avenue for AD [163].
CHOLESTEROL-LOWERING AGENTS (STATINS). ARE COGNITIVE IMPROVEMENTS THE BOTTLENECK?
Extensive critical revisions of AD treatments have been published by a number of authors in recent years [160–162] but none of them puts emphasis on the potential role of statins in AD based on the existing molecular and clinical evidence. Here, we have briefly summarized the main current pharmacological treatments. Clinical data regarding the use of statins and thier controversial conclusions are also presented in order to overview the problem and analyze its potential impact on AD treatment (Table 2).
Table 2.
Summary of the main clinical studies that have provided statins efficacy results in AD or other types of dementias. Dur: duration of the study (months), N: number of patients, SD: study design, PO: main primary outcomes or biological effects, AO: additional outcomes, SO: main secondary outcomes, Ref: references.
| Drug dosage | SD | N | Dur | PO | SO | AO | Results | Ref |
| Different statins | Case control | 284 | 72 | Relative risk of AD (odd ratio) | — | — | Decrease relative AD risk (0.29) | [56] |
| Atorvastatin calcium 80 mg/day | Randomized, double-blind, placebo-controlled | 63 | 12 | ADAS cog and CGIC change score | ADAS cog, CGI, and NPI Scales | — | Improvements in PO, trends to improving in SO | [154] |
| Simvastatin 20 mg/day | Uncontrolled, open trial | 19 | 3 | CSF levels of βsAPP, αsAPP, Tau, phospho-Tau, Aβ1−42, and plasma levels of Aβ1−42 | ADAS cog | — | βsAPP, αsAPP decreased, ADAS-cog slightly increased | [153] |
| Simvastatin 20 mg/day | Uncontrolled, open | 19 | 12 | CSF levels of Aβ1−42, βsAPP, αsAPP, totAPP, and total Tau, plasma levels of Aβ1−42 ADAS cog | MMSE | — | No changes in CSF levels of Aβ1−42, βsAPP, totAPP, total Tau, plasma levels of Aβ1−42, ADAS cog, and MMSE, αsAPP increased | [158] |
| Pravastatin 40 mg/day | Randomized, placebo-controlled | 5804 | 38.4 | Coronary death, nonfatal myocardial infarction, fatal and nonfatal stroke | — | MMSE | No differences between treatment and controlled group | [155] |
| Simvastatin 40 mg/day | Randomized, placebo-controlled | 20.536 | 60 | Plasma levels of LDL, major coronary events, strokes, and revascularizations (separated into prior and not prior cerebrovascular disease) | Ischaemic and/or hemorrhagic stroke (separated into prior and not prior cerebrovascular disease) | TICS-m | No differences in the cognitive score between treatment and control groups | [156] |
The epidemiological data regarding the relationship between the use of statins and the risk of AD is controversial. Most of the preclinical results derived from cells and animal studies show a direct relationship between elevated cholesterol levels and molecular or cognitive markers of AD [15]. However, the current state of research in human studies is not solid enough to establish a certain and reliable causal relationship between the use of cholesterol lowering agents and the incidence of AD and other types of dementias [45, 160, 163, 164]. Concrete clinical evidence has been provided by Wolozin et al indicating that there is a 60 to 73% lower prevalence (P < .001) of probable diagnosed AD in patients taking lovastatin or pravastatin [165, 166]. Jick et al reported that patients of 50 years and older had lower risk of developing dementia, though not specifically Alzheimer's, when treated with statins. This lower relative risk was neither related to the presence or the absence of hyperlipidemia [56].
An open uncontrolled clinical trial with the simbastatin in 19 patients with AD settled that 20 mg/d of simbastatin for 12 weeks significantly reduced the cerebrospinal fluid (CSF) levels of β-secretase-cleaved amyloid precursor protein (βsAPP) and α-secretase-cleaved amyloid precursor protein (αsAPP). Conversely, CSF levels of tau, phosphotau, Aβ1−42, and plasma levels of Aβ1−42 remained unchanged. Cognitive capacity was measured using the disease assessment scale cognition (ADAS-cog). In this study, ADAS-cog score was just slightly increased in less than 50% of all patients [167]. Even though, this open observational study concluded that the simbastatin reduces Aβ1−42 formation in AD patients, results were not consistent with a cognitive improvement due to the use of simbastatin.
A recently published pilot double-blind, placebo-controlled trial with a one year exposure to atorvastatin calcium 80 mg/d in 63 AD patients with an MMSE of 12–28 demonstrated that atorvastatin produces a significant beneficial effect at 6 months when evaluated using both geriatric depression scale and the ADAS cog. However, the secondary outcome measures ADAS cog, CGI, and neuropsychiatric inventory scales, evaluated at 12 months, were not consistent with certain protection against AD [168].
Another study demonstrated that simbastatin 20 mg/d for 12 months produces no changes in CSF levels of β-amyloid (Aβ1−42), βsAPP, totAPP, and total tau as compared to the baseline, nor were there changes in Aβ1−42 plasma levels. However, αsAPP was significantly increased suggesting that simbastatin could favor the nonamyloidogenic APP processing pathway [169]. Studies indicate that significantly more research is needed in order to clarify the roles of cholesterol therapeutic agents in the AD treatment. At the present stage of this research, there is cumulative evidence that the search of reliable biomarkers for AD, of importance for early AD diagnosis and monitoring the course of the disease, will allow the growth of therapeutical approaches, including both pharmacological treatment and cognitive rehabilitation of patients.
ACKNOWLEDGMENTS
This research was supported by the Millennium Institute for Advanced Studies (CBB), the International Center for Biomedicine (ICC), and Fondecyt Grant 1050198.
References
- 1.Maccioni RB, Muñoz JP, Barbeito L. The molecular bases of Alzheimer's disease and other neurodegenerative disorders. Archives of Medical Research. 2001;32(5):367–381. doi: 10.1016/s0188-4409(01)00316-2. [DOI] [PubMed] [Google Scholar]
- 2.Klein WL, Stine WB, Jr, Teplow DB. Small assemblies of unmodified amyloid β-protein are the proximate neurotoxin in Alzheimer's disease. Neurobiology of Aging. 2004;25(5):569–580. doi: 10.1016/j.neurobiolaging.2004.02.010. [DOI] [PubMed] [Google Scholar]
- 3.Zambrano CA, Egaña JT, Núñez MT, Maccioni RB, González-Billault C. Oxidative stress promotes τ dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radical Biology and Medicine. 2004;36(11):1393–1402. doi: 10.1016/j.freeradbiomed.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 4.Moreira PI, Smith MA, Zhu X, Nunomura A, Castellani RJ, Perry G. Oxidative stress and neurodegeneration. Annals of the New York Academy of Sciences. 2005;1043:545–552. doi: 10.1196/annals.1333.062. [DOI] [PubMed] [Google Scholar]
- 5.Sayre LM, Zelasko DA, Harris PLR, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. Journal of Neurochemistry. 1997;68(5):2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 6.Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Experimental Cell Research. 2004;295(1):245–257. doi: 10.1016/j.yexcr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Saez ET, Pehar M, Vargas M, Barbeito L, Maccioni RB. Astrocytic nitric oxide triggers tau hyperphosphorylation in hippocampal neurons. In Vivo. 2004;18(3):275–280. [PubMed] [Google Scholar]
- 8.Koudinov AR, Berezov TT. Cholesterol, statins, and Alzheimer disease. PLoS Medicine. 2005;2(3):e81. doi: 10.1371/journal.pmed.0020081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li L, Cao D, Garber DW, Kim H, Fukuchi K-I. Association of aortic atherosclerosis with cerebral β-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. American Journal of Pathology. 2003;163(6):2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahrle S, Das P, Nyborg AC, et al. Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiology of Disease. 2002;9(1):11–23. doi: 10.1006/nbdi.2001.0470. [DOI] [PubMed] [Google Scholar]
- 11.Maccioni RB, Otth C, Concha II, Muñoz JP. The protein kinase cdk5: structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. European Journal of Biochemistry. 2001;268(6):1518–1527. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- 12.Otth C, Concha II, Arendt T, et al. AβPP induces cdk5-dependent tau hyperphosphorylation in transgenic mice Tg2576. Journal of Alzheimer's Disease. 2002;4(5):417–430. doi: 10.3233/jad-2002-4508. [DOI] [PubMed] [Google Scholar]
- 13.Binder LI, Guillozet-Bongaarts AL, Garcia-Sierra F, Berry RW. Tau, tangles, and Alzheimer's disease. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2005;1739(2):216–223. doi: 10.1016/j.bbadis.2004.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Lavados M, Farías G, Rothhammer F, et al. ApoE alleles and tau markers in patients with different levels of cognitive impairment. Archives of Medical Research. 2005;36(5):474–479. doi: 10.1016/j.arcmed.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 15.Maccioni RB, Lavados M, Maccioni CB, Mendoza-Naranjo A. Biological markers of Alzheimer's disease and mild cognitive impairment. Current Alzheimer Research. 2004;1(4):307–314. doi: 10.2174/1567205043332018. [DOI] [PubMed] [Google Scholar]
- 16.Lee G, Thangavel R, Sharma VM, et al. Phosphorylation of tau by fyn: implications for Alzheimer's disease. Journal of Neuroscience. 2004;24(9):2304–2312. doi: 10.1523/JNEUROSCI.4162-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akiyama H, McGeer PL. Specificity of mechanisms for plaque removal after Aβ immunotherapy for Alzheimer disease. Nature Medicine. 2004;10(2):117–118. doi: 10.1038/nm0204-117. [DOI] [PubMed] [Google Scholar]
- 18.Orellana DI, Quintanilla RA, Gonzalez-Billault C, Maccioni RB. Role of the JAKs/STATs pathway in the intracellular calcium changes induced by interleukin-6 in hippocampal neurons. Neurotoxicity Research. 2005;8(3-4):295–304. doi: 10.1007/BF03033983. [DOI] [PubMed] [Google Scholar]
- 19.Armstrong J, Boada M, Rey MJ, Vidal N, Ferrer I. Familial Alzheimer disease associated with A713T mutation in APP. Neuroscience Letters. 2004;370(2-3):241–243. doi: 10.1016/j.neulet.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 20.Ishikawa A, Piao Y-S, Miyashita A, et al. A mutant PSEN1 causes dementia with Lewy bodies and variant Alzheimer's disease. Annals of Neurology. 2005;57(3):429–434. doi: 10.1002/ana.20393. [DOI] [PubMed] [Google Scholar]
- 21.Ezquerra M, Lleó A, Castellví M, et al. A novel mutation in the PSEN2 gene (T430M) associated with variable expression in a family with early-onset Alzheimer disease. Archives of Neurology. 2003;60(8):1149–1151. doi: 10.1001/archneur.60.8.1149. [DOI] [PubMed] [Google Scholar]
- 22.Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR. Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology. 2004;62(10):1869–1871. doi: 10.1212/01.wnl.0000125323.15458.3f. [DOI] [PubMed] [Google Scholar]
- 23.Zubenko GS, Hughes HB III, Stiffler JS. D10S1423 identifies a susceptibility locus for Alzheimer's disease in a prospective, longitudinal, double-blind study of asymptomatic individuals. Molecular Psychiatry. 2001;6(4):413–419. doi: 10.1038/sj.mp.4000900. [DOI] [PubMed] [Google Scholar]
- 24.Olson JM, Goddard KAB, Dudek DM. A second locus for very-late-onset Alzheimer disease: a genome scan reveals linkage to 20p and epistasis between 20p and the Amyloid Precursor Protein region. American Journal of Human Genetics. 2002;71(1):154–161. doi: 10.1086/341034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Davis RE, Miller S, Herrnstadt C, et al. Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(9):4526–4531. doi: 10.1073/pnas.94.9.4526. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Hutchin T, Cortopassi G. A mitochondrial DNA clone is associated with increased risk for Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(15):6892–6895. doi: 10.1073/pnas.92.15.6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hope C, Mettenburg J, Gonias SL, DeKosky ST, Kamboh MI, Chu CT. Functional analysis of plasma α2-macroglobulin from Alzheimer's disease patients with the A2M intronic deletion. Neurobiology of Disease. 2003;14(3):504–512. doi: 10.1016/j.nbd.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Zappia M, Manna I, Serra P, et al. Increased risk for Alzheimer disease with the interaction of MPO and A2M polymorphisms. Archives of Neurology. 2004;61(3):341–344. doi: 10.1001/archneur.61.3.341. [DOI] [PubMed] [Google Scholar]
- 29.Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow E-M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. Journal of Cell Biology. 2002;156(6):1051–1063. doi: 10.1083/jcb.200108057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Augustinack JC, Schneider A, Mandelkow E-M, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer's disease. Acta Neuropathologica. 2002;103(1):26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 31.Mandelkow E-M, Thies E, Trinczek B, Biernat J, Mandelkow E. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. Journal of Cell Biology. 2004;167(1):99–110. doi: 10.1083/jcb.200401085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 33.Gervais FG, Xu D, Robertson GS, et al. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-β precursor protein and amyloidogenic Aβ peptide formation. Cell. 1999;97(3):395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 34.Coskun PE, Beal MF, Wallace DC. Alzheimer's brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simon DK, Lin MT, Zheng L, et al. Somatic mitochondrial DNA mutations in cortex and substantia nigra in aging and Parkinson's disease. Neurobiology of Aging. 2004;25(1):71–81. doi: 10.1016/s0197-4580(03)00037-x. [DOI] [PubMed] [Google Scholar]
- 36.Beal MF. Mitochondria, oxidative damage, and inflammation in Parkinson's disease. Annals of the New York Academy of Sciences. 2003;991:120–131. doi: 10.1111/j.1749-6632.2003.tb07470.x. [DOI] [PubMed] [Google Scholar]
- 37.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Annals of Neurology. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 38.Honer WG. Pathology of presynaptic proteins in Alzheimer's disease: more than simple loss of terminals. Neurobiology of Aging. 2003;24(8):1047–1062. doi: 10.1016/j.neurobiolaging.2003.04.005. [DOI] [PubMed] [Google Scholar]
- 39.Yao PJ, Zhu M, Pyun EI, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiology of Disease. 2003;12(2):97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 40.Kelly BL, Vassar R, Ferreira A. β-amyloid-induced dynamin 1 depletion in hippocampal neurons: a potential mechanism for early cognitive decline in Alzheimer disease. Journal of Biological Chemistry. 2005;280(36):31746–31753. doi: 10.1074/jbc.M503259200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Archives of Neurology. 2002;59(11):1737–1746. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 42.Von Strauss E, Viitanen M, De Ronchi D, Winblad B, Fratiglioni L. Aging and the occurrence of dementia: findings from a population-based cohort with a large sample of nonagenarians. Archives of Neurology. 1999;56(5):587–592. doi: 10.1001/archneur.56.5.587. [DOI] [PubMed] [Google Scholar]
- 43.Gao S, Hendrie HC, Hall KS, Hui S. The relationships between age, sex, and the incidence of dementia and Alzheimer disease: a meta-analysis. Archives of General Psychiatry. 1998;55(9):809–815. doi: 10.1001/archpsyc.55.9.809. [DOI] [PubMed] [Google Scholar]
- 44.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 45.Cummings JL, Cole G. Alzheimer disease. Journal of the American Medical Association. 2002;287(18):2335–2338. doi: 10.1001/jama.287.18.2335. [DOI] [PubMed] [Google Scholar]
- 46.Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. Journal of the American Medical Association. 1997;278(16):1349–1356. [PubMed] [Google Scholar]
- 47.Skoog I. Hypertension and cognition. International Psychogeriatrics. 2003;15(suppl 1):139–146. doi: 10.1017/S1041610203009104. [DOI] [PubMed] [Google Scholar]
- 48.Skoog I, Lernfelt B, Landahl S, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347(9009):1141–1145. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 49.Ott A, Breteler MMB, De Bruyne MC, van Harskamp F, Grobbee DE, Hofman A. Atrial fibrillation and dementia in a population-based study: the Rotterdam study. Stroke. 1997;28(2):316–321. doi: 10.1161/01.str.28.2.316. [DOI] [PubMed] [Google Scholar]
- 50.Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Archives of Neurology. 2005;62(10):1556–1560. doi: 10.1001/archneur.62.10.1556. [DOI] [PubMed] [Google Scholar]
- 51.Merchant C, Tang M-X, Albert S, Manly J, Stern Y, Mayeux R. The influence of smoking on the risk of Alzheimer's disease. Neurology. 1999;52(7):1408–1412. doi: 10.1212/wnl.52.7.1408. [DOI] [PubMed] [Google Scholar]
- 52.Seshadri S, Beiser A, Selhub J, et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. New England Journal of Medicine. 2002;346(7):476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 53.Grossman H. Does diabetes protect or provoke Alzheimer's disease? Insights into the pathobiology and future treatment of Alzheimer's disease. CNS Spectrums. 2003;8(11):815–823. doi: 10.1017/s1092852900019258. [DOI] [PubMed] [Google Scholar]
- 54.Nicolls MR. The clinical and biological relationship between Type II diabetes mellitus and Alzheimer's disease. Current Alzheimer Research. 2004;1(1):47–54. doi: 10.2174/1567205043480555. [DOI] [PubMed] [Google Scholar]
- 55.Luchsinger JA, Reitz C, Honig LS, Tang M-X, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65(4):545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet. 2000;356(9242):1627–1631. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- 57.Pappolla MA, Bryant-Thomas TK, Herbert D, et al. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003;61(2):199–205. doi: 10.1212/01.wnl.0000070182.02537.84. [DOI] [PubMed] [Google Scholar]
- 58.Kuo Y-M, Emmerling MR, Bisgaier CL, et al. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain Aβ 1–42 levels. Biochemical and Biophysical Research Communications. 1998;252(3):711–715. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- 59.Papassotiropoulos A, Wollmer MA, Tsolaki M, et al. A cluster of cholesterol-related genes confers susceptibility for Alzheimer's disease. Journal of Clinical Psychiatry. 2005;66(7):940–947. [PubMed] [Google Scholar]
- 60.Sjögren M, Blennow K. The link between cholesterol and Alzheimer's disease. World Journal of Biological Psychiatry. 2005;6(2):85–97. doi: 10.1080/15622970510029795. [DOI] [PubMed] [Google Scholar]
- 61.Buxbaum JD, Cullen EI, Friedhoff LT. Pharmacological concentrations of the HMG-CoA reductase inhibitor lovastatin decrease the formation of the Alzheimer β-amyloid peptide in vitro and in patients. Frontiers in Bioscience. 2002;7:a50–a59. doi: 10.2741/A739. [DOI] [PubMed] [Google Scholar]
- 62.Kalvodova L, Kahya N, Schwille P, et al. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. Journal of Biological Chemistry. 2005;280(44):36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 63.Pahan K, Sheikh FG, Namboodiri MS, Singh I. Lovastatin and phenylacetate inhibit the induction of nitric oxide synthase and cytokines in rat primary astrocytes, microglia, and macrophages. Journal of Clinical Investigation. 1997;100(11):2671–2679. doi: 10.1172/JCI119812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grip O, Janciauskiene S, Lindgren S. Atorvastatin activates PPAR-γ and attenuates the inflammatory response in human monocytes. Inflammation Research. 2002;51(2):58–62. doi: 10.1007/BF02684000. [DOI] [PubMed] [Google Scholar]
- 65.Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate β-amyloid-induced microglial inflammatory responses. Journal of Neuroscience. 2005;25(2):299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang FL, Casey PJ. Protein prenylation: molecular mechanisms and functional consequences. Annual Review of Biochemistry. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 67.Liao JK. Isoprenoids as mediators of the biological effects of statins. Journal of Clinical Investigation. 2002;110(3):285–288. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. Journal of Biological Chemistry. 2005;280(40):34202–34209. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- 69.Désiré L, Bourdin J, Loiseau N, et al. RAC1 inhibition targets amyloid precursor protein processing by γ-secretase and decreases Aβ production in vitro and in vivo. Journal of Biological Chemistry. 2005;280(45):37516–37525. doi: 10.1074/jbc.M507913200. [DOI] [PubMed] [Google Scholar]
- 70.Thelen KM, Rentsch KM, Gutteck U, et al. Brain cholesterol synthesis in mice is affected by high dose of simvastatin but not of pravastatin. Journal of Pharmacology and Experimental Therapeutics. 2006;316(3):1146–1152. doi: 10.1124/jpet.105.094136. [DOI] [PubMed] [Google Scholar]
- 71.Banerjee P, Joo JB, Buse JT, Dawson G. Differential solubilization of lipids along with membrane proteins by different classes of detergents. Chemistry and Physics of Lipids. 1995;77(1):65–78. doi: 10.1016/0009-3084(95)02455-r. [DOI] [PubMed] [Google Scholar]
- 72.Harder T, Simons K. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Current Opinion in Cell Biology. 1997;9(4):534–542. doi: 10.1016/s0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- 73.Rajendran L, Simons K. Lipid rafts and membrane dynamics. Journal of Cell Science. 2005;118(6):1099–1102. doi: 10.1242/jcs.01681. [DOI] [PubMed] [Google Scholar]
- 74.Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiological Reviews. 1995;75(4):835–864. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- 75.Andreadis A. Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2005;1739(2):91–103. doi: 10.1016/j.bbadis.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 76.Buée-Scherrer V, Goedert M. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases in intact cells. FEBS Letters. 2002;515(1–3):151–154. doi: 10.1016/s0014-5793(02)02460-2. [DOI] [PubMed] [Google Scholar]
- 77.Mandelkow E-M, Biernat J, Drewes G, Gustke N, Trinczek B, Mandelkow E. Tau domains, phosphorylation, and interactions with microtubules. Neurobiology of Aging. 1995;16(3):355–362. doi: 10.1016/0197-4580(95)00025-a. [DOI] [PubMed] [Google Scholar]
- 78.Biernat J, Gustke N, Drewes G, Mandelkow E-M, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11(1):153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 79.Godemann R, Biernat J, Mandelkow E, Mandelkow E-M. Phosphorylation of tau protein by recombinant GSK-3β: pronounced phosphorylation at select Ser/Thr-Pro motifs but no phosphorylation at Ser262 in the repeat domain. FEBS Letters. 1999;454(1-2):157–164. doi: 10.1016/s0014-5793(99)00741-3. [DOI] [PubMed] [Google Scholar]
- 80.Alvarez A, Muñoz JP, Maccioni RB. A cdk5-p35 stable complex is involved in the β-amyloid-induced deregulation of cdk5 activity in hippocampal neurons. Experimental Cell Research. 2001;264(2):266–274. doi: 10.1006/excr.2001.5152. [DOI] [PubMed] [Google Scholar]
- 81.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochemical Journal. 1997;323(3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maas T, Eidenmüller J, Brandt R. Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. Journal of Biological Chemistry. 2000;275(21):15733–15740. doi: 10.1074/jbc.M000389200. [DOI] [PubMed] [Google Scholar]
- 83.Lee G. Tau and src family tyrosine kinases. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2005;1739(2):323–330. doi: 10.1016/j.bbadis.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 84.Saito Y, Murayama S. Expression of tau immunoreactivity in the spinal motor neurons of Alzheimer's disease. Neurology. 2000;55(11):1727–1729. doi: 10.1212/wnl.55.11.1727. [DOI] [PubMed] [Google Scholar]
- 85.Bhaskar K, Yen S-H, Lee G. Disease-related modifications in tau affect the interaction between Fyn and tau. Journal of Biological Chemistry. 2005;280(42):35119–35125. doi: 10.1074/jbc.M505895200. [DOI] [PubMed] [Google Scholar]
- 86.Derkinderen P, Scales TME, Hanger DP, et al. Tyrosine 394 is phosphorylated in Alzheimer's paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. Journal of Neuroscience. 2005;25(28):6584–6593. doi: 10.1523/JNEUROSCI.1487-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brandt R, Lee G. Orientation, assembly, and stability of microtubule bundles induced by a fragment of tau protein. Cell Motility and the Cytoskeleton. 1994;28(2):143–154. doi: 10.1002/cm.970280206. [DOI] [PubMed] [Google Scholar]
- 88.Moraga DM, Nunez P, Garrido J, Maccioni RB. A tau fragment containing a repetitive sequence induces bundling of actin filaments. Journal of Neurochemistry. 1993;61(3):979–986. doi: 10.1111/j.1471-4159.1993.tb03611.x. [DOI] [PubMed] [Google Scholar]
- 89.Shea TB. Phospholipids alter tau conformation, phosphorylation, proteolysis, and association with microtubules: implication for tau function under normal and degenerative conditions. Journal of Neuroscience Research. 1997;50(1):114–122. doi: 10.1002/(SICI)1097-4547(19971001)50:1<114::AID-JNR12>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 90.Tatulian SA, Qin S, Pande AH, He X. Positioning membrane proteins by novel protein engineering and biophysical approaches. Journal of Molecular Biology. 2005;351(5):939–947. doi: 10.1016/j.jmb.2005.06.080. [DOI] [PubMed] [Google Scholar]
- 91.Klein C, Krämer E-M, Cardine A-M, Schraven B, Brandt R, Trotter J. Process outgrowth of oligodendrocytes is promoted by interaction of Fyn kinase with the cytoskeletal protein Tau. Journal of Neuroscience. 2002;22(3):698–707. doi: 10.1523/JNEUROSCI.22-03-00698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stellaard F, Kuipers F. Assessment of modes of action and efficacy of plasma cholesterol-lowering drugs: measurement of cholesterol absorption, cholesterol synthesis and bile acid synthesis and turnover using novel stable isotope techniques. Current Drug Targets: Immune, Endocrine and Metabolic Disorders. 2005;5(2):209–218. doi: 10.2174/1568008054064913. [DOI] [PubMed] [Google Scholar]
- 93.Dietschy JM, Turley SD. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. Journal of Lipid Research. 2004;45(8):1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 94.Ishibashi S, Herz J, Maeda N, Goldstein JL, Brown MS. The two-receptor model of lipoprotein clearance: tests of the hypothesis in “knockout” mice lacking the low density lipoprotein receptor, apolipoprotein E, or both proteins. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(10):4431–4435. doi: 10.1073/pnas.91.10.4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nixon RA. Niemann-Pick type C disease and Alzheimer's disease: the APP-endosome connection fattens up. American Journal of Pathology. 2004;164(3):757–761. doi: 10.1016/S0002-9440(10)63163-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Papassotiropoulos A, Lütjohann D, Bagli M, et al. Plasma 24S-hydroxycholesterol: a peripheral indicator of neuronal degeneration and potential state marker for Alzheimer's disease. NeuroReport. 2000;11(9):1959–1961. doi: 10.1097/00001756-200006260-00030. [DOI] [PubMed] [Google Scholar]
- 97.Schönknecht P, Lütjohann D, Pantel J, et al. Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer's disease compared to healthy controls. Neuroscience Letters. 2002;324(1):83–85. doi: 10.1016/s0304-3940(02)00164-7. [DOI] [PubMed] [Google Scholar]
- 98.Park I-H, Hwang EM, Hong HS, et al. Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiology of Aging. 2003;24(5):637–643. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 99.Abad-Rodriguez J, Ledesma MD, Craessaerts K, et al. Neuronal membrane cholesterol loss enhances amyloid peptide generation. Journal of Cell Biology. 2004;167(5):953–960. doi: 10.1083/jcb.200404149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kirsch C, Eckert GP, Mueller WE. Statin effects on cholesterol micro-domains in brain plasma membranes. Biochemical Pharmacology. 2003;65(5):843–856. doi: 10.1016/s0006-2952(02)01654-4. [DOI] [PubMed] [Google Scholar]
- 101.Vanier MT, Suzuki K. Recent advances in elucidating Niemann-Pick C disease. Brain Pathology. 1998;8(1):163–174. doi: 10.1111/j.1750-3639.1998.tb00143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Treiber-Held S, Distl R, Meske V, Albert F, Ohm TG. Spatial and temporal distribution of intracellular free cholesterol in brains of a Niemann-Pick type C mouse model showing hyperphosphorylated tau protein. Implications for Alzheimer's disease. Journal of Pathology. 2003;200(1):95–103. doi: 10.1002/path.1345. [DOI] [PubMed] [Google Scholar]
- 103.Gallegos AM, McIntosh AL, Atshaves BP, Schroeder F. Structure and cholesterol domain dynamics of an enriched caveolae/raft isolate. Biochemical Journal. 2004;382(2):451–461. doi: 10.1042/BJ20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arvanitis DN, Min W, Gong Y, Heng YM, Boggs JM. Two types of detergent-insoluble, glycosphingolipid/cholesterol-rich membrane domains from isolated myelin. Journal of Neurochemistry. 2005;94(6):1696–1710. doi: 10.1111/j.1471-4159.2005.03331.x. [DOI] [PubMed] [Google Scholar]
- 105.Pike LJ. Lipid rafts: heterogeneity on the high seas. Biochemical Journal. 2004;378(2):281–292. doi: 10.1042/BJ20031672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pralle A, Keller P, Florin E-L, Simons K, Hörber JKH. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. Journal of Cell Biology. 2000;148(5):997–1008. doi: 10.1083/jcb.148.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387(6633):569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 108.Edidin M. The state of lipid rafts: from model membranes to cells. Annual Review of Biophysics and Biomolecular Structure. 2003;32:257–283. doi: 10.1146/annurev.biophys.32.110601.142439. [DOI] [PubMed] [Google Scholar]
- 109.Ipsen JH, Karlström G, Mouritsen OG, Wennerström H, Zuckermann MJ. Phase equilibria in the phosphatidylcholine-cholesterol system. Biochimica et Biophysica Acta - Molecular Basis of Disease. 1987;905(1):162–172. doi: 10.1016/0005-2736(87)90020-4. [DOI] [PubMed] [Google Scholar]
- 110.Brown DA. Seeing is believing: visualization of rafts in model membranes. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10517–10518. doi: 10.1073/pnas.191386898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schwille P, Kettling U. Analyzing single protein molecules using optical methods. Current Opinion in Biotechnology. 2001;12(4):382–386. doi: 10.1016/s0958-1669(00)00231-7. [DOI] [PubMed] [Google Scholar]
- 112.Kahya N, Scherfeld D, Bacia K, Poolman B, Schwille P. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. Journal of Biological Chemistry. 2003;278(30):28109–28115. doi: 10.1074/jbc.M302969200. [DOI] [PubMed] [Google Scholar]
- 113.Kahya N, Pécheur E-I, de Boeij WP, Wiersma DA, Hoekstra D. Reconstitution of membrane proteins into giant unilamellar vesicles via peptide-induced fusion. Biophysical Journal. 2001;81(3):1464–1474. doi: 10.1016/S0006-3495(01)75801-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kahya N, Scherfeld D, Bacia K, Schwille P. Lipid domain formation and dynamics in giant unilamellar vesicles explored by fluorescence correlation spectroscopy. Journal of Structural Biology. 2004;147(1):77–89. doi: 10.1016/j.jsb.2003.09.021. [DOI] [PubMed] [Google Scholar]
- 115.Megha, London E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts) Journal of Biological Chemistry. 2004;279(11):9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 116.Anderson RGW, Jacobson K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science. 2002;296(5574):1821–1825. doi: 10.1126/science.1068886. [DOI] [PubMed] [Google Scholar]
- 117.Pankov R, Markovska T, Hazarosova R, Antonov P, Ivanova L, Momchilova A. Cholesterol distribution in plasma membranes of β1 integrin-expressing and β1 integrin-deficient fibroblasts. Archives of Biochemistry and Biophysics. 2005;442(2):160–168. doi: 10.1016/j.abb.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 118.Blonder J, Terunuma A, Conrads TP, et al. A proteomic characterization of the plasma membrane of human epidermis by high-throughput mass spectrometry. Journal of Investigative Dermatology. 2004;123(4):691–699. doi: 10.1111/j.0022-202X.2004.23421.x. [DOI] [PubMed] [Google Scholar]
- 119.Brown DA, London E. Structure and function of sphingolipid- and cholesterol-rich membrane rafts. Journal of Biological Chemistry. 2000;275(23):17221–17224. doi: 10.1074/jbc.R000005200. [DOI] [PubMed] [Google Scholar]
- 120.Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. Journal of Cell Biology. 2003;160(1):113–123. doi: 10.1083/jcb.200207113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wada S, Morishima-Kawashima M, Qi Y, et al. γ-secretase activity is present in rafts but is not cholesterol-dependent. Biochemistry. 2003;42(47):13977–13986. doi: 10.1021/bi034904j. [DOI] [PubMed] [Google Scholar]
- 122.Wischik CM, Crowther RA. Subunit structure of the Alzheimer tangle. British Medical Bulletin. 1986;42(1):51–56. doi: 10.1093/oxfordjournals.bmb.a072098. [DOI] [PubMed] [Google Scholar]
- 123.Brigger D, Muckle TJ. Comparison of Sirius red and Congo red as stains for amyloid in animal tissues. Journal of Histochemistry and Cytochemistry. 1975;23(1):84–88. doi: 10.1177/23.1.46874. [DOI] [PubMed] [Google Scholar]
- 124.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochemical and Biophysical Research Communications. 1984;122(3):1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 125.Kang J, Lemaire H-G, Unterbeck A, et al. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 126.Tun H, Marlow L, Pinnix I, Kinsey R, Sambamurti K. Lipid rafts play an important role in Aβ biogenesis by regulating the β-secretase pathway. Journal of Molecular Neuroscience. 2002;19(1-2):31–35. doi: 10.1007/s12031-002-0007-5. [DOI] [PubMed] [Google Scholar]
- 127.Montine TJ, Amarnath V, Martin ME, Strittmatter WJ, Graham DG. E-4-hydroxy-2-nonenal is cytotoxic and cross-links cytoskeletal proteins in P19 neuroglial cultures. American Journal of Pathology. 1996;148(1):89–93. [PMC free article] [PubMed] [Google Scholar]
- 128.Sayre LM, Zelasko DA, Harris PLR, Perry G, Salomon RG, Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. Journal of Neurochemistry. 1997;68(5):2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 129.Uchida K, Kanematsu M, Morimitsu Y, Osawa T, Noguchi N, Niki E. Acrolein is a product of lipid peroxidation reaction: formation of free acrolein and its conjugate with lysine residues in oxidized low density lipoproteins. Journal of Biological Chemistry. 1998;273(26):16058–16066. doi: 10.1074/jbc.273.26.16058. [DOI] [PubMed] [Google Scholar]
- 130.Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiology of Aging. 2001;22(2):187–194. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- 131.McGrath LT, McGleenon BM, Brennan S, McColl D, McIlroy S, Passmore AP. Increased oxidative stress in Alzheimer's disease as assessed with 4-hydroxynonenal but not malondialdehyde. QJM. 2001;94(9):485–490. doi: 10.1093/qjmed/94.9.485. [DOI] [PubMed] [Google Scholar]
- 132.Selley ML, Close DR, Stern SE. The effect of increased concentrations of homocysteine on the concentration of (E)-4-hydroxy-2-nonenal in the plasma and cerebrospinal fluid of patients with Alzheimer's disease. Neurobiology of Aging. 2002;23(3):383–388. doi: 10.1016/s0197-4580(01)00327-x. [DOI] [PubMed] [Google Scholar]
- 133.Bourdel-Marchasson I, Delmas-Beauvieux M-C, Peuchant E, et al. Antioxidant defences and oxidative stress markers in erythrocytes and plasma from normally nourished elderly Alzheimer patients. Age and Ageing. 2001;30(3):235–241. doi: 10.1093/ageing/30.3.235. [DOI] [PubMed] [Google Scholar]
- 134.Sinclair AJ, Bayer AJ, Johnston JO, Warner C, Maxwell SRJ. Altered plasma antioxidant status in subjects with Alzheimer's disease and vascular dementia. International Journal of Geriatric Psychiatry. 1998;13(12):840–845. doi: 10.1002/(sici)1099-1166(1998120)13:12<840::aid-gps877>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 135.Lütjohann D, Papassotiropoulos A, Björkhem I, et al. Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. Journal of Lipid Research. 2000;41(2):195–198. [PubMed] [Google Scholar]
- 136.Vega GL, Weiner M, Kölsch H, et al. The effects of gender and CYP46 and apo E polymorphism on 24S-hydroxycholesterol levels in Alzheimer's patients treated with statins. Current Alzheimer Research. 2004;1(1):71–77. doi: 10.2174/1567205043480546. [DOI] [PubMed] [Google Scholar]
- 137.Vega GL, Weiner MF, Lipton AM, et al. Reduction in levels of 24S-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Archives of Neurology. 2003;60(4):510–515. doi: 10.1001/archneur.60.4.510. [DOI] [PubMed] [Google Scholar]
- 138.Locatelli S, Lütjohann D, Schmidt HH-J, Otto C, Beisiegel U, von Bergmann K. Reduction of plasma 24S-hydroxycholesterol (cerebrosterol) levels using high-dosage simvastatin in patients with hypercholesterolemia: evidence that simvastatin affects cholesterol metabolism in the human brain. Archives of Neurology. 2002;59(2):213–216. doi: 10.1001/archneur.59.2.213. [DOI] [PubMed] [Google Scholar]
- 139.Rafael H. Cerebral atherosclerosis and oxidative stress in Alzheimer's disease. Journal of Alzheimer's Disease. 2003;5(6):479–480. doi: 10.3233/jad-2003-5609. [DOI] [PubMed] [Google Scholar]
- 140.Aliyev A, Chen SG, Seyidova D, et al. Mitochondria DNA deletions in atherosclerotic hypoperfused brain microvessels as a primary target for the development of Alzheimer's disease. Journal of the Neurological Sciences. 2005;229-230:285–292. doi: 10.1016/j.jns.2004.11.040. [DOI] [PubMed] [Google Scholar]
- 141.Nanetti L, Vignini A, Moroni C, et al. Peroxynitrite production and NOS expression in astrocytes U373MG incubated with lipoproteins from Alzheimer patients. Brain Research. 2005;1054(1):38–44. doi: 10.1016/j.brainres.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 142.Puglielli L, Friedlich AL, Setchell KDR, et al. Alzheimer disease β-amyloid activity mimics cholesterol oxidase. Journal of Clinical Investigation. 2005;115(9):2556–2563. doi: 10.1172/JCI23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and β-amyloid peptide. Journal of Biological Chemistry. 2005;280(8):7377–7387. doi: 10.1074/jbc.M409071200. [DOI] [PubMed] [Google Scholar]
- 144.Han X, Fagan AM, Cheng H, Morris JC, Xiong C, Holtzman DM. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Annals of Neurology. 2003;54(1):115–119. doi: 10.1002/ana.10618. [DOI] [PubMed] [Google Scholar]
- 145.Cheng H, Xu J, McKeel DW Jr, Han X. Specificity and potential mechanism of sulfatide deficiency in Alzheimer's disease: an electrospray ionization mass spectrometric study. Cellular and Molecular Biology. 2003;49(5):809–818. [PubMed] [Google Scholar]
- 146.Jarvik GP, Wijsman EM, Kukull WA, Schellenberg GD, Yu C, Larson EB. Interactions of apolipoprotein E genotype, total cholesterol level, age, and sex in prediction of Alzheimer's disease: a case-control study. Neurology. 1995;45(6):1092–1096. doi: 10.1212/wnl.45.6.1092. [DOI] [PubMed] [Google Scholar]
- 147.Kalvodova L, Kahya N, Schwille P, et al. Lipids as modulators of proteolytic activity of BACE: involvement of cholesterol, glycosphingolipids, and anionic phospholipids in vitro. Journal of Biological Chemistry. 2005;280(44):36815–36823. doi: 10.1074/jbc.M504484200. [DOI] [PubMed] [Google Scholar]
- 148.George AJ, Holsinger RMD, McLean CA, et al. APP intracellular domain is increased and soluble Aβ is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiology of Disease. 2004;16(1):124–132. doi: 10.1016/j.nbd.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 149.Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Park I-H, Hwang EM, Hong HS, et al. Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiology of Aging. 2003;24(5):637–643. doi: 10.1016/s0197-4580(02)00155-0. [DOI] [PubMed] [Google Scholar]
- 151.Li L, Cao D, Garber DW, Kim H, Fukuchi K-I. Association of aortic atherosclerosis with cerebral β-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. American Journal of Pathology. 2003;163(6):2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cordle A, Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate β-amyloid-induced microglial inflammatory responses. Journal of Neuroscience. 2005;25(2):299–307. doi: 10.1523/JNEUROSCI.2544-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Cordle A, Koenigsknecht-Talboo J, Wilkinson B, Limpert A, Landreth G. Mechanisms of statin-mediated inhibition of small G-protein function. Journal of Biological Chemistry. 2005;280(40):34202–34209. doi: 10.1074/jbc.M505268200. [DOI] [PubMed] [Google Scholar]
- 154.Ellis JM. Cholinesterase inhibitors in the treatment of dementia. Journal of the American Osteopathic Association. 2005;105(3):145–158. [PubMed] [Google Scholar]
- 155.Gauthier SG. Alzheimer's disease: the benefits of early treatment. European Journal of Neurology. 2005;12(suppl 3):11–16. doi: 10.1111/j.1468-1331.2005.01322.x. [DOI] [PubMed] [Google Scholar]
- 156.Wesnes KA, McKeith IG, Ferrara R, et al. Effects of rivastigmine on cognitive function in dementia with lewy bodies: a randomised placebo-controlled international study using the cognitive drug research computerised assessment system. Dementia and Geriatric Cognitive Disorders. 2002;13(3):183–192. doi: 10.1159/000048651. [DOI] [PubMed] [Google Scholar]
- 157.Harry RD, Zakzanis KK. A comparison of donepezil and galantamine in the treatment of cognitive symptoms of Alzheimer's disease: a meta-analysis. Human Psychopharmacology. 2005;20(3):183–187. doi: 10.1002/hup.676. [DOI] [PubMed] [Google Scholar]
- 158.Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt H-P, van den Bussche H. Cholinesterase inhibitors for patients with Alzheimer's disease: systematic review of randomised clinical trials. British Medical Journal. 2005;331(7512):321–323. doi: 10.1136/bmj.331.7512.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Butterfield DA, Pocernich CB. The glutamatergic system and Alzheimer's disease: therapeutic implications. CNS Drugs. 2003;17(9):641–652. doi: 10.2165/00023210-200317090-00004. [DOI] [PubMed] [Google Scholar]
- 160.Potyk D. Treatments for Alzheimer disease. Southern Medical Journal. 2005;98(6):628–635. doi: 10.1097/01.SMJ.0000166671.86815.C1. [DOI] [PubMed] [Google Scholar]
- 161.Citron M. Alzheimer's disease: treatments in discovery and development. Nature Neuroscience. 2002;5(suppl):1055–1057. doi: 10.1038/nn940. [DOI] [PubMed] [Google Scholar]
- 162.Citron M. β-Secretase inhibition for the treatment of Alzheimer's disease—promise and challenge. Trends in Pharmacological Sciences. 2004;25(2):92–97. doi: 10.1016/j.tips.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 163.Lahiri DK. Advances in the four major pillars of Alzheimer's disease research: pathobiology, genetics, diagnosis, and treatment. Current Alzheimer Research. 2004;1(3):i–ii. doi: 10.2174/1567205043480537. [DOI] [PubMed] [Google Scholar]
- 164.Samuels SC, Grossman H. Emerging therapeutics for Alzheimer's disease: an avenue of hope. CNS Spectrums. 2003;8(11):834–845. doi: 10.1017/s1092852900019271. [DOI] [PubMed] [Google Scholar]
- 165.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Archives of Neurology. 2000;57(10):1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 166.Shepherd J, Blauw GJ, Murphy MB, et al. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet. 2002;360(9346):1623–1630. doi: 10.1016/s0140-6736(02)11600-x. [DOI] [PubMed] [Google Scholar]
- 167.Sjögren M, Gustafsson K, Syversen S, et al. Treatment with simvastatin in patients with Alzheimer's disease lowers both α- and β-cleaved amyloid precursor protein. Dementia and Geriatric Cognitive Disorders. 2003;16(1):25–30. doi: 10.1159/000069989. [DOI] [PubMed] [Google Scholar]
- 168.Sparks DL, Sabbagh MN, Connor DJ, et al. Atorvastatin for the treatment of mild to moderate Alzheimer disease: preliminary results. Archives of Neurology. 2005;62(5):753–757. doi: 10.1001/archneur.62.5.753. [DOI] [PubMed] [Google Scholar]
- 169.Höglund K, Thelen KM, Syversen S, et al. The effect of simvastatin treatment on the amyloid precursor protein and brain cholesterol metabolism in patients with Alzheimer's disease. Dementia and Geriatric Cognitive Disorders. 2005;19(5-6):256–265. doi: 10.1159/000084550. [DOI] [PubMed] [Google Scholar]