

Abstract
Insulin stimulates glucose uptake in muscle and adipose cells by mobilizing intracellular membrane vesicles containing GLUT4 glucose transporter proteins to the plasma membrane. Here we show in live cultured adipocytes that intracellular membranes containing GLUT4–yellow fluorescent protein (YFP) move along tubulin–cyan fluorescent protein-labeled microtubules in response to insulin by a mechanism that is insensitive to the phosphatidylinositol 3 (PI3)-kinase inhibitor wortmannin. Insulin increased by several fold the observed frequencies, but not velocities, of long-range movements of GLUT4–YFP on microtubules, both away from and towards the perinuclear region. Genomics screens show conventional kinesin KIF5B is highly expressed in adipocytes and this kinesin is partially co-localized with perinuclear GLUT4. Dominant-negative mutants of conventional kinesin light chain blocked outward GLUT4 vesicle movements and translocation of exofacial Myc-tagged GLUT4–green fluorescent protein to the plasma membrane in response to insulin. These data reveal that insulin signaling targets the engagement or initiates the movement of GLUT4-containing membranes on microtubules via conventional kinesin through a PI3-kinase-independent mechanism. This insulin signaling pathway regulating KIF5B function appears to be required for GLUT4 translocation to the plasma membrane.
Keywords: adipocytes/GLUT4/insulin/kinesin/microtubules
Introduction
Glucose homeostasis in humans is dependent upon the catalytic activity of a glucose transporter protein, GLUT4, that is highly expressed in fat and muscle and responds to insulin by translocation from intracellular vesicles to the plasma membrane (Olson and Pessin, 1996; Czech and Corvera, 1999; Bryant et al., 2002). Ablation of GLUT4 expression in either adipocytes (Abel et al., 2001) or skeletal muscle (Zisman et al., 2000) of mice can lead to glucose intolerance, insulin resistance and diabetes. The mechanism whereby insulin signaling causes intracellular membranes containing GLUT4 to move and fuse with the plasma membrane requires the p85/p110 type phosphatidylinositol 3 (PI3)-kinase (Cheatham et al., 1994; Okada et al., 1994; Sharma et al., 1998) and appears to involve the downstream protein kinase Akt2 (Hill et al., 1999; Cho et al., 2001). Some components involved in membrane trafficking of GLUT4 have also been identified, including the SNARE proteins VAMP2 (Cheatham et al., 1996; Martin et al., 1996) and syntaxin-4 (Volchuk et al., 1996; Olson et al., 1997; Pessin et al., 1999), as well as SNAP25 (Jagadish et al., 1996) and SNAP23 (Kawanishi et al., 2000). In both primary and cultured adipocytes, GLUT4 recycles slowly between intracellular membranes and the plasma membrane even in the absence of insulin, and recycling is markedly enhanced by activation of GLUT4 exocytosis by the hormone (Jhun et al., 1992; Yang and Holman, 1993). A PI3-kinase-dependent step in this process may be fusion of GLUT4-containing membranes with the plasma membrane, based on the identification of the insulin-regulated protein synip as an apparent modulator of syntaxin-4 function (Min et al., 1999).
Evidence has been steadily accumulating in favor of the hypothesis that GLUT4 movements in adipocytes and muscle cells are also dependent on cytoskeletal structures. Many laboratories have confirmed that optimal GLUT4 translocation in response to insulin requires intact actin filaments (Wang et al., 1998; Omata et al., 2000; Bose et al., 2001; Emoto et al., 2001; Patki et al., 2001; Jiang et al., 2002), and co-localization of GLUT4 and polymerized actin is observed in insulin-treated muscle cells (Asahi et al., 1999; Khayat et al., 2000) and adipocytes (unpublished data). We found that depolymerization of intermediate filaments and microtubules also disrupted perinuclear GLUT4 localization, as well impairing GLUT4 responsiveness to insulin (Guilherme et al., 2000; Emoto et al., 2001). Numerous laboratories have confirmed that depolymerization of microtubules partially inhibits GLUT4 recycling and insulin-mediated GLUT4 translocation (Fletcher et al., 2000; Emoto et al., 2001; Molero et al., 2001; Olson et al., 2001). Live cell imaging of 3T3-L1 adipocytes expressing GLUT4–green fluorescent protein (GFP) has revealed short sporadic GLUT4 movements, as well as longer range movements, that are disrupted by colchicine (Fletcher et al., 2000). However, while there is agreement that endocytosis of GLUT4 to perinuclear membranes is driven by dynein motors on microtubule tracks, a recent report has questioned the involvement of microtubules and kinesins in the outward process (Molero et al., 2001). Another report concludes outright that microtubules are not required for GLUT4 translocation (Shigematsu et al., 2002). Thus, the issue of microtubule and kinesin involvement in insulin-mediated GLUT4 movements has remained unresolved.
The aim of this study was to visualize directly GLUT4– yellow fluorescent protein (YFP) movements in live insulin-sensitive 3T3-L1 adipocytes under conditions where microtubules could also be observed. Here we report images showing fluorescence from GLUT4–YFP moving along microtubules labeled with tubulin–cyan fluorescent protein (CFP), providing direct evidence that membranes containing GLUT4 move on microtubule tracks. Remarkably, insulin treatment is shown to increase the frequency of these long-range movements both away from and towards the perinuclear region. The outward movements of GLUT4 towards the plus ends of microtubules appear to be physiologically relevant because these movements as well as insulin-mediated GLUT4 translocation to the plasma membrane was blocked by dominant inhibitory kinesin light chain proteins. These new data are consistent with the hypothesis that GLUT4-containing membranes are cargo for conventional kinesin, which moves these membranes towards the cell periphery in response to insulin.
Results
GLUT4-containing membranes move on microtubules
Preliminary imaging performed on live 3T3-L1 adipocytes expressing GLUT4–GFP or GLUT4–YFP confirmed two earlier reports that showed very active movements of fluorescent vesicles which were mostly randomly directed and short range (Oatey et al., 1997; Patki et al., 2001). Occasional linear movements over distances of 10 µm or more could be observed as well, as noted previously (Oatey et al., 1997). To examine whether these long-range movements are microtubule based, 3T3-L1 adipocytes electroporated with plasmids encoding both GLUT4–YFP and tubulin–CFP were examined by time-lapse microscopy at a single optical plane. Under these conditions, complex networks of labeled microtubules could be visualized throughout the cultured adipocytes (Figure 1). Remarkably, the majority of the long-range GLUT4–YFP-containing vesicle movements occurred along the microtubule tracks within this network (Figures 1 and 2). Figure 1 shows a typical single long-range movement of GLUT4–YFP fluorescence over a distance of ∼10 µm in 40 s. Moreover, some vesicles could be observed to travel over a total distance of 20 µm by transiting onto several different microtubules. These data directly demonstrate that the long-range movements of GLUT4–YFP vesicles are associated with microtubule-based motor activity rather than being random, Brownian or based on other cytoskeletal components.

Fig. 1. GLUT4–YFP vesicles move on microtubules in cultured adipocytes (a time-lapse movie is presented in the Supplementary data available at The EMBO Journal Online). (A) 3T3-L1 adipocytes were electroporated with plasmids encoding GLUT4–YFP and tubulin–CFP. After serum starvation, cells were treated with 100 nM insulin for 15 min, followed by time-lapse image recording with a 5 s interval up to 15 min. Shown here is a merged image of the first and the last frames of a 40 s sequence, showing that a GLUT4–YFP-containing vesicle (red) adjacent to microtubule (green) moves from right to left as indicated by arrows with time points (seconds). N, nucleus. (B) Nine sequential frames of boxed area in (A) are enlarged and merged. Positions of GLUT4–YFP vesicles are indicated by arrows with elapsed time (seconds).

Fig. 2. A single GLUT4–YFP vesicle moves bi-directionally on microtubules. Three different GLUT4–YFP vesicles from time-lapse images that displayed bi-directional movements are shown. The movements were categorized as ‘inward’ and ‘outward’, as described in the text. The numbers inside images indicate elapsed time (seconds) from the first frame of each sequence. Right and left panels show bi-directional movements of the identical vesicles in the same field of cells. The numbers below images indicate maximal velocity of vesicles. Bars, 4 µm.
Using the microtubule organizing center (MTOC) and/or the nuclei as visual indicators, we were able to distinguish the directionality of many of the GLUT4– YFP movements. Figure 2 shows three examples in which the identical GLUT4–YFP-containing vesicles were capable of moving both towards (right panels) and away from (left panels) the perinuclear region of the adipocyte. These directions are referred to as ‘inward’ and ‘outward’, respectively, in Figure 2. Analysis of the velocities of these long-range movements in many cells (Figures 2 and 3A) showed that the speeds of outward and inward movements were not significantly different. These data suggest that an individual GLUT4–YFP-containing vesicle may be bound to and carried by both minus end- and plus end-directed motor proteins.

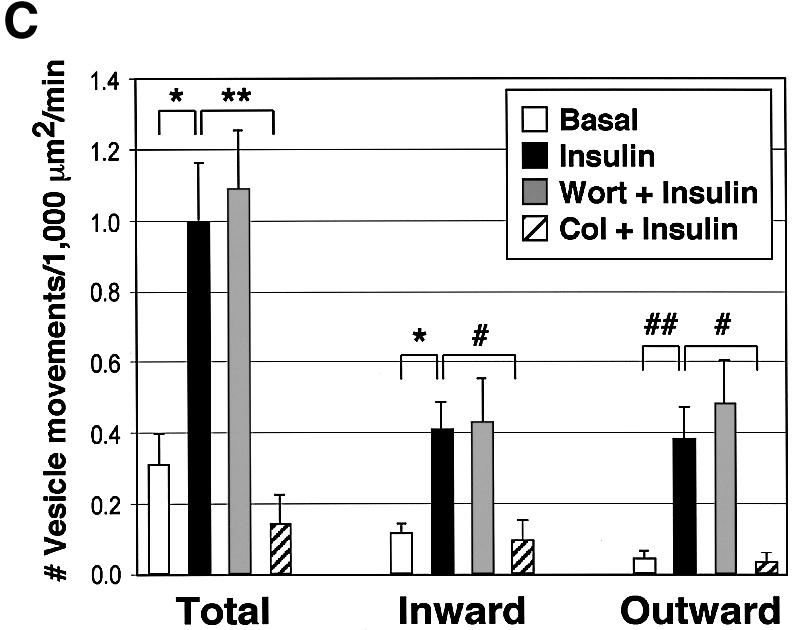
Fig. 3. Insulin increases the number, but not velocities, of long-range GLUT4–YFP vesicle movements on microtubules in a wortmannin-insensitive manner. 3T3-L1 adipocytes expressing GLUT4–YFP and tubulin–CFP were starved and either left untreated (Basal), or treated with either 100 nM insulin (Insulin), 100 nM wortmannin for 15 min followed by a 15 min insulin treatment (Wort + Insulin), or 50 µM colchicine for 2 h followed by a 15 min insulin treatment (Col + Insulin). Long-range movements of GLUT4–YFP vesicles from 6 to 16 time-lapse images for each condition were analyzed for their velocities and number of events. (A) Average maximal velocities of GLUT4– YFP vesicles moving either towards (inward) or away from (outward) the perinuclear region. Total vesicle counts represent the sum of all long-range GLUT4–YFP vesicle movements observed, including those where directionality could not be unequivocally determined. Error bars represent the SEM. (B) Distribution of maximal velocities of GLUT4–YFP vesicles. (C) The number of GLUT4–YFP vesicle movements in a unit area is shown. Error bars represent the SEM. *P = 0.0006; **P = 0.002; #P = 0.01; ##P = 0.004.
Insulin signaling increases the frequency of long-range GLUT4–YFP movements
Experiments were conducted to determine whether insulin action modulates long-range movements of GLUT4–YFP-containing vesicles on microtubules. Analysis of numerous movies of control versus insulin-treated 3T3-L1 adipocytes expressing both GLUT4–YFP and tubulin– CFP revealed no significant effect of the hormone on the mean velocities of GLUT4–YFP movements in either inward or outward directions (Figure 3A). There was also no consistent change in the distribution of velocities exhibited by various groups of GLUT4–YFP-containing vesicles (Figure 3B). However, insulin signaling in these cultured adipocytes caused a marked increase in the observed frequency of total long-range movements of GLUT4–YFP on microtubules (Figure 3C). This increase in the number of such events per unit time was highly significant when either inward (∼3-fold increase) or outward (∼8-fold increase) directions were monitored. Thus insulin-treated adipocytes display much higher numbers of GLUT4–YFP-containing vesicles engaged in extended linear motion along microtubules than untreated cells at any given time.
To further confirm that these long-range movements represent microtubule-based GLUT4–YFP transport, microtubules were depolymerized by treatment with colchicine. As expected, the colchicine treatment resulted in the loss of our ability to visualize microtubules with tubulin–CFP (unpublished data) and the abolition of insulin-stimulated long-range GLUT4–YFP movements (Figure 3C). Interestingly, very active short-range random movements of GLUT4–YFP-containing vesicles could still be observed in the periphery of the colchicine-treated cells, which may represent movements on other cytoskeletal structures, such as F-actin. These results demonstrate the specific requirement of intact microtubules for long-range movements of GLUT4–YFP in cultured adipocytes in response to insulin.
Insulin signaling to increase long-range movements of GLUT4–YFP on microtubules is PI3-kinase independent
It is likely that there are multiple steps in the regulated movements and fusion of GLUT4-containing membranes with the plasma membrane. These steps might include membrane budding or release of membranes from tethering in the perinuclear region, movement along microtubules (Figures 1–3), movement along actin filaments (Omata et al., 2000; Bose et al., 2001; Jiang et al., 2002), and docking and fusion with the plasma membrane (Cheatham et al., 1996; Martin et al., 1996; Volchuk et al., 1996; Min et al., 1999). Insulin regulation of this overall process resulting in increased GLUT4 proteins in the plasma membrane and enhanced glucose transport into adipocytes is totally inhibited by agents that disrupt PI3-kinase (Cheatham et al., 1994; Okada et al., 1994; Sharma et al., 1998). This could mean that: (i) all the above regulated steps depend on PI3-kinase; (ii) as few as one step in the pathway exhibits such dependency; or (iii) none of the regulated steps are PI3-kinase dependent, but part of the machinery of forming the regulatable sequestration compartment or other aspect of the cell machinery necessary for the response is PI3-kinase dependent. In order to examine whether insulin regulates linear GLUT4– YFP movements on microtubules through a PI3-kinase-dependent mechanism, we treated 3T3-L1 adipocytes with wortmannin prior to addition of insulin. Under the conditions of these experiments wortmannin abolished the strong increase in phosphorylation of the protein kinase Akt in response to insulin (data not shown). Surprisingly, this treatment did not affect the number of long-range GLUT4–YFP movements per unit time in insulin-treated cells (Figure 3C), the average maximal velocities in either inward or outward GLUT4 movements (Figure 3A) or the distribution of the velocities among vesicle populations (Figure 3B).
Based on this failure of wortmannin to inhibit linear GLUT4–YFP movements, we wanted to confirm the known effect of the drug to inhibit GLUT4 translocation to the plasma membrane in response to insulin under these same experimental conditions (Figure 4). Using a GLUT4–GFP construct with a Myc tag on the first exofacial loop as reporter, it was found that the insulin-stimulated increase in cell-surface Myc-GLUT4–GFP was indeed nearly, but not completely, inhibited (74%) by 100 nM wortmannin, similar to a previous report (Hausdorff et al., 1999). Interestingly, however, 46% of these wortmannin- and insulin-treated cells did display a strong rim of GLUT4–GFP fluorescence around the cell periphery (Figure 4B). These data are consistent with the hypothesis that insulin acts to move GLUT4-containing vesicles towards the cell surface on microtubules in the presence of wortmannin. According to this model, GLUT4-containing vesicles in cells treated with insulin plus wortmannin can then dock with the plasma membrane, leading to the rim of Myc-GLUT4–GFP at the sub-plasma membrane region observed by monitoring the GFP signal but not the exofacial Myc signal (Figure 4A). Some of these vesicles are perhaps returned to the perinuclear region, while others accumulate as docked vesicles. This hypothesis suggests that the fusion step whereby Myc-GLUT4–GFP is inserted into the plasma membrane to become accessible to Myc antibody in unpermeabilized cells requires PI3-kinase.


Fig. 4. Accumulation of GLUT–YFP-containing vesicles near the cell periphery in adipocytes treated with wortmannin plus insulin. (A) 3T3-L1 cells were electroporated with the Myc-GLUT4–EGFP plasmid, starved, and then either treated with wortmannin for 15 min and then 100 nM insulin for 30 min (Wort + Insulin), or 100 nM insulin alone for 30 min (Insulin). Cells were then fixed and stained for plasma membrane GLUT4 (red) with anti-Myc antibody and rhodamine-conjugated secondary antibody without permeabilization. EGFP signal is shown in green. Bar, 10 µm. (B) Myc-GLUT4–EGFP-electroporated 3T3-L1 adipocytes were starved and left untreated (Basal), treated with either 100 nM wortmannin for 45 min (Wort) or 100 nM insulin for 30 min (Insulin), or pre-treated with 100 nM wortmannin for 15 min followed by a 30 min 100 nM insulin treatment (Wort + Ins). For each set, 50 cells were counted for the EGFP and the Myc rims.
Insulin-stimulated GLUT4 translocation requires conventional kinesin KIF5B
Plus end-directed transport of vesicles and organelles on microtubules is mediated by the large family of kinesin motor proteins. In a screen for genes highly expressed in 3T3-L1 adipocytes using Affymetrix GeneChip arrays (U74 series), oligonucleotides derived from the gene sequence of the conventional kinesin heavy chain KIF5B were found to yield the highest signal of all the kinesins represented on the arrays (Table I). Furthermore, higher expression of conventional heavy chain in cultured adipocytes compared with fibroblasts was observed by western blot analysis (Figure 5A). Conventional kinesin KIF5B is composed of two heavy chains and two light chains in a heterotetrameric configuration (Figure 6A) and is expressed in many tissues, while the other conventional kinesin isotypes KIF5A and KIF5C function mainly in neuronal tissues (Goldstein and Yang, 2000). Since the anti-KIF5 antibody (H2) we used recognizes all three isotypes but has lowest affinity for KIF5B (Kanai et al., 2000; Cai et al., 2001), we performed immunoprecipitation with the H2 antibody followed by mass spectrometry in order to confirm the expression of KIF5B. As shown in Figure 5B and C, a single major band was detected at ∼120–130 kDa after an NP-40 extraction in both starved and insulin-stimulated conditions. Mass spectrometry analysis of the band identified peptides with sequences that exactly match the murine kinesin heavy chain KIF5B, indicating that only this isotype is present in 3T3-L1 adipocytes (Figure 5C). Experiments were therefore designed to test the hypothesis that conventional kinesin KIF5B function is required for GLUT4 translocation to the plasma membrane in response to insulin. First, the intracellular localizations of endogenous conventional kinesin and GLUT4 were analyzed in 3T3-L1 adipocytes by immunostaining. The kinesin heavy chain is mainly present in the perinuclear region of 3T3-L1 adipocytes and partially co-localizes with GLUT4-containing vesicles (Figure 5D). These data are consistent with the hypothesis that this kinesin is associated with intracellular GLUT4-containing membranes in intact cultured adipocytes.
Table I. Expression of kinesins in 3T3-L1 adipocytes was analyzed by Affymetrix GeneChips.
| Kinesin type | Average signal | SEM | Chip set | Probe set |
|---|---|---|---|---|
| KIF5B | 20732 | 2220 | B | 113588_f_at |
| KIF21B | 4063 | 666 | B | 114821_at |
| KIF3B | 3520 | 996 | B | 115208_at |
| KIF3A | 2829 | 334 | B | 115929_at |
| KIF21A | 2041 | 572 | B | 117231_at |
| KIF1B | 1579 | 192 | B | 115895_at |
| KIF13A | 853 | 334 | B | 109305_at |
| KIF2 | 574 | 168 | A | 99962_at |
| KIF3C | 377 | 303 | A | 93635_at |
| KIF4 | 274 | 42 | A | 104644_at |
| KIFC2 | 123 | 68 | A | 93891_at |
| KIFC3 | 73 | 105 | A | 104335_at |
| KIF1A | 52 | 119 | A | 92890_at |
| KIFC1 | –978 | 461 | A | 98471_f_at |
| KLC1 | 786 | 90 | A | 93565_at |
| KLC2 | 242 | 439 | A | 102636_at |
Shown here are the average signal representing the mean of average differences and the SEM from three independent experiments. The names of murine genome chip sets (U74A and U74B) and individual probe sets giving the highest signal for a particular kinesin are shown. Expression signals of KLC1 and KLC2 are also presented.
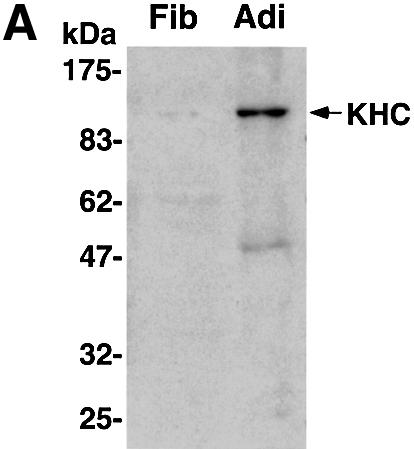
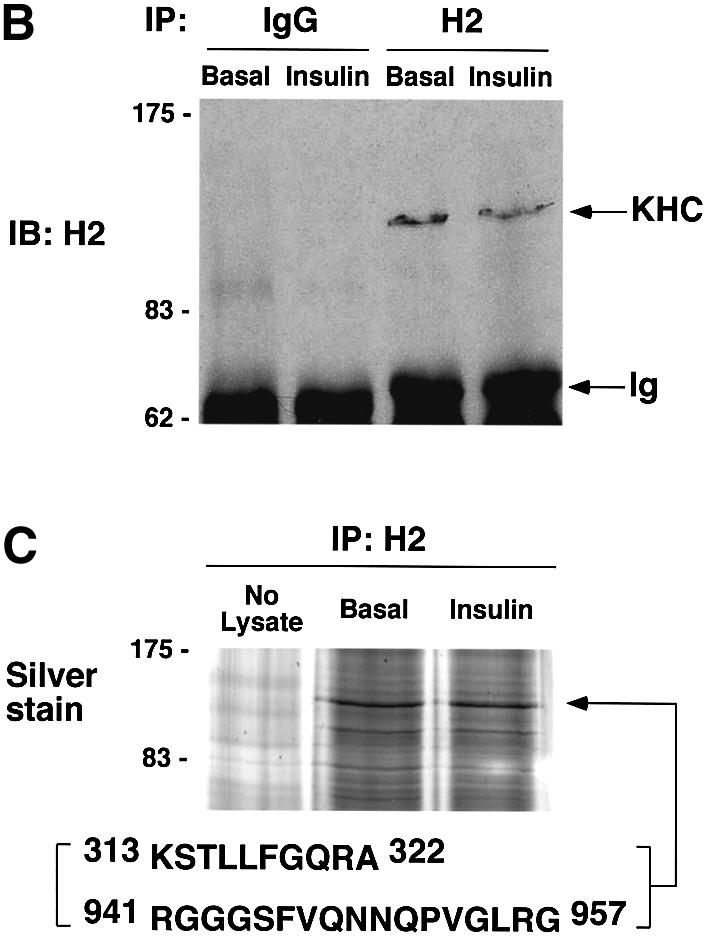

Fig. 5. Endogenous conventional kinesin KIF5B partially co-localizes with GLUT4 in 3T3-L1 adipocytes. (A) Confluent fibroblasts (Fib) and fully differentiated 3T3-L1 adipocytes (Adi) were analyzed by immunoblotting using anti-kinesin heavy chain H2 antibodies. (B) Starved (Basal) and insulin-stimulated 3T3-L1 adipocytes were lysed and subject to immunoprecipitation (IP) with either non-immune mouse IgG (IgG) and anti-kinesin heavy chain H2 antibody. Ig, immunoglobulin. (C) A gel was prepared as above and silver stained. The 120 kDa band was excised and analyzed by mass spectrometry. Shown are two peptides for which the amino acid sequences were matched to the murine conventional kinesin heavy chain KIF5B. (D) 3T3-L1 adipocytes were immunostained with anti-kinesin heavy chain H2 antibody (αKHC; red) and anti-GLUT4 antibody (αGLUT4; green). Two different cells are shown with merged images (Merge) of both channels. Bars, 10 µm.
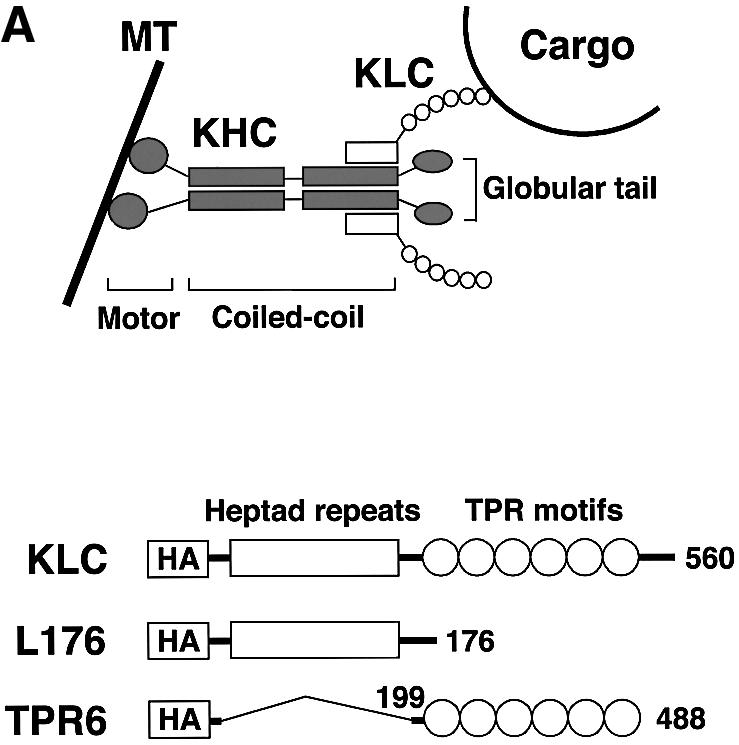


Fig. 6. Dominant-negative KLC mutants inhibit insulin-stimulated GLUT4–YFP vesicle movements. (A) Top diagram shows heterotetrameric structure of conventional kinesin and a proposed model for cargo binding (MT, microtubule). Structures of wild type (KLC), and C-terminal (L176) and an N-terminal deletion (TPR6) mutants of kinesin light chain are shown in the bottom diagram. All constructs have HA tags within their N-termini. The numbers represent amino acid sequences. These diagrams are adapted from Verhey et al. (2001). (B) Plasmids containing the full-length (KLC) and deletion mutant (L176 and TPR6) constructs were electroporated into 3T3-L1 adipocytes. Cells were starved and either left untreated (Basal) or stimulated with insulin for 30 min. Electroporated cells were identified by immunostaining with anti-HA antibody (αHA). F-actin and mitochondria (Mito) were visualized with rhodamine–phalloidin and Mitotracker, respectively. Bar, 10 µm. (C) 3T3-L1 adipocytes were electroporated with plasmids containing GLUT4–YFP and the kinesin light chain–CFP constructs. The number of GLUT4–YFP vesicles moving away from the nucleus in either basal or insulin-stimulated cells was counted. Five to seven cells were counted for each condition. Error bars represent the SEM. *P = 0.019; **P = 0.016.
Next we employed two dominant inhibitory kinesin light chain mutant proteins, based on the KLC1 isoform, to disrupt conventional kinesin KIF5B function (Figure 6A). The KLC1 isoform was found to be expressed in adipocytes, while KLC-2 is not (Table I). One of the constructs, hemagglutinin (HA)-tagged KLC-TPR6 (or CFP–KLC-TPR6), lacks the heptad repeats domain that functions as the kinesin heavy chain binding region (Diefenbach et al., 1998; Verhey et al., 1998; Kamal and Goldstein, 2002). Its expression is predicted to block interaction between endogenous kinesin and cargo proteins by saturating kinesin receptors such as those that might be present in GLUT4-containing vesicles. The other construct, HA-tagged KLC-L176 (or CFP–KLC-L176), lacks the C-terminal TPR6 domain involved in KLC–cargo binding (Skoufias et al., 1994; Bi et al., 1997). This mutant is predicted to incorporate into heterotetrameric kinesin complex and block the kinesin–cargo binding. Both the HA-KLC-L176 and HA-KLC-TPR6 constructs have been shown to disrupt conventional kinesin function when expressed in other cell types (Verhey et al., 2001). We also employed expression of the native kinesin light chain in these experiments as a control. After electroporation with these HA- or CFP-conjugated constructs, 3T3-L1 adipocytes remained intact and showed no phenotypic defect. All three KLC constructs localized mostly in the perinuclear region, while two dominant-negative KLC mutants often mislocalized into the nucleus upon higher expression (Figures 6B, and 7A and B). Consistent with the known function of conventional kinesin in mitochondrial dispersion (Tanaka et al., 1998), cells expressing the dominant-negative KLC mutants displayed reduced mitochondrial staining in the peripheral cytoplasm (Figure 6B). Confirming the specificity of the dominant-negative mutant function, expression of either the full-length or the dominant-negative KLC proteins did not affect intermediate filament distribution (data not shown) or insulin-stimulated cortical actin rearrangement (Figure 6B). To test whether vesicle movements stimulated by insulin are mediated by KIF5B, 3T3-L1 adipocytes were electroporated with the plasmid containing CFP-conjugated KLC constructs and GLUT4–YFP, and counted for the number of vesicle movements toward the cell periphery by live cell imaging. Figure 6C showed that the dominant-negative KLC mutants, but not the full-length KLC, significantly inhibited insulin-stimulated outward long-range movements of GLUT4-containing vesicles. Together with high expression of KIF5B (Figure 5), these data suggest that KIF5B is required for mobilizing GLUT4-containing vesicles in response to insulin in 3T3-L1 adipocytes.


Fig. 7. Dominant-negative kinesin light chain mutants block insulin-stimulated GLUT4 translocation to the cell surface. (A) 3T3-L1 cells were electroporated with the Myc-GLUT4–EGFP plasmid and plasmid containing either the wild-type or dominant-negative KLC constructs. Cells were starved and either left untreated (Basal) or incubated with 100 nM insulin (Insulin), and immunostained for the plasma membrane-fused GLUT4 with anti-Myc antibody (αMyc) without permeabilization. Cells were then permeabilized and immunostained with anti-HA antibody (αHA). Low magnification images show single-transfected (Myc-GLUT4–EGFP) cells and double-transfected (GLUT4-Myc–EGFP and KLC constructs) cells in the same viewing fields. Bars, 20 µm. (B) High magnification images of insulin-treated 3T3-L1 adipocytes that were either single- (–) or double-transfected as described in (A). Bars, 10 µm.
We next tested whether these outward GLUT4-containing vesicle movements mediated by KIF5B are required for insulin-stimulated translocation of GLUT4 to the plasma membrane. As shown in Figure 7A, expression of either dominant-negative mutant KLC abolished the ability of insulin to stimulate translocation of exofacial Myc-GLUT4–GFP to the cell surface, as reflected by the absence of anti-Myc antibody binding to adipocytes under these conditions. In contrast, intact adipocytes expressing Myc-GLUT4–GFP and the native kinesin light chain displayed strong anti-Myc antibody binding to the cell surface in response to insulin (Figure 7A and B). Quantitation of the results from a large number of adipocytes in these experiments was performed by visually counting the number of cells that display cell-surface binding of anti-Myc antibody (Figure 8A), as well as by quantifying the ratio of signal intensity from the anti-Myc antibody binding (cell-surface Myc-GLUT4–GFP) versus the signal from the total GFP fluorescence (total expressed Myc-GLUT4–GFP) (Figure 8B). These techniques for estimating the action of insulin on translocation of Myc-GLUT4–GFP have been described in detail previously (Park et al., 2001; Jiang et al., 2002). By both these measurements, expression of the inhibitory kinesin mutant proteins exerted virtually complete abolition of cell-surface Myc-GLUT4–GFP display in response to insulin (Figures 7 and 8), consistent with a requirement of conventional kinesin for insulin action on GLUT4 translocation.

Fig. 8. Dominant-negative kinesin light chain mutants block insulin-stimulated GLUT4 translocation to the cell surface. (A) The cell- surface anti-Myc contents of basal and insulin-treated Myc-GLUT4– EGFP-transfected adipocytes (–) and the double-transfected adipocytes (KLC, L176 and TPR6) were quantified. The bars represent the mean of the Myc/EGFP signal ratio of 20 cells for each condition. *P < 0.0001. (B) The percentage of cells displaying the cell-surface Myc rim of basal and insulin-treated single- and double-transfected 3T3-L1 adipocytes (50 cells each).
Discussion
Long-range transport of GLUT4 vesicles on microtubules
A key finding of this study is the observed movement of fluorescence signal from GLUT4–YFP along tracks delineated by microtubules labeled with tubulin–CFP (Figures 1 and 2). Furthermore, our data document such linear movements of GLUT4–YFP over distances as long as 20 µm, directed towards both the plus and minus ends of microtubules (Figures 2 and 3). Remarkably, insulin stimulates the number of these events observed per unit time in cultured adipocytes (Figure 3). Depolymerization of microtubules by colchicine results in complete inhibition of long-range GLUT4–YFP-containing vesicle movements (Figure 3C). Only a relatively small number of GLUT4 vesicles can be seen traveling over long distances after insulin stimulation relative to the total number of GLUT4-containing membranes observed in a single field. However, our live cell microscopy is performed at a single optical plane, which probably provides an underestimate of the total events per cell. These data extend previous indirect evidence suggesting that GLUT4-containing membranes are mobile on microtubules based on the distance and linearity of their movements (Oatey et al., 1997) as well as on the inhibitory actions of microtubule depolymerizing agents on GLUT4 translocation (Guilherme et al., 2000; Emoto et al., 2001). The present findings directly establish the conclusion that GLUT4-containing membranes move on microtubules in an outward direction towards the periphery of cultured adipocytes, as well as in an inward direction towards the perinuclear region.
KIF5B mediates GLUT4 vesicle movements on microtubules
A second significant finding reported here is the identification of conventional kinesin KIF5B as a molecular motor required for GLUT4 translocation in response to insulin (Figures 7 and 8). Conventional kinesin was first identified as a processive molecular motor involved in driving vesicles and organelles in axons of vertebrate and squid brain (Brady, 1985; Vale et al., 1985). Kinesin heavy chain has a microtubule plus end-directed motor domain, while kinesin light chain is generally thought to be involved in kinesin binding to cargos, mediating their motility toward the cell periphery (Hirokawa, 1993; Skoufias and Scholey, 1993). Recent reports demonstrated that conventional kinesin is essential for mitochondrial (Tanaka et al., 1998), lysosomal (Tanaka et al., 1998) and endosomal (Nielsen et al., 1999) transport, as well as for the microtubule-based motility of virus particles in infected cells (Rietdorf et al., 2001). Of the three isotypes of conventional kinesin known, only KIF5B could be detected in adipocytes. Using the same dominant inhibitory kinesin light chain constructs and methods as those previously validated for disrupting conventional kinesin function in other cells (Verhey et al., 2001), we observed profound inhibition of insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes (Figures 7 and 8) and GLUT4–YFP vesicle movements in live cells (Figure 6C). Although it is not known which isotypes of kinesin light chain interact with KIF5B heavy chains in adipocytes, the TPR6 domain and the heterodimerization region of the heptad repeats are highly conserved among the various kinesin isoforms, and no preference of binding is observed between kinesin heavy chain and different light chain isotypes (Rahman et al., 1998). Therefore, the dominant-negative mutants constructed from kinesin light chain KLC1-C used in this study are able to block efficiently conventional kinesin-mediated cargo transport. Taken together, our data are consistent with the hypothesis that KIF5B drives GLUT4 movements along microtubules as a required step in insulin’s ability to stimulate GLUT4 translocation to the plasma membrane.
Effect of microtubule disrupting agents on GLUT4 translocation
The data presented here raise an interesting paradox. If long-range GLUT4 movements on microtubules are required for transit and fusion of GLUT4-containing membranes with the plasma membrane, why is the complete disruption of microtubules by agents such as colchicine only partially effective in inhibiting GLUT4 translocation? One explanation for this may be related to the change in GLUT4 localization observed in response to disruption of microtubules. Dispersion of perinuclear GLUT4 towards the cell periphery is quite dramatic under these conditions, even in the absence of insulin, reflecting the inhibition of dynein-mediated transport of GLUT4-containing vesicles towards the minus ends of microtubules (Emoto et al., 2001; Patki et al., 2001). Thus, depolymerization of microtubules actually mimics the action of insulin to relocate perinuclear GLUT4-containing membranes throughout the cell and towards the actin-rich cell cortex. Insulin appears to mediate this effect through kinesin-directed GLUT4 movements on microtubules, while colchicine mediates the effect through blocking the return of GLUT4-containing vesicles to the perinuclear region, and perhaps by disrupting their retention in this region.
Another explanation for the paradoxical incomplete inhibition of GLUT4 translocation by microtubule depolymerizing agents may be that the acute action of insulin on GLUT4 translocation is only partially dependent on GLUT4 movements on microtubules. Insulin may act at several steps in the GLUT4 trafficking pathway, each of which can elicit an incremental increase in the overall rate of exocytosis. It is also possible that microtubule-based GLUT4 movements are mostly involved in replenishing GLUT4-containing vesicles near the cell periphery that have undergone regulation by insulin. According to this model, the initial acute action of insulin is targeted to GLUT4-containing vesicles already near the cell periphery, and then the relatively slow long-range movements from the perinuclear region increase the GLUT4-containing membranes in this insulin-sensitive pool. This concept is similar to models of synaptic vesicle regulation by neurotransmitters (Terada and Hirokawa, 2000). Our experiments designed to disrupt KIF5B function are performed over 24 h to allow for adequate expression of the dominant inhibitory constructs. Thus it is not possible to determine whether the requirements for kinesin function on GLUT4 trafficking is acute or long term. Future experiments are needed to address this issue.
Role of PI3-kinase in GLUT4 translocation
Glucose transport regulation by insulin is completely dependent on functional p85/p110-type PI3-kinase (Okada et al., 1994). It was therefore surprising to observe that microtubule-based movements of GLUT4 are apparently totally wortmannin insensitive (Figure 3). This remarkable finding raises two important questions. First, the data suggest the possibility that the PI3-kinase-dependent step or steps in the GLUT4 trafficking pathway are late steps, perhaps related to the fusion of GLUT4-containing membranes with the plasma membrane. This is also suggested by a report showing that synip is released from syntaxin-4 upon insulin stimulation (Min et al., 1999). This regulation of syntaxin-4, which in turn is thought to mediate membrane fusion at the plasma membrane, is wortmannin sensitive. Secondly, our data raise the question of the identity of the other pathway(s) involved in mediating insulin’s effect on kinesin-directed movements of GLUT4. It would seem that the possibility of insulin signaling through p21Ras is ruled out, given that a dominant inhibitory mutant of p21Ras is ineffective in modulating GLUT4 translocation (Hausdorff et al., 1994; Haruta et al., 1995), as is inhibition of the MEK protein kinase downstream of p21Ras (Usui et al., 1999). This question related to the molecular elements involved in insulin signaling to modulate GLUT4 movements on microtubules will be important to address in future studies.
KIF5B receptors on GLUT4 vesicles
Another related issue is the mechanism whereby conventional kinesin KIF5B engages GLUT4-containing vesicles. Recent work has identified potential receptors for the TPR domains of kinesin light chains, including JIP1 (Verhey et al., 2001), Sunday Driver (SYD or JIP3) (Bowman et al., 2000) and amyloid precursor protein (APP) (Kamal et al., 2001). Furthermore, the interaction between kinesin and cargo can be regulated with other proteins, such as heat shock chaperone 70 (Tsai et al., 2000). Also, recent work has implicated the protein kinase GSK3 as a regulator of kinesin light chain interaction with membranes (Morfini et al., 2002). These findings suggest a mechanism whereby phosphorylation of kinesin light chain by GSK3 disrupts its interaction with receptors on membrane organelles. The role of phosphorylation in regulating kinesin function was also suggested by others (Lee and Hollenbeck, 1995; De Vos et al., 2000; Morfini et al., 2001). This model, coupled with our data presented here, suggest there may be one or more receptors on GLUT4-containing vesicles that bind the kinesin light chain TPR domain and are potentially regulated by insulin signaling. Future experiments in our laboratory are directed to test this hypothesis.
Materials and methods
Materials
Plasmids encoding HA-tagged rat KLC1-C and its truncated constructs, HA-KLC-176 and HA-KLC-TPR6, were kindly provided by Dr K.J.Verhey (Harvard Medical School). Myc-GLUT4–EGFP construct was prepared as described previously (Jiang et al., 2002). GLUT4–YFP was constructed by inserting Myc-GLUT4 into pBluescript containing a linker with unique sites (NheI and AgeI), and then by inserting it into the same sites of pECFP-C1 vector (Clontech). For the tubulin–CFP construct, rat tubulin cDNA (DDBJ/EMBL/GenBank accession No. NM_011655) was isolated and amplified from total RNA using standard RT–PCR. Anti-kinesin heavy chain (H2) monoclonal (Chemicon International) and anti-Myc (clone 9E10) monoclonal (Santa Cruz Biotechnology) antibodies were used. Rabbit polyclonal anti-GLUT4 antibody and anti-HA polyclonal antibodies were produced as described previously (Langille et al., 1999). The Alexa 350-conjugated anti-rabbit antibody was from Molecular Probes. Human insulin was obtained from Eli Lilly Co. All other chemicals were from Sigma unless otherwise stated.
Cell culture, transfection and treatment
3T3-L1 fibroblasts were cultured and differentiated into adipocytes as described previously (Park et al., 2001). Four days after starting differentiation, 3T3-L1 adipocytes were transfected by electroporating (0.18 kV and 950 µF) 50 µg of DNA. All experiments were performed between 24 and 48 h after the electroporation. As indicated in each corresponding figure, the cells were starved and then treated either with 100 nM insulin only or with combinational treatment that involved 100 nM wortmannin for 15 min or 50 µM colchicine for 2 h, followed by stimulation with 100 nM insulin for 30 min.
Imaging of live adipocytes
3T3-L1 adipocytes, expressing appropriate cDNA constructs, were seeded in glass-bottomed dishes (MatTek Corporation). During the treatment and recording, cells were kept in Krebs–Ringer/HEPES buffer (pH 7.4) supplemented with 2% BSA and 0.22 mg/ml sodium pyruvate. Throughout the 15 min time lapse, images were taken every 5 s using an Olympus IX-70 inverted microscope with CCD camera. Deconvolution and image reconstruction of the image stacks was performed using Metamorph software (Universal Imaging). The movements of GLUT4–YFP vesicles over 2 µm for three consecutive frames (10 s) were counted blindly and categorized into inward (toward the nucleus and/or MTOC), outward (away from the nucleus and/or MTOC) or parallel (undistinguishable) movements.
Affymetrix GeneChip analysis
Expression analysis was carried out as suggested by the manufacturer (Affymetrix). Briefly, from three different sets of 3T3-L1 adipocytes, mRNA was isolated using Oligotex mRNA kit (Qiagen). Double-stranded cDNA was synthesized from 5 µg mRNA, purified and biotinylated. The cDNA was hybridized to the murine genome U74 arrays (A and B) for 16 h at 45 °C. The arrays were stained with streptavidin phycoerythrin solution and scanned in an HP GeneArray scanner. Average signal was calculated by subtracting signals from 11–20 overlapping 25mer probes by those from corresponding mismatch probes.
Immunoprecipitation, mass spectrometry and western blotting
Immunoprecipitation and mass spectrometry were performed as described previously (Park et al., 2001) with 1 mg total protein and 4 µg antibodies. For western blotting, cell lysates were collected in SDS lysis buffer (20 mM HEPES, pH 7.2, 1% sodium dodecyl sulfate, 1 mM sodium vanadate) supplemented with protease inhibitors and loaded onto 10% polyacrylamide gels.
Immunofluorescence microscopy
3T3-L1 adipocytes were fixed with 4% formaldehyde in phosphate-buffered saline (PBS), permeabilized and blocked with 0.5% Triton X-100 and 1% fetal bovine serum in PBS for 20 min. Cells were incubated with primary antibodies for 2 h and with rhodamine- or FITC-conjugated secondary antibodies for 30 min. To analyze Myc-GLUT4– EGFP translocation in adipocytes, cells were immunostained as described previously (Jiang et al., 2002). Briefly, the cell-surface Myc-GLUT4– GFP was visualized with anti-Myc antibody and rhodamine-labeled anti-mouse secondary antibody without permeabilization. Where applicable, the adipocytes were then fixed, permeabilized and immunostained for HA-tagged proteins, using polyclonal anti-HA antibody and Alexa-350-conjugated secondary antibody. Images were taken with an Olympus IX-70 microscope with CCD camera and then processed using Metamorph software. The specific plasma membrane content of Myc-GLUT4–GFP was measured as described previously (Jiang et al., 2002). For F-actin and mitochondrial staining, cells were immunostained with anti-HA antibody and FITC-conjugated secondary antibody. Rhodamine–phalloidin was added together with the secondary antibody. Mitotracker (Molecular Probes) was added before initial fixation.
Supplementary data
Supplementary data are available at The EMBO Journal Online.
Acknowledgments
Acknowledgements
This work was supported by grants from the National Institutes of Health (DK 30898 and DK 60837) to M.P.C. and a Postdoctoral Fellowship from the American Diabetes Association to S.S.
References
- Abel E.D., Peroni,O., Kim,J.K., Kim,Y.B., Boss,O., Hadro,E., Minnemann,T., Shulman,G.I. and Kahn,B.B. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature, 409, 729–733. [DOI] [PubMed] [Google Scholar]
- Asahi Y., Hayashi,H., Wang,L. and Ebina,Y. (1999) Fluoromicroscopic detection of Myc-tagged GLUT4 on the cell surface. Co-localization of the translocated GLUT4 with rearranged actin by insulin treatment in CHO cells and L6 myotubes. J. Med. Invest., 46, 192–199. [PubMed] [Google Scholar]
- Bi G.Q., Morris,R.L., Liao,G., Alderton,J.M., Scholey,J.M. and Steinhardt,R.A. (1997) Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J. Cell Biol., 138, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose A., Cherniack,A.D., Langille,S.E., Nicoloro,S.M., Buxton,J.M., Park,J.G., Chawla,A. and Czech,M.P. (2001) Gα11 signaling through ARF6 regulates F-actin mobilization and GLUT4 glucose transporter translocation to the plasma membrane. Mol. Cell. Biol., 21, 5262–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman A.B., Kamal,A., Ritchings,B.W., Philp,A.V., McGrail,M., Gindhart,J.G. and Goldstein,L.S. (2000) Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell, 103, 583–594. [DOI] [PubMed] [Google Scholar]
- Brady S.T. (1985) A novel brain ATPase with properties expected for the fast axonal transport motor. Nature, 317, 73–75. [DOI] [PubMed] [Google Scholar]
- Bryant N.J., Govers,R. and James,D.E. (2002) Regulated transport of the glucose transporter GLUT4. Nat. Rev. Mol. Cell. Biol., 3, 267–277. [DOI] [PubMed] [Google Scholar]
- Cai Y., Singh,B.B., Aslanukov,A., Zhao,H. and Ferreira,P.A. (2001) The docking of kinesins, KIF5B and KIF5C, to Ran-binding protein 2 (RanBP2) is mediated via a novel RanBP2 domain. J. Biol. Chem., 276, 41594–41602. [DOI] [PubMed] [Google Scholar]
- Cheatham B., Vlahos,C.J., Cheatham,L., Wang,L., Blenis,J. and Kahn,C.R. (1994) Phosphatidylinositol 3-kinase activation is required for insulin stimulation of pp70 S6 kinase, DNA synthesis and glucose transporter translocation. Mol. Cell. Biol., 14, 4902–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham B., Volchuk,A., Kahn,C.R., Wang,L., Rhodes,C.J. and Klip,A. (1996) Insulin-stimulated translocation of GLUT4 glucose transporters requires SNARE-complex proteins. Proc. Natl Acad. Sci. USA, 93, 15169–15173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. et al. (2001) Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKB beta). Science, 292, 1728–1731. [DOI] [PubMed] [Google Scholar]
- Czech M.P. and Corvera,S. (1999) Signaling mechanisms that regulate glucose transport. J. Biol. Chem., 274, 1865–1868. [DOI] [PubMed] [Google Scholar]
- De Vos K., Severin,F., Van Herreweghe,F., Vancompernolle,K., Goossens,V., Hyman,A. and Grooten,J. (2000) Tumor necrosis factor induces hyperphosphorylation of kinesin light chain and inhibits kinesin-mediated transport of mitochondria. J. Cell Biol., 149, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diefenbach R.J., Mackay,J.P., Armati,P.J. and Cunningham,A.L. (1998) The C-terminal region of the stalk domain of ubiquitous human kinesin heavy chain contains the binding site for kinesin light chain. Biochemistry, 37, 16663–16670. [DOI] [PubMed] [Google Scholar]
- Emoto M., Langille,S.E. and Czech,M.P. (2001) A role for kinesin in insulin-stimulated GLUT4 glucose transporter translocation in 3T3-L1 adipocytes. J. Biol. Chem., 276, 10677–10682. [DOI] [PubMed] [Google Scholar]
- Fletcher L.M., Welsh,G.I., Oatey,P.B. and Tavare,J.M. (2000) Role for the microtubule cytoskeleton in GLUT4 vesicle trafficking and in the regulation of insulin-stimulated glucose uptake. Biochem. J., 352, 267–276. [PMC free article] [PubMed] [Google Scholar]
- Goldstein L.S. and Yang,Z. (2000) Microtubule-based transport systems in neurons: the roles of kinesins and dyneins. Annu. Rev. Neurosci., 23, 39–71. [DOI] [PubMed] [Google Scholar]
- Guilherme A., Emoto,M., Buxton,J.M., Bose,S., Sabini,R., Theurkauf,W.E., Leszyk,J. and Czech,M.P. (2000) Perinuclear localization and insulin responsiveness of GLUT4 requires cytoskeletal integrity in 3T3-L1 adipocytes. J. Biol. Chem., 275, 38151–38159. [DOI] [PubMed] [Google Scholar]
- Haruta T., Morris,A.J., Rose,D.W., Nelson,J.G., Mueckler,M. and Olefsky,J.M. (1995) Insulin-stimulated GLUT4 translocation is mediated by a divergent intracellular signaling pathway. J. Biol. Chem., 270, 27991–27994. [DOI] [PubMed] [Google Scholar]
- Hausdorff S.F., Frangioni,J.V. and Birnbaum,M.J. (1994) Role of p21ras in insulin-stimulated glucose transport in 3T3-L1 adipocytes. J. Biol. Chem., 269, 21391–21394. [PubMed] [Google Scholar]
- Hausdorff S.F., Fingar,D.C., Morioka,K., Garza,L.A., Whiteman,E.L., Summers,S.A. and Birnbaum,M.J. (1999) Identification of wortmannin-sensitive targets in 3T3-L1 adipocytes. Dissociation of insulin-stimulated glucose uptake and glut4 translocation. J. Biol. Chem., 274, 24677–24684. [DOI] [PubMed] [Google Scholar]
- Hill M.M., Clark,S.F., Tucker,D.F., Birnbaum,M.J., James,D.E. and Macaulay,S.L. (1999) A role for protein kinase Bβ/Akt2 in insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol., 19, 7771–7781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N. (1993) Axonal transport and the cytoskeleton. Curr. Opin. Neurobiol., 3, 724–731. [DOI] [PubMed] [Google Scholar]
- Jagadish M.N. et al. (1996) Insulin-responsive tissues contain the core complex protein SNAP-25 (synaptosomal-associated protein 25) A and B isoforms in addition to syntaxin 4 and synaptobrevins 1 and 2. Biochem. J., 317, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhun B.H., Rampal,A.L., Liu,H., Lachaal,M. and Jung,C.Y. (1992) Effects of insulin on steady state kinetics of GLUT4 subcellular distribution in rat adipocytes. Evidence of constitutive GLUT4 recycling. J. Biol. Chem., 267, 17710–17715. [PubMed] [Google Scholar]
- Jiang Z.Y., Chawla,A., Bose,A., Way,M. and Czech,M.P. (2002) A phosphatidylinositol 3-kinase-independent insulin signaling pathway to N-WASP/Arp2/3/F-actin required for GLUT4 glucose transporter recycling. J. Biol. Chem., 277, 509–515. [DOI] [PubMed] [Google Scholar]
- Kamal A. and Goldstein,L.S. (2002) Principles of cargo attachment to cytoplasmic motor proteins. Curr. Opin. Cell Biol., 14, 63–68. [DOI] [PubMed] [Google Scholar]
- Kamal A., Almenar-Queralt,A., LeBlanc,J.F., Roberts,E.A. and Goldstein,L.S. (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature, 414, 643–648. [DOI] [PubMed] [Google Scholar]
- Kanai Y., Okada,Y., Tanaka,Y., Harada,A., Terada,S. and Hirokawa,N. (2000) KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci., 20, 6374–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanishi M., Tamori,Y., Okazawa,H., Araki,S., Shinoda,H. and Kasuga,M. (2000) Role of SNAP23 in insulin-induced translocation of GLUT4 in 3T3-L1 adipocytes. Mediation of complex formation between syntaxin4 and VAMP2. J. Biol. Chem., 275, 8240–8247. [DOI] [PubMed] [Google Scholar]
- Khayat Z.A., Tong,P., Yaworsky,K., Bloch,R.J. and Klip,A. (2000) Insulin-induced actin filament remodeling colocalizes actin with phosphatidylinositol 3-kinase and GLUT4 in L6 myotubes. J. Cell Sci., 113, 279–290. [DOI] [PubMed] [Google Scholar]
- Langille S.E., Patki,V., Klarlund,J.K., Buxton,J.M., Holik,J.J., Chawla,A., Corvera,S. and Czech,M.P. (1999) ADP-ribosylation factor 6 as a target of guanine nucleotide exchange factor GRP1. J. Biol. Chem., 274, 27099–27104. [DOI] [PubMed] [Google Scholar]
- Lee K.D. and Hollenbeck,P.J. (1995) Phosphorylation of kinesin in vivo correlates with organelle association and neurite outgrowth. J. Biol. Chem., 270, 5600–5605. [DOI] [PubMed] [Google Scholar]
- Martin S., Tellam,J., Livingstone,C., Slot,J.W., Gould,G.W. and James,D.E. (1996) The glucose transporter (GLUT-4) and vesicle-associated membrane protein-2 (VAMP-2) are segregated from recycling endosomes in insulin-sensitive cells. J. Cell Biol., 134, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J. et al. (1999) Synip: a novel insulin-regulated syntaxin 4-binding protein mediating GLUT4 translocation in adipocytes. Mol. Cell, 3, 751–760. [DOI] [PubMed] [Google Scholar]
- Molero J.C., Whitehead,J.P., Meerloo,T. and James,D.E. (2001) Nocodazole inhibits insulin-stimulated glucose transport in 3T3-L1 adipocytes via a microtubule-independent mechanism. J. Biol. Chem., 276, 43829–43835. [DOI] [PubMed] [Google Scholar]
- Morfini G., Szebenyi,G., Richards,B. and Brady,S.T. (2001) Regulation of kinesin: implications for neuronal development. Dev. Neurosci., 23, 364–376. [DOI] [PubMed] [Google Scholar]
- Morfini G., Szebenyi,G., Elluru,R., Ratner,N. and Brady,S.T. (2002) Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J., 21, 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen E., Severin,F., Backer,J.M., Hyman,A.A. and Zerial,M. (1999) Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol., 1, 376–382. [DOI] [PubMed] [Google Scholar]
- Oatey P.B., Van Weering,D.H., Dobson,S.P., Gould,G.W. and Tavare,J.M. (1997) GLUT4 vesicle dynamics in living 3T3 L1 adipocytes visualized with green-fluorescent protein. Biochem. J., 327, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T., Kawano,Y., Sakakibara,T., Hazeki,O. and Ui,M. (1994) Essential role of phosphatidylinositol 3-kinase in insulin-induced glucose transport and antilipolysis in rat adipocytes. Studies with a selective inhibitor wortmannin. J. Biol. Chem., 269, 3568–3573. [PubMed] [Google Scholar]
- Olson A.L. and Pessin,J.E. (1996) Structure, function and regulation of the mammalian facilitative glucose transporter gene family. Annu. Rev. Nutr., 16, 235–256. [DOI] [PubMed] [Google Scholar]
- Olson A.L., Knight,J.B. and Pessin,J.E. (1997) Syntaxin 4, VAMP2 and/or VAMP3/cellubrevin are functional target membrane and vesicle SNAP receptors for insulin-stimulated GLUT4 translocation in adipocytes. Mol. Cell. Biol., 17, 2425–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson A.L., Trumbly,A.R. and Gibson,G.V. (2001) Insulin-mediated GLUT4 translocation is dependent on the microtubule network. J. Biol. Chem., 276, 10706–10714. [DOI] [PubMed] [Google Scholar]
- Omata W., Shibata,H., Li,L., Takata,K. and Kojima,I. (2000) Actin filaments play a critical role in insulin-induced exocytotic recruitment but not in endocytosis of GLUT4 in isolated rat adipocytes. Biochem. J., 346, 321–328. [PMC free article] [PubMed] [Google Scholar]
- Park J.G., Bose,A., Leszyk,J. and Czech,M.P. (2001) PYK2 as a mediator of endothelin-1/G alpha 11 signaling to GLUT4 glucose transporters. J. Biol. Chem., 276, 47751–47754. [DOI] [PubMed] [Google Scholar]
- Patki V., Buxton,J., Chawla,A., Lifshitz,L., Fogarty,K., Carrington,W., Tuft,R. and Corvera,S. (2001) Insulin action on GLUT4 traffic visualized in single 3T3-l1 adipocytes by using ultra-fast microscopy. Mol. Biol. Cell, 12, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessin J.E., Thurmond,D.C., Elmendorf,J.S., Coker,K.J. and Okada,S. (1999) Molecular basis of insulin-stimulated GLUT4 vesicle trafficking. Location! Location! Location! J. Biol. Chem., 274, 2593–2596. [DOI] [PubMed] [Google Scholar]
- Rahman A., Friedman,D.S. and Goldstein,L.S. (1998) Two kinesin light chain genes in mice. Identification and characterization of the encoded proteins. J. Biol. Chem., 273, 15395–15403. [DOI] [PubMed] [Google Scholar]
- Rietdorf J., Ploubidou,A., Reckmann,I., Holmstrom,A., Frischknecht,F., Zettl,M., Zimmermann,T. and Way,M. (2001) Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol., 3, 992–1000. [DOI] [PubMed] [Google Scholar]
- Sharma P.M., Egawa,K., Huang,Y., Martin,J.L., Huvar,I., Boss,G.R. and Olefsky,J.M. (1998) Inhibition of phosphatidylinositol 3-kinase activity by adenovirus-mediated gene transfer and its effect on insulin action. J. Biol. Chem., 273, 18528–18537. [DOI] [PubMed] [Google Scholar]
- Shigematsu S., Khan,A.H., Kanzaki,M. and Pessin,J.E. (2002) Intracellular insulin-responsive glucose transporter (GLUT4) distribution but not insulin-stimulated GLUT4 exocytosis and recycling are microtubule dependent. Mol. Endocrinol., 16, 1060–1068. [DOI] [PubMed] [Google Scholar]
- Skoufias D.A. and Scholey,J.M. (1993) Cytoplasmic microtubule-based motor proteins. Curr. Opin. Cell Biol., 5, 95–104. [DOI] [PubMed] [Google Scholar]
- Skoufias D.A., Cole,D.G., Wedaman,K.P. and Scholey,J.M. (1994) The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J. Biol. Chem., 269, 1477–1485. [PubMed] [Google Scholar]
- Tanaka Y., Kanai,Y., Okada,Y., Nonaka,S., Takeda,S., Harada,A. and Hirokawa,N. (1998) Targeted disruption of mouse conventional kinesin heavy chain, KIF5B, results in abnormal perinuclear clustering of mitochondria. Cell, 93, 1147–1158. [DOI] [PubMed] [Google Scholar]
- Terada S. and Hirokawa,N. (2000) Moving on to the cargo problem of microtubule-dependent motors in neurons. Curr. Opin. Neurobiol., 10, 566–573. [DOI] [PubMed] [Google Scholar]
- Tsai M.Y., Morfini,G., Szebenyi,G. and Brady,S.T. (2000) Release of kinesin from vesicles by hsc70 and regulation of fast axonal transport. Mol. Biol. Cell, 11, 2161–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui I. et al. (1999) Differential effects of palmitate on glucose uptake in rat-1 fibroblasts and 3T3-L1 adipocytes. Horm. Metab. Res., 31, 546–552. [DOI] [PubMed] [Google Scholar]
- Vale R.D., Reese,T.S. and Sheetz,M.P. (1985) Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell, 42, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey K.J., Lizotte,D.L., Abramson,T., Barenboim,L., Schnapp,B.J. and Rapoport,T.A. (1998) Light chain-dependent regulation of kinesin’s interaction with microtubules. J. Biol. Chem., 143, 1053–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey K.J., Meyer,D., Deehan,R., Blenis,J., Schnapp,B.J., Rapoport,T.A. and Margolis,B. (2001) Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J. Cell Biol., 152, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volchuk A., Wang,Q., Ewart,H.S., Liu,Z., He,L., Bennett,M.K. and Klip,A. (1996) Syntaxin 4 in 3T3-L1 adipocytes: regulation by insulin and participation in insulin-dependent glucose transport. Mol. Biol. Cell, 7, 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Bilan,P.J., Tsakiridis,T., Hinek,A. and Klip,A. (1998) Actin filaments participate in the relocalization of phosphatidylinositol 3-kinase to glucose transporter-containing compartments and in the stimulation of glucose uptake in 3T3-L1 adipocytes. Biochem. J., 331, 917–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. and Holman,G.D. (1993) Comparison of GLUT4 and GLUT1 subcellular trafficking in basal and insulin-stimulated 3T3-L1 cells. J. Biol. Chem., 268, 4600–4603. [PubMed] [Google Scholar]
- Zisman A. et al. (2000) Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nat. Med., 6, 924–928. [DOI] [PubMed] [Google Scholar]