

Abstract
RNA polymerase (pol) III transcription increases within minutes of serum addition to growth-arrested fibroblasts. We show that ERK mitogen-activated protein kinases regulate pol III output by directly binding and phosphorylating the BRF1 subunit of transcription factor TFIIIB. Blocking the ERK signalling cascade inhibits TFIIIB binding to pol III and to transcription factor TFIIIC2. Chromatin immunoprecipitation shows that the association of BRF1 and pol III with tRNALeu genes in cells decreases when ERK is inactivated. Furthermore, mutation of an ERK docking domain or phosphoacceptor site in BRF1 prevents serum induction of pol III transcription. These data identify a novel target for ERK, and suggest that its ability to stimulate biosynthetic capacity and growth involves direct transcriptional activation of tRNA and 5S rRNA genes.
Keywords: ERK/pol III/TFIIIB/TFIIIC/transcription
Introduction
RNA polymerase (pol) III output is tightly linked to growth (Larminie et al., 1998). Thus, pol III transcription decreases when cells are deprived of serum or nutrients and increases again upon mitogenic stimulation (Johnson et al., 1974; Mauck and Green, 1974; Tower and Sollner-Webb, 1988; Sethy et al., 1995; Scott et al., 2001). It is also subject to cell cycle control in mammals (White et al., 1995b). The pol III-specific factor TFIIIB is regulated by the retinoblastoma protein RB (Larminie et al., 1997). Only in its hypophosphorylated form, found during the G0 and early G1 phases, can RB bind and repress TFIIIB (Scott et al., 2001). A major increase in tRNA synthesis occurs at the G1–S phase transition (Johnson et al., 1974; Mauck and Green, 1974; White et al., 1995b), which coincides with the hyperphosphorylation of RB by cyclin-dependent kinases (CDKs) (Scott et al., 2001). Hyper phosphorylated RB releases TFIIIB (Scott et al., 2001), allowing it to interact with TFIIIC2 at the promoter of class III genes and recruit pol III to these templates (Sutcliffe et al., 2000). The RB-related pocket protein p130 also binds TFIIIB during G0 and early G1, contributing to its repression in serum-starved cells (Sutcliffe et al., 1999; Scott et al., 2001). Although RB and p130 play major roles in the growth factor sensitivity of pol III transcription in mammalian cells, it is likely that other mechanisms are also involved since a rapid increase in pol III transcription occurs in serum-stimulated fibroblasts before RB dissociates from TFIIIB (Johnson et al., 1974; Scott et al., 2001).
The ERK MAP kinase cascade promotes growth in several ways, including activation of translational capacity (Whitmarsh and Davis, 2000). Growth requires ribosome production (Montagne, 2000; Brandenburger et al., 2001) and the ERK pathway links growth factor signalling to ribosome biogenesis in mammalian cells. It has been shown (Stefanovsky et al., 2001) that synthesis of rRNA by pol I is regulated by ERK through phosphorylation of the upstream binding factor (UBF). For this to be biologically significant, one might predict a concomitant rise in 5S rRNA synthesis by pol III, since ribosomes require an equimolar ratio of rRNAs. We show that the ERK cascade is indeed involved in serum induction of pol III transcription. The effects of this pathway are independent of RB and the cell cycle. Activated ERK interacts with and phosphorylates the BRF1 subunit of TFIIIB in vitro and in vivo. Furthermore, mutation of an ERK docking domain or phosphoacceptor site in BRF1 blocks pol III induction. We conclude that ERK activation in response to mitogens stimulates pol III transcription through a mechanism dependent on BRF1 phosphorylation. This provides a further connection between growth factor signalling and the biosynthetic pathways that underlie cell growth.
Results
The ERK pathway regulates pol III transcription
An initial increase in pol III transcription precedes the substantial rise in late G1 that accompanies inactivation of RB and p130 (Mauck and Green, 1974; Scott et al., 2001). This can be clearly seen by monitoring pol III transcripts derived from the B2 middle repetitive gene family, which have a short half-life and so provide a reliable indication of transcriptional output. Thus, when quiescent fibroblasts are stimulated to re-enter the cell cycle by addition of serum, B2 transcripts increase 3-fold within 10 min of serum addition (Figure 1A, upper panel). This effect is specific, since levels of a pol II transcript encoding acidic ribosomal phosphoprotein P0 (ARPP P0) do not respond to serum (Figure 1A, lower panel). B2 expression increases further as cells pass the restriction point later in G1 phase (Figure 1A; Scott et al., 2001).

Fig. 1. Manipulation of the Ras signalling cascade affects serum-stimulated pol III activity in 3T3 fibroblasts. (A) Northern blot of total RNA (20 µg) from 3T3 cells cultured in 0.5% serum for 24 h (lane 1) and then stimulated with 10% serum for the times indicated (lanes 2–8). The upper panel shows the blot probed with a B2 gene; the lower panel shows the same blot that has been stripped and reprobed with the ARPP P0 gene. (B) Northern blot of total RNA (20 µg) from 3T3 cells cultured in 0.5% serum for 24 h (lanes 1, 3 and 5) or in medium containing 10% serum (lanes 2, 4 and 6) and treated for 12 h with vehicle (lanes 1 and 2) or with PD98059 (50 µM, lanes 3 and 4) or FTI-277 (1 µM, lanes 5 and 6). Upper and lower panels show blots probed with B2 and ARPP P0 as for (A). (C) The B2 signals from (B) were quantified by PhosphorImager (Molecular Dynamics) and normalized against the ARPP P0 signal. The graph shows means and standard deviations from three independent experiments; values obtained for cells grown in serum in the absence of inhibitor (lane 2) were set as 100 and other values were calculated as a percentage of this. (D) 3T3 cells growing in 10% serum were transfected with pVA1 (0.5 µg; all lanes), pCAT (0.5 µg; all lanes), pCMV vector (3 µg, lane 1; 2 µg, lane 2), pCMV-RasV12 (1 µg, lane 2; 3 µg, lane 3), pRSV-LTR (3 µg, lane 4; 1 µg, lane 5) and pRSV-LTR-RafΔ2–334 (2 µg, lane 5; 3 µg, lane 6). VA1 (upper panel) and CAT levels (lower panel) assayed by primer extension are shown. (E) Results from (D) were quantified as above. Graphs show means and standard deviations from three independent experiments for VA1 expression after normalization to levels of CAT RNA; the activities obtained for cells transfected with pCMV alone (lane 1) were set at 100 and other values were calculated as a percentage of this.
We tested the effect on pol III activity of blocking the ERK pathway using the specific MEK inhibitor PD98059 (50 µM) or the farnesyltransferase inhibitor FTI 277 (1 µM), which blocks activation of Ras (Bernhard et al., 1996). Both inhibitors markedly reduce the serum induction of pol III transcripts without affecting ARPP P0 mRNA (Figure 1B). After normalization to the ARPP P0 control, B2 induction was reduced 2-fold by PD98059 and 2.1-fold by FTI 277 (Figure 1C). In contrast, neither compound had any significant effect on the basal B2 expression in serum-deprived cells. We also investigated whether pol III transcription responds to constitutively active forms of Ras (RasV12) or Raf (RafΔ2–334), which stimulate the ERK pathway. Fibroblasts were transfected with vectors encoding these kinases, along with the adenovirus VA1 gene, as a pol III reporter, and a control CAT gene driven by the SV40 early promoter to normalize for transfection efficiency. Both RasV12 and RafΔ2–334 increase VA1 expression in a dose-dependent manner (Figure 1D). After normalization for CAT RNA levels, both stimulate VA1 by up to 3.5-fold (Figure 1E).
Clearly, a pathway involving Ras/Raf/MEK can influence pol III transcription, but is it sufficient to induce pol III in the absence of other mitogenic signals? To test this, we used the CCL39-ΔRaf-1:ER cell system, in which treatment with estradiol directly induces Raf and subsequently ERK1/2 activity (Lenormand et al., 1996). After only 2 h of estradiol treatment, the abundance of pol III transcripts increased above basal levels and continued to rise through S phase, whereas ARPP P0 did not respond (Figure 2A).

Fig. 2. The Ras/ERK signalling cascade affects pol III transcription independently of RB. (A) CCL39-ΔRaf-1:ER cells were arrested by serum starvation for 24 h. RNA was extracted from cells that were left untreated (lane 1) or stimulated either with 10% serum for 24 h (lane 5) or with 1 µM estradiol for 2 h (lane 2), 10 h (lane 3) or 24 h (lane 4). The upper and lower panels show northern blots probed with B2 and ARPP P0 as above. (B) Protein (10 µg) extracted in parallel from the above cells was resolved by SDS–PAGE and then immunoblotted with antibodies against RB phosphorylated at serine 780 (upper panel), cyclin D1 (second panel), TBP (third panel) or actin (lower panel). (C) Templates (500 ng) VA1 (upper panel) and tRNALeu (lower panel) were transcribed using 15 µg of whole-cell extract prepared from 3T3 cells grown continuously in 10% serum in the presence of vehicle (lane 1) or 1 µM U0126 for the times indicated. (D) Whole-cell extracts from the above experiment were resolved by SDS–PAGE and analysed by western blotting with antibodies against RB phosphorylated at serine 780 (upper panel), cyclin D1 (second panel) or actin (lower panel). (E) Wild-type (wt; lanes 1 and 2) or Rb–/–p130–/–p107–/– triple knockout mouse embryo fibroblasts (TKO; lanes 3 and 4) were transfected with pVA1 (0.5 µg, all lanes), pCAT (0.5 µg, all lanes), pCMV vector (3 µg, lanes 1 and 3) or pCMV-RasV12 (3 µg, lanes 2 and 4). VA1 and CAT RNA levels were assayed by primer extension. VA1 RNA levels are shown; CAT levels remained constant (data not shown). (F) VA1 RNA levels are shown; CAT levels remained constant (data not shown). VA1 and CAT levels from (E) were quantified by PhosphorImager as before. Values presented graphically are means and standard deviations for VA1 expression after normalization to the levels of CAT RNA to correct for transfection efficiency. The activities obtained for wt cells transfected with pCMV alone (lane 1) were set at 100 and other values were calculated as a percentage of this.
Constitutive activation of the Ras/Raf signalling pathway can raise TATA-binding protein (TBP) levels and thereby enhance pol III transcription in certain cell types (Wang et al., 1995). However, we detected little or no change in the level of TBP in CCL39-ΔRaf-1:ER cells even after treatment with estradiol for 24 h (Figure 2B), although longer exposure to the hormone does raise TBP levels (data not shown). Similar results were obtained with serum-induced CCL39-ΔRaf-1:ER or 3T3 cells (Figure 2B; data not shown). We conclude that ERK activation is sufficient to stimulate pol III transcription in fibroblasts and that this can precede TBP induction.
RB is not required for pol III activation by the ERK pathway
Ras-mediated activation of ERK can induce expression of cyclin D1 leading to RB phosphorylation (Aktas et al., 1997; Cheng et al., 1998). Since pol III transcription is sensitive to the phosphorylation status of RB (Scott et al., 2001), cyclin D1 induction via ERK could raise pol III output. However, activation of Raf in CCL39-ΔRaf-1:ER cells induces pol III transcription prior to induction of cyclin D1 and phosphorylation of RB (Figure 2A and B). Similarly, pol III responds more rapidly than RB when the MEK inhibitor U0126 (1 µM) is added to asynchronously growing 3T3 cells. Extracts of fibroblasts incubated for just 1 h with U0126 transcribe the adenovirus VA1 and tRNALeu genes significantly less actively than extracts prepared from control cells treated with vehicle alone (Figure 2C). In contrast, little or no change in the levels of cyclin D1 or RB phosphorylation were observed until after 3 h incubation with the MEK inhibitor (Figure 2D).
A further way of addressing whether the Ras pathway requires RB in order to influence pol III transcription is to study its effect in RB-null cells. Mouse embryonic fibroblasts (MEFs) from wild-type and RB–/–/p107–/–/p130–/– triple knockout (TKO) mice (Dannenberg et al., 2000; Sage et al., 2000) were transiently transfected with or without RasV12 along with the VA1 pol III reporter and SV40-CAT control. As in the 3T3 cells, transfecting wild-type MEFs with RasV12 increases VA1 expression (Figure 2E). VA1 is more rapidly expressed in TKO cells than in wild-type MEFs, consistent with the fact that RB, p107 and p130 can all repress pol III transcription (White et al., 1996; Sutcliffe et al., 1999). However, Ras V12 can still stimulate VA1 expression in the TKO cells. After normalization, RasV12 was found to activate VA1 ∼2-fold in the TKO MEFs (Figure 2F).
ERK2 inhibition or immunodepletion reduces pol III transcription in vitro
Since the ERK signalling cascade can stimulate pol III transcription independently of RB and the cell cycle, it may act directly on the pol III machinery. Consistent with this, VA1 transcription in vitro was reduced by up to 87% in a dose-dependent manner by a substrate peptide containing an ERK consensus phosphoacceptor site, which can act as a competitive inhibitor. This response was specific, since a protein kinase A (PKA) substrate peptide had minimal effect (Figure 3A). To confirm the influence of ERK on pol III transcription in vitro, 3T3 cell extracts were immunodepleted using anti-ERK antibody and compared with extracts immunodepleted with irrelevant control antibodies against viral oncoproteins. As a positive control, extracts were also immunodepleted using antibody against the essential factor TBP. Pol III transcription was substantially reduced in extracts depleted of either TBP or ERK compared with extracts immunodepleted using the negative control antibodies (Figure 3B). These data suggest that ERK can contribute significantly to the level of pol III transcription in vitro.
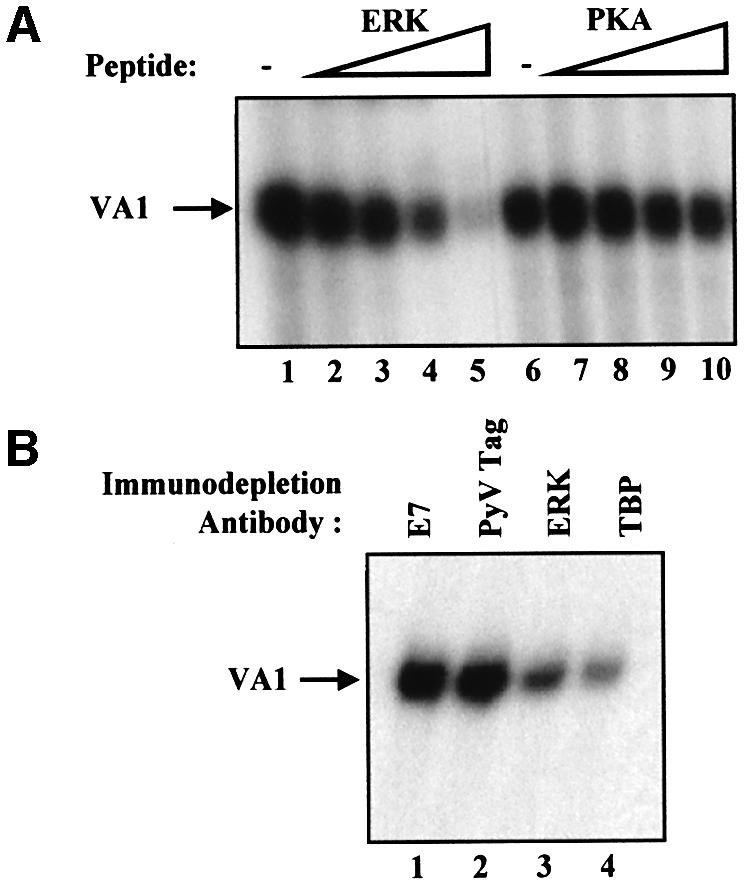
Fig. 3. Pol III transcription is blocked specifically by a peptide inhibitor of ERK. (A) In vitro transcription was carried out using 20 µg of 3T3 whole-cell extract and a VA1 template (250 ng) after preincubation for 15 min at 30°C with buffer (lanes 1 and 6) or with 10, 20, 30 or 40 µg of ERK phosphoacceptor peptide (APRTPGGRR, lanes 2–5, respectively) or PKA phosphoacceptor peptide (LRRASLG, lanes 7–10, respectively). (B) Whole-cell extracts (145 µg) prepared from Py3T3 fibroblasts were immunodepleted using anti-E7 antibody TVG710Y (E7, lane 1), F4 antibody against T antigen of polyomavirus (PyV Tag, lane 2), anti-ERK2 antibody (ERK, lane 3) or anti-TBP antibody MTBP-6 (TBP, lane 4). Then, 20 µg of each of the immunodepleted extracts was used for in vitro transcription assay with 250 ng of pVA1.
ERK interacts with TFIIIB both in vitro and in vivo
We tested whether ERK associates with TFIIIB, which is limiting for pol III transcription in fibroblasts (Scott et al., 2001). Immunoprecipitations were carried out using serum-starved and growing fibroblasts that stably overexpress an HA-tagged version of the BRF1 subunit of TFIIIB. Expression of HA.BRF1 in these cells was not affected by serum (Figure 4A, lanes 1 and 2). RB or ERK2 immunoprecipitates were probed by western blotting for HA.BRF1, using an antibody that recognizes the HA tag. As shown previously (Scott et al., 2001), BRF1 associates with RB in quiescent cells, but this interaction is substantially reduced by mitogenic stimulation (Figure 4A, lanes 4 and 5). A small amount of ERK2 associates with BRF1 in starved cells, but the interaction is greatly increased in the presence of serum (Figure 4A, lanes 7 and 8). This coprecipitation reflects a specific interaction with ERK2, since BRF1 is not co-immunoprecipitated with a control antiserum against the TAFI48 subunit of the pol I factor SL1 (Figure 4A, lane 9). Immunofluorescence reveals that BRF1 is localized to the nucleus of these cells whether or not serum is present in the medium (data not shown). This suggests that ERK2 activated in the cytoplasm must translocate to the nucleus to interact with TFIIIB. The increase in binding of ERK2 to BRF1 in response to serum is blocked by the MEK inhibitor U0126 (1 µM) (Figure 4B). This suggests that activation of ERK2 is required for it to bind TFIIIB. To test this directly, binding assays were carried out in vitro using GST.ERK2 that was either unphosphorylated or activated by phosphorylation with MEK (Marais et al., 1997). These were incubated with extracts from growing cells overexpressing HA.BRF1, in which endogenous MEK was inactivated by heat treatment. Proteins that remained bound to the beads after extensive washing were visualized by western blotting using anti-HA antibody. Only beads that carried active ERK2 were found to retain HA.BRF1 with high efficiency (Figure 4C). This confirms the immunoprecipitation data indicating that only the activated form of ERK2 associates with TFIIIB.

Fig. 4. Activated ERK2 is co-immunoprecipitated with TFIIIB both in vitro and in vivo. (A) Cell extract (500 µg) was prepared from Rat1A fibroblasts expressing pCDNA3HA.BRF1 either cultured for 24 h in serum-free conditions (lanes 1, 4 and 7) or left growing in 5% serum (lanes 2, 3, 5, 6, 8 and 9). These were immunoprecipitated (IP) with anti-HA (lanes 1 and 2), anti-RB antibody C-15 (lanes 4 and 5), anti-ERK2 antibody (lanes 7 and 8) or anti-TAFI48 antibody M-19 (lanes 3, 6 and 9). Precipitates were resolved by SDS–PAGE and then analysed by western blotting with anti-HA antibody. (B) Rat1A fibroblasts expressing pCDNA3HA.BRF1 (500 µg) were cultured for 24 h in serum-free conditions (lanes 1, 2, 6 and 7) or left growing in 5% serum (lanes 3–5 and 8–10) and in the presence (lanes 2, 4, 7 and 9) or absence (lanes 1, 3, 5, 6, 8 and 10) of 1 µM U0126 for a further 2 h. Precipitates were resolved by SDS–PAGE and then blotted with anti-HA antibody. (C) Cell extract prepared from growing Rat 1A cells expressing pCDNA3HA.BRF1 was heat treated at 65°C for 30 min to inactivate endogenous ERK2. Then, 250 µg of this extract was incubated in the presence of glutathione beads carrying equal amounts of GST (lanes 1 and 3) or GST-ERK2 that was left inactive (lane 2) or was activated by MEK (lane 4). Proteins retained after extensive washing were resolved by SDS–PAGE and immunoblotted with anti-HA antibody to detect binding of HA.BRF1.
The BRF1 subunit of TFIIIB is phosphorylated by ERK2 in vitro and in vivo
As it is bound by ERK2, TFIIIB may be phosphorylated by this kinase. We focused on BRF1, since transfection assays showed that this subunit of TFIIIB is limiting for pol III transcription in both quiescent and proliferating 3T3 fibroblasts (Figure 5A). In the presence of [γ-32P]ATP and activated recombinant ERK2, recombinant BRF1 was phosphorylated, consistent with the presence of several Ser/Thr-Pro consensus phosphoacceptor sites in the BRF1 sequence (Figure 5A). We next asked whether BRF1 is also phosphorylated by the ERK signalling cascade in vivo. HA-tagged BRF1 was immunoprecipitated from transfected cells labelled with [32P]orthophosphate. As shown previously (Johnston et al., 2002), serum stimulation results in an ∼5-fold increase in BRF1 phosphorylation (Figure 5C and D). However, addition of U0126 (1 µM) for 1 h decreased the serum-induced phospho-labelling by 60% without significantly altering basal phosphorylation of BRF1 in serum-starved cells. Western blotting confirmed that neither serum nor inhibitor affected the amount of BRF1 that was immunoprecipitated (Figure 5C, lower panel).

Fig. 5. BRF is phosphorylated in vitro and in vivo by ERK2. (A) 3T3 cells incubated in the presence (lanes 1–3) or absence (lanes 4–6) of serum were transfected with pVA1 (0.5 µg, all lanes), pCAT (0.5 µg, all lanes), pCDNA3HA (3 µg, lanes 1 and 4; 2 µg, lanes 2 and 5) or pCDNA3HA.BRF1 (1 µg, lanes 2 and 5; 3 µg, lanes 3 and 6). VA1 and CAT levels were assayed by primer extension and then quantified by PhosphorImager. VA1 RNA levels are shown; CAT levels remained constant (data not shown). (B) Glutathione beads carrying equal amounts of GST (lane 2) or GST.BRF1 (lane 1) and GST.ERK2 activated by constitutively active MEK (lanes 1–3) were incubated in the presence of [γ-32P]ATP for 30 min at 30°C. Laemmli sample buffer was added to stop the reaction and samples were boiled for 5 min before being subjected to SDS–PAGE and autoradiography. (C) CHO cells growing in 10% FCS were transiently transfected with pCDNA3HA.BRF1 and labelled 48 h later with [32P]orthophosphate for 3 h in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of U0126 (1 µM). Cells in lanes 1 and 2 were transferred to serum-free medium for 24 h prior to labelling. Cell extracts were prepared and BRF1 was immunoprecipitated with an anti-HA antibody F-7, resolved by SDS–PAGE, transferred to PVDF membrane and visualized by autoradiography (top panel) followed by western blotting with F-7 (lower panel). (D) Phosphorylated BRF1 was quantified and normalized to total immunoprecipitated BRF1. Serum-starved untreated values (lane 1) were assigned as 100. Means and standard deviations from three independent experiments are represented graphically.
The ERK pathway promotes binding of TFIIIB to TFIIIC2 and pol III
For most genes transcribed by pol III, TFIIIB is brought to the promoter by protein–protein interactions with TFIIIC2; in turn, TFIIIB recruits pol III, placing it over the start site so that transcription can commence (Paule and White, 2000; Geiduschek and Kassavetis, 2001). Therefore, TFIIIB needs to bind both TFIIIC2 and pol III. To investigate whether ERK can influence these interactions in vivo, co-immunoprecipitations were carried out from HA.BRF1-expressing cells treated with or without U0126. When an anti-HA antibody was used to immunoprecipitate proteins from serum-starved cells, only a small amount of TFIIIC2 was found bound to BRF1; however, this increased substantially when cells were grown in the presence of serum (Figure 6A). The binding of TFIIIC2 to BRF1 was specific, since an interaction was not observed when an irrelevant control antibody was used. Binding was substantially reduced when these growing cells were incubated for 2 h with the MEK inhibitor (Figure 6A). When normalized to the amount of BRF1 present in each immunoprecipitate, U0126 reduced by 90% the 7.5-fold induction of TFIIIC2–BRF1 binding observed in response to serum (Figure 6B). A similar pattern was found for the interaction between BRF1 and pol III. Growing fibroblasts in the presence of serum resulted in a 15-fold increase of binding of TFIIIB to pol III (Figure 6C and D), and this was reduced by 85% when these cells were incubated with U0126. The effect is specific, since BRF and TBP were found to coprecipitate irrespective of the presence of either serum or the MEK inhibitor (Figure 6E). These data suggest that ERK activity promotes the interaction of TFIIIB with TFIIIC2 and also with pol III in growing cells.

Fig. 6. Inactivation of ERK compromises the serum-stimulated binding of TFIIIB to TFIIIC2 and of TFIIIB to pol III. (A) Rat1A cells stably transfected with pCDNA3HA.BRF1 were incubated in the absence of serum for 24 h (lanes 1 and 2) or were left growing in 5% FCS (lanes 3–5), and were treated with either vehicle (lanes 1, 3 and 5) or U0126 (1 µM, lanes 2 and 4) for 2 h. Cell extracts were immunoprecipitated with anti-TAFI48 antibody M-19 (lane 5) or anti-HA antibody F-7 (lanes 1–4), resolved by SDS–PAGE and analysed by western blotting with either antiserum 4286 against the TFIIICβ subunit of TFIIIC2 (upper panel) or antibody F-7 against the HA tag on transfected BRF1 (lower panel). (B) Coprecipitated TFIIIC2 was quantified by densitometry and normalized against the amount of HA.BRF1. The value for vehicle-treated serum-starved cells was assigned as 100. Values are the means and standard deviations of three experiments. (C) Cell extracts from Rat1A cells treated as in (A) were immunoprecipitated with anti-TAFI48 antibody M-19 (lane 5) or anti-HA antibody F-7 (lanes 1–4), resolved by SDS–PAGE and analysed by western blotting with antiserum BN51 against pol III (upper panel) or antibody F-7 against the HA tag on transfected BRF1 (lower panel). (D) Coprecipitated pol III was quantified by densitometry and normalized against the amount of HA.BRF1. The value for vehicle-treated serum-starved cells was assigned as 100. Values are the means and standard deviations of three experiments. (E) Cell extracts prepared as above were immunoprecipitated with anti-TAFI48 antibody (lane 5) or anti-TBP antibody (lanes 1–4) and analysed by western blotting with anti-HA antibody F-7. (F) Asynchronous Rat1A fibroblasts were treated without (lanes 1 and 3) and with (lanes 2 and 4) PD98059 (50 µM) for 3 h. Association of TFIIICβ, BRF1 and pol III (lower three panels, respectively) with tRNALeu genes was then determined by ChIP, and quantitative PCR was performed with equivalent DNA input amounts determined by PCR on undiluted, 1:5 diluted and 1:25 diluted input chromatin (upper panel). Control ChIPs were carried out in the absence of antibody. PCR products were resolved by agarose gel electrophoresis and revealed by staining with ethidium bromide. The results shown are representative of three independent experiments.
To investigate in proliferating fibroblasts whether blocking ERK activation affects occupancy of a pol III promoter, we performed chromatin immunoprecipitation (ChIP) experiments. Formaldehyde cross-linked soluble chromatin was prepared from asynchronous cells treated for 3 h with vehicle (Figure 6F, lanes 1 and 3) or the MEK inhibitor PD98059 (Figure 6F, lanes 2 and 4). This was normalized for DNA content and immunoprecipitated with antibodies against TFIIIC2 as well as BRF1 or pol III. Quantitative PCR analysis of precipitated DNA showed that TFIIIC2 occupancy of tRNALeu genes is essentially unaffected when ERK activity is blocked (Figure 6F). However, BRF1 ChIP signals from PD98059-treated cells were decreased by 55% compared with untreated cells, suggesting a partial loss of promoter-bound BRF1 when ERK is inactivated. Furthermore, the MEK inhibitor decreased promoter-bound pol III by 80%. The greater decrease in pol III occupancy may reflect the fact that it depends on both TFIIIB–pol III and TFIIIB–TFIIIC2 interactions, both of which are sensitive to ERK activity (Figure 6A and C). Similar results were seen at 5S rRNA genes (data not shown).
Mutating an ERK docking domain or phosphoacceptor site in BRF1 compromises its binding to ERK2 and reduces serum-induced pol III transcription
BRF1 is limiting for pol III transcription in both quiescent and growing fibroblasts (Figure 5A), and appears to be a target for phosphorylation by ERK. The BRF1 sequence contains seven Ser/Thr-Pro sites that might be utilized by ERK. Members of the MAP kinase family interact with their substrates via docking sites called D-domains and FXFP motifs (Holland and Cooper, 1999; Sharrocks et al., 2000). Several such D-domains and one imperfect FXFP motif exist in BRF1. The crystal structure of the BRF1-related factor TFIIB (Tsai and Sigler, 2000) suggests that only one of the potential docking domains may lie near a potential phosphoacceptor site on the surface of BRF1 (Figure 7A). This occurs in the first direct repeat, where a putative docking domain between α-helices 1 and 2 is predicted to be close in space to a Thr/Pro sequence between α-helices 3 and 4. This region of TFIIB adopts a cyclin fold, whilst ERK is related to the cyclin-dependent kinases (Noble et al., 1997). Since a crystal structure is available for the cyclin A–CDK2 complex, we overlaid the cyclin fold of TFIIB onto cyclin A. When these two structures were compared, the potential docking domains superimposed. These modelling data suggested that the putative docking domain in BRF1 (90-RRHIHH LGNQLQL-102) might be a good target for binding ERK.

Fig. 7. Mutating ERK2 docking and phosphoacceptor sites on BRF1 prevent serum induction of pol III transcription. (A) Structure of TFIIB showing where the putative docking domain and phosphoacceptor sites are predicted to lie on BRF1. The docking domain (green) lies on a loop between the first and second α-helices, whilst the phosphoacceptor site (red) lies on the loop between the third and fourth α-helices. (B) 3T3 cells were transiently transfected with 3 µg of pCDNA3HA (lane 1), pBRF1 (lane 2) or pBRF1-L>A (lane 3). Cell extracts were resolved by SDS–PAGE and immunoblotted with antibody F-7 against the HA tag on BRF1 to compare expression levels. (C) 3T3 cells growing in 10% serum were transiently transfected with pVA1 (0.5 µg, all lanes), pCAT (0.5 µg, all lanes), pCDNA3HA vector (3 µg, lane 1), pCDNA3HA.BRF1 (pBRF1) (3 µg, lane 2) or pBRF1-L>A (3µg, lane 3). VA1 and CAT levels were assayed by primer extension and VA1 levels are shown. (D) 3T3 cells growing in 10% serum were transiently transfected for 48 h with pVA1 (0.5 µg, all lanes), pCAT (0.5 µg, all lanes), pCDNA3HA vector (3 µg, lanes 1 and 4; 2 µg, all other lanes), pCDNA3HA.BRF1 (pBRF1) (1 µg, lanes 2 and 5) or pBRF1-L>A (1 µg, lanes 3 and 6). During the final 16 h of transfection, the cells were either maintained in the same medium (lanes 4–6) or in medium containing 0.5% serum (lanes 1–3). VA1 and CAT levels were assayed by primer extension and VA1 levels are shown. In all cases, CAT levels remained constant. (E) VA1 and CAT levels were quantified by PhosphorImager. Values presented graphically are means and standard deviations for VA1 expression after normalization to the levels of CAT RNA to correct for transfection efficiency; the activities obtained for cells transfected with pCDNA3HA alone (lane 1) were set at 100 and other values were calculated as a percentage of this. (F) Extracts from cells transfected as in (B) were immunoprecipitated with anti-TAFI48 antibody M-19 (all panels, lane 4), anti-TBP antibody (lower panel, lanes 1–3), anti-HA antibody F-7 (upper panel, lanes 1–3), or antibody D-2 against ERK2 (middle panel, lanes 1–3). The samples were resolved by SDS–PAGE and analysed by western blotting with antibody F-7 against the HA tag on transfected BRF1. (G) 3T3 cells were transiently transfected for 48 h with pVA1 (0.5 µg, all lanes), pCAT (0.5 µg, all lanes), pCDNA3HA vector (3 µg, lane 1; 2 µg, all other lanes), pCDNA3HA.BRF1 (pBRF1) (1 µg, lane 2), pBRF1-P146A (1 µg, lane 3), pBRF1-T145D (1 µg, lane 4) or pBRF1-T145A (1 µg, lane 5). VA1 and CAT levels were assayed by primer extension and VA1 levels are shown. In all cases, CAT levels remain constant. (H) VA1 and CAT levels were quantified by PhosphorImager. Values from three independent experiments are presented graphically, as before.
To test this, we generated a BRF1 construct where leucines at positions 100 and 102 were mutated to alanine (pBRF1-L>A). The abilities of wild-type or mutant BRF1 to stimulate pol III transcription were compared following transient transfection. Figure 7B shows that equal amounts of the constructs were expressed in transfected cells, indicating that the mutation has not destabilized BRF1. Whereas transfecting cells with the wild-type construct stimulates VA1 transcription by pol III, mutation of the ERK docking domain prevents this increase (Figure 7C). Primer extension of CAT showed that transfection efficiency was the same in all reactions (data not shown). Although unable to support mitogen-activated transcription, pBRF1-L>A can stimulate VA1 expression to a similar extent to wild type when the ERKs are switched off by serum deprivation (Figure 7D and E, lanes 1–3). Therefore, it is primarily the serum-induced increase in pol III activity that is lost when the ERK docking domain is mutated. Co-immunoprecipitation showed that mutation of the docking site in BRF1 substantially reduced its ability to bind ERK2 (Figure 7F, middle panel, lanes 2 and 3). This inability of pBRF1-L>A to support serum-activated transcription was not due to denaturation of BRF1, since the mutant remains able to support basal transcription in the absence of serum (Figure 7D and E, lanes 1–3). Furthermore, co-immunoprecipitation shows that it can still bind TBP (Figure 7F, lower panel). The continued integrity of this mutant is to be expected because of the conservative nature of the L to A substitutions introduced at surface residues.
The effect of mutating the T145 putative phosphoacceptor site in BRF1 was also investigated. This site is predicted to lie between the third and fourth α-helices of the first repeat (Figure 7A), and was mutated to alanine (T145A) or aspartate (T145D) to prevent or simulate phosphorylation, respectively. Whilst the ERK docking domain and T145 site in BRF1 are highly conserved from yeast to humans, the proline at 146 is only found in mammals and hence is unlikely to be important for structure. Therefore, a further construct was made in which the proline, required to specify an ERK site, was mutated to alanine (P146A). These constructs were cotransfected into 3T3 cells along with VA1 and CAT as control. Primer extension analysis shows that the increase in VA1 transcription observed in response to wild-type BRF1 (Figure 7G and H, lane 2) is abolished when the ERK phosphorylation site is mutated to alanine (lane 5) or proline (lane 3). In contrast, the T to D mutation does not prevent VA1 activation (lane 4). Thus, mutation of either a putative ERK docking domain or a phosphoacceptor site in BRF1 significantly reduced pol III activation in fibroblasts.
Discussion
Our data suggest that serum induces an immediate increase in pol III transcription that is regulated by an ERK signalling cascade. Specific inhibitors of the ERK pathway diminish expression of class III genes in proliferating fibroblasts, and pol III transcription in fibroblast extracts is decreased specifically by a peptide inhibitor of ERK. Activation of an estradiol-regulated form of Raf-1 is sufficient to induce pol III transcription in the absence of other mitogens. Manipulating ERK activity in mammalian cells can regulate pol III transcription independently of cyclin D1 levels and phosphorylation of RB. This can be explained by the ability of ERK2 to bind and phosphorylate TFIIIB, thereby enhancing its activity. Mutation of an ERK docking site in the putative cyclin fold domain of the BRF1 subunit of TFIIIB reduces its ability to bind ERK2 and support serum-induced pol III transcription. Furthermore, blocking ERK activity reduces promoter recruitment of BRF1 and pol III without affecting TFIIIC2 binding. Therefore, we suggest that rapid mitogenic induction of pol III transcription is mediated by ERK. Since BRF1 and TFIIIC2 are used by most class III genes, this is likely to be a very general effect. However, 7SK and some U6 snRNA genes may respond differently, since they do not utilize BRF1 or TFIIIC2 (Mital et al., 1996; Teichmann et al., 2000). These genes use BRF2, which does not contain an ERK phosphoacceptor site in its N-terminal region.
Oncogenic Ras has previously been found to stimulate pol III activity through an increase in the level of TBP (Wang et al., 1995). Indeed, a MEK-dependent pathway was shown to induce TBP promoter activity through putative ETS recognition sequences (Johnson et al., 2000). Our data indicate that exposure of fibroblasts to mitogens for less than 24 h does not significantly raise TBP levels. However, there was a slight increase in TBP after this time, in agreement with the previous observations in a different cell type (Wang et al., 1997; Johnson et al., 2000). Changes in the amount of TBP are not responsible for the immediate induction of pol III activity when serum is added to quiescent fibroblasts.
The increase in pol III activity at the G1–S phase transition reflects dissociation of TFIIIB from RB and p130 when these pocket proteins are inactivated through hyperphosphorylation by CDKs (Scott et al., 2001). Although RB and p130 are key regulators of TFIIIB, other control mechanisms also exist since the immediate early induction of pol III transcription precedes hyperphosphorylation of the pocket proteins. Since not all TFIIIB is bound and repressed by RB and p130 (Scott et al., 2001), the pool of free TFIIIB is probably involved in this rapid activation. The immediate early response of pol III transcription to serum coincides with an increase in pol I activity (Stefanovsky et al., 2001). This rapid and concerted increase in pol I and pol III transcription to raise tRNA and rRNA production may be central for regulating protein synthesis and ribosome biogenesis in mammalian cells.
ERK may be a key player in coordinating this process. Upon activation, ERK can translocate to the nucleus to phosphorylate transcription factors, including the pol I factor UBF (Stefanovsky et al., 2001). TFIIIB is another mitogen-regulated transcription factor since ERK phosphorylates its subunit BRF1. Mutation of a putative ERK docking domain on BRF1 impairs the serum-induced rise in pol III transcription. Although docking sites exhibit variability in their distances from phosphoacceptor motifs, in transcription factors these are typically located 40–100 residues downstream (Holland and Cooper, 1999; Sharrocks et al., 2000). In BRF1 there is a Thr/Pro site 45 residues downstream of the docking domain. In the folded protein, this site is likely to lie within close spatial proximity to the ERK docking region, as predicted from the location of equivalent sites in the crystal structure of the related factor TFIIB. It is notable that this region adopts a cyclin fold and therefore may make an excellent site for binding the CDK-related kinase ERK. Although mutating the docking site prevented interaction with the kinase, it did not interfere with the ability of BRF1 to bind TBP, arguing against a general disruption of protein structure. This conclusion is supported by the ability of the mutant to support basal transcription in serum-starved cells.
Our data suggest that ERK phosphorylation of BRF1 stimulates initiation complex assembly. ERK activation enhances interaction between TFIIIB and TFIIIC2 and also between TFIIIB and pol III. This mechanism contrasts with that for activation of pol I transcription; Stefanovsky et al. (2001) proposed that UBF phosphorylation by ERK may release the grip of UBF on DNA to allow promoter clearance. ERK is not the only kinase involved in regulating TFIIIB. Work in both yeast and mammalian cells has shown that CK2 can regulate pol III transcription via an interaction with TFIIIB. In Saccharomyces, only the TBP component of TFIIIB is phosphorylated efficiently by CK2 in vitro; however, all three human TFIIIB subunits could be phosphorylated directly by CK2 in vitro (Ghavidel and Schultz, 1997; Ghavidel et al., 1999; Johnston et al., 2002). Since phosphorylation of BRF1 is only partially reduced by blocking ERK activity in vivo, it is likely that phosphorylation by both CK2 and ERK are required for maximal pol III transcription.
ERK stimulates transcription by both pol I and pol III, and may help to coordinate their activities. In this way, ribosomal production in mammals would be regulated to ensure that levels of large rRNA and 5S rRNA are appropriately balanced, along with supplies of tRNA. It is well established that mitogens trigger a rapid increase in ribosome biosynthesis that is necessary for growth and cell cycle progression (Thomas, 2000; Stefanovsky et al., 2001). Our results provide insight into how this can be achieved in a coordinated manner.
Materials and methods
Cell culture
Rat1A fibroblasts stably overexpressing pCDNA3HA.BRF1 and Balb/C 3T3 mouse fibroblasts were grown in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies Inc.) supplemented with 10% fetal calf serum (FCS), 400 µg/ml Geneticin, 100 IU/ml penicillin and 100 µg/ml streptomycin. Chinese hamster lung fibroblasts CCL39- ΔRaf-1:ER were maintained in H21 medium, as described previously (Lenormand et al., 1996). CHO cells were cultured in Ham’s F12 medium supplemented with 10% heat-inactivated FCS and antibiotics. Primary cultures of wild-type and Rb–/–p130–/–p107–/– (TKO) mouse embryo fibroblasts were grown in BHK-21 medium supplemented with 1 µM sodium pyruvate, 1× non-essential amino acids, 0.1 µM 2-mercaptoethanol, 10% FCS and antibiotics. Proliferation was arrested by reducing serum concentration to 0.5% for 24 h, and mitogenic stimulation was induced with 10% serum.
Transient transfection
Transient transfections used Lipofectamine (Life Technologies Inc.) for CHO cells and Superfect (Qiagen) for fibroblasts. After 48 h, total RNA was extracted using TRI reagent (Sigma) according to the manufacturer’s instructions. It was analysed by primer extension using primers for VA1 (5′-CACGCGGGCGGTAACCGCATG-3′) and CAT (5′-CGATGC CATTGGGATATATCA-3′) as described previously (White et al., 1996).
Phosphate labelling in vivo
Labelling was carried out with 0.5 mCi/ml [32P]orthophosphate for 3 h in phosphate-free medium. After incubation, cells were washed twice in ice-cold phosphate-buffered saline (PBS) and then solubilized in 0.5 ml of IP buffer (50 mM HEPES pH 7.5, 5 mM EDTA, 10 mM NaF, 150 mM NaCl, 25% glycerol, 0.5% Triton X-100, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), 0.5 µg/ml leupeptin, 0.7 µg/ml pepstatin, 0.5 µg/ml aprotinin, 40 µg/ml bestatin, 1 mM sodium vanadate and 50 mM β-glycerophosphate). After 60 min on a rotating wheel, insoluble material was removed by centrifugation at 14 000 g for 15 min prior to immunoprecipitation.
Northern blotting
Northern blotting was carried out as described previously (Cairns and White, 1998). B2 and ARPP P0 probes have been described elsewhere (White et al., 1989; Hurford et al., 1997).
Extracts, recombinant proteins and transcription assays
Whole-cell extracts were prepared for transcription assays using a freeze–thaw procedure described previously (White et al., 1995a). For immunoprecipitations, cells were washed twice with ice-cold PBS and then scraped into IP buffer. After incubation on ice for 15 min, the lysates were cleared by centrifugation at 4°C for 10 min prior to immunoprecipitation.
Bacterially expressed recombinant ERK2 was prepared and activated by MEK as described (Marais et al., 1997). Recombinant BRF1 was expressed in bacteria and purified as described previously (Schramm et al., 2000). Transcription reactions were carried out as described (White et al., 1995a).
In vitro phosphorylation assays
Recombinant BRF1 (100 ng) was phosphorylated by incubating 250 U recombinant ERK2 with 2 µCi of [γ-32P]ATP (3000 µCi/mmol; Amersham) for 30 min at 30°C in 20 mM Tris–HCl pH 7.5, 50 mM KCl, 10 mM MgCl2 and 100 µM ATP. The phosphorylated product was separated by SDS–PAGE and analysed by autoradiography.
Plasmids
pVA1 plasmid contains the adenovirus VA1 gene (Dean and Berk, 1988). pCAT (Promega) contains the CAT gene driven by the SV40 promoter and enhancer. Human HA.BRF1 in the mammalian pCDNA3 expression vector has been described (Sutcliffe et al., 2000). Mutations (T145A, T145D, P146A and pBRF1-L>A) were introduced into pCDNA3HA.BRF1 (designated pBRF1) by PCR using the QuikChange Site-Directed Mutagenesis kit (Stratagene) according to the manufacturer’s instructions, and each was then sequenced. pCMV-RasV12 and pRSV-LTR-RafΔ2–334 were gifts from Dr D.Johnson.
Antibodies and western blotting
Peptide antiserum 4286 against the TFIIICβ subunit of TFIIIC2 has been characterized previously (Sutcliffe et al., 2000). Antiserum against BN51 was generously provided by Michael Ittmann (Ittman et al., 1993). ERK2 antibody D-2, RB antibody C-15, cyclin D1 antibody HD11, TBP antibody 58C9, HA tag antibody F-7 and TAFI48 antibody M-19 were obtained from Santa Cruz Biotechnology. The phospho-RB (ser-795) antibody was purchased from New England Biolabs. Western immunoblots were performed as described previously (White et al., 1995a).
Immunoprecipitation
Cell extract (500 µg) was incubated at 4°C on an orbital shaker with 20 µl of protein A–Sepharose beads carrying equivalent amounts of prebound IgG. Samples were then pelleted, supernatants were removed and the beads were washed five times with 300 µl TBS. The bound material was analysed by western blotting. When cells were labelled with [32P]orthophosphate, immunoprecipitation was carried out as above and the bound material was analysed by both autoradiography and western blotting.
GST pulldown assay
Five hundred micrograms of extract from Rat1A cells stably overexpressing pCDNA3HA.BRF was incubated at 4°C on an orbital shaker with glutathione beads carrying equivalent amounts of immobilized GST, inactive GST-ERK or active GST-ERK. After 3 h, the samples were pelleted, supernatants were removed and the beads were washed five times in 1× TBS. Bound material was analysed by western blotting.
Chromatin immunoprecipitation assay
Asynchronously growing Rat1A fibroblasts treated for 3 h with either vehicle or PD98059 (50 µM) were washed with ice-cold PBS and chromatin immunoprecipitation was performed as previously (Gomez-Roman et al., 2003). Immunoprecipitated DNA was quantitated by PCR performed using previously described primers and amplification procedures (Winter et al., 2000).
Acknowledgments
Acknowledgements
We thank Deborah Johnson for the RasV12 and RafΔ2–334 constructs as well as the BRF1-overexpressing Rat1A cells, Michael Ittmann for antisera against BN51 and Nouria Hernandez for constructs encoding human BRF1. We also thank Jane Endicott for advice in modelling kinase interaction sites. PHS is a Wellcome Trust Research Fellow (fellowship number 055409). This work has been supported by project grant 17/C11067 from the Biotechnology and Biological Sciences Research Council.
References
- Aktas H., Cai,H. and Cooper,G.M. (1997) Ras links growth factor signalling to the cell cycle machinery via regulation of cyclin D1 and the cdk inhibitor p27KIP1. Mol. Cell. Biol., 17, 3850–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhard E.J., Kao,G., Cox,A.D., Sebti,S.M., Hamilton,A.D., Muschel,R.J. and McKenna,W.G. (1996) The farnesyltransferase inhibitor FTI-277 radiosensitizes H-ras-transformed rat embryo fibroblasts. Cancer Res., 56, 1727–1730. [PubMed] [Google Scholar]
- Brandenburger Y., Jenkins,A., Autelitano,D.J. and Hannan,R.D. (2001) Increased expression of UBF is a critical determinant for rRNA synthesis and hypertrophic growth of cardiac myocytes. FASEB J., 15, 2051–2053. [DOI] [PubMed] [Google Scholar]
- Cairns C.A. and White,R.J. (1998) p53 is a general repressor of RNA polymerase III transcription. EMBO J., 17, 3112–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng M., Sexl,V., Sherr,C.J. and Roussel,M.F. (1998) Assembly of cyclin D-dependent kinase and titration of p27KIP1 regulated by mitogen-activated protein kinase kinase (MEK1). Proc. Natl Acad. Sci. USA, 95, 1091–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenberg J.-H., van Rossum,A., Schuijff,L. and te Riele,H. (2000) Ablation of the retinoblastoma gene family deregulates G1 control causing immortalisation and increased cell turnover under growth-restricting conditions. Genes Dev., 14, 3051–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean N. and Berk,A.J. (1988) Ordering promoter binding of class III transcription factors TFIIIC1 and TFIIIC2. Mol. Cell. Biol., 8, 3017–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiduschek E.P. and Kassavetis,G.A. (2001) The RNA polymerase III transcription apparatus. J. Mol. Biol., 310, 1–26. [DOI] [PubMed] [Google Scholar]
- Ghavidel A. and Schultz,M.C. (1997) Casein kinase II regulation of yeast TFIIIB is mediated by the TATA-binding protein. Genes Dev., 11, 2780–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavidel A., Hockman,D.J. and Schultz,M.C. (1999) A review of progress towards elucidating the role of protein kinase CK2 in polymerase III transcription: regulation of the TATA binding protein. Mol. Cell. Biochem., 191, 143–148. [PubMed] [Google Scholar]
- Gomez-Roman N., Grandori,C., Eisenman,R.N. and White,R.J. (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature, 421, 290–294. [DOI] [PubMed] [Google Scholar]
- Holland P.M. and Cooper,J.A. (1999) Protein modification: docking sites for kinases. Curr. Biol., 9, R329–R331. [DOI] [PubMed] [Google Scholar]
- Hurford R.K., Cobrinik,D., Lee,M.-H. and Dyson,N. (1997) pRB and p107/p130 are required for the regulated expression of different sets of E2F responsive genes. Genes Dev., 11, 1447–1463. [DOI] [PubMed] [Google Scholar]
- Ittman M., Ali,J., Greco,A. and Basilico,C. (1993) The gene complementing a temperature-sensitive cell cycle mutant of BHK cells is the human homologue of the yeast RPC53 gene, which encodes a subunit of RNA polymerase C (III). Cell Growth Differ., 4, 503–511. [PubMed] [Google Scholar]
- Johnson L.F., Abelson,H.T., Green,H. and Penman,S. (1974) Changes in RNA in relation to growth of the fibroblast. Amounts of mRNA, rRNA and tRNA in resting and growing cells. Cell, 1, 95–100. [DOI] [PubMed] [Google Scholar]
- Johnson S.A.S., Mandavia,N., Wang,H.-D. and Johnson,D.L. (2000) Transcriptional regulation of the TATA-binding protein by ras cellular signalling. Mol. Cell. Biol., 20, 5000–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston I.M., Allison,S.J., Morton,J.P., Schramm,L., Scott,P.H. and White,R.J. (2002) CK2 forms a stable complex with TFIIIB and activates RNA polymerase III transcription in human cells. Mol. Cell. Biol., 22, 3757–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larminie C.G.C., Cairns,C.A., Mital,R., Kouzarides,T., Jackson,S.P. and White,R.J. (1997) Mechanistic analysis of RNA polymerase III regulation by the retinoblastoma protein. EMBO J. 16, 2061–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larminie C.G.C., Alzuherri,H., Cairns,C.A., McLees,A. and White,R.J. (1998) Transcription by RNA polymerases I and III: a potential link between cell growth, protein synthesis and the retinoblastoma protein. J. Mol. Med., 76, 94–103. [DOI] [PubMed] [Google Scholar]
- Lenormand P., McMahon,M. and Pouyssegur,J. (1996) Oncogenic Raf-1 activates p70 S6 kinase via a mitogen-activated protein kinase-independent pathway. J. Biol. Chem., 271, 15762–15768. [DOI] [PubMed] [Google Scholar]
- Marais R., Light,Y., Paterson,H.F., Mason,C.S. and Marshall,C.J. (1997) Differential regulation of Raf-1, A-Raf and B-Raf by oncogenic Ras and tyrosine kinases. J. Biol. Chem., 272, 4378–4383. [DOI] [PubMed] [Google Scholar]
- Mauck J.C. and Green,H. (1974) Regulation of pre-transfer RNA synthesis during transition from resting to growing state. Cell, 3, 171–177. [DOI] [PubMed] [Google Scholar]
- Mital R., Kobayashi,R. and Hernandez,N. (1996) RNA polymerase III transcription from the human U6 and adenovirus type 2 VA1 promoters has different requirements for human BRF, a subunit of human TFIIIB. Mol. Cell. Biol., 16, 7031–7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagne J. (2000) Genetic and molecular mechanisms of cell size control. Mol. Cell. Biol. Res. Commun., 4, 195–202. [DOI] [PubMed] [Google Scholar]
- Noble M.E.M., Endicott,J.A., Brown,N.R. and Johnson,L.N. (1997) The cyclin box fold: protein recognition in cell cycle and transcription control. Trends Biochem. Sci., 22, 482–487. [DOI] [PubMed] [Google Scholar]
- Paule M.R. and White,R.J. (2000) Transcription by RNA polymerases I and III. Nucleic Acids Res., 28, 1283–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage J., Mulligan,G.J., Attardi,L.D., Miller,A., Chen,S., Williams,B., Theodorou,E. and Jacks,T. (2000) Targetted disruption of the three Rb-related genes leads to loss of G1 control and immortalisation. Genes Dev., 14, 3037–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L., Pendergrast,P.S., Sun,Y. and Hernandez,N. (2000) Different human TFIIIB activities direct RNA polymerase III transcription from TATA-containing and TATA-less promoters. Genes Dev., 14, 2650–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott P.H., Cairns,C.A., Sutcliffe,J.E., Alzuherri,H., McLees,A., Winter,A.G. and White,R.J. (2001) Regulation of RNA polymerase III transcription during cell cycle entry. J. Biol. Chem., 276, 1005–1014. [DOI] [PubMed] [Google Scholar]
- Sethy I., Moir,R.D., Librizzi,M.D. and Willis,I.M. (1995) In vitro evidence for growth factor regulation of tRNA gene transcription in yeast. J. Biol. Chem., 270, 28463–28470. [DOI] [PubMed] [Google Scholar]
- Sharrocks A.D., Yang,S.-H. and Galanis,A. (2000) Docking domains and substrate specificity determination for MAP kinases. Trends Biochem. Sci., 25, 448–453. [DOI] [PubMed] [Google Scholar]
- Stefanovsky V.Y., Pelletier,G., Hannan,R.D., Gagnon-Kugler,T., Rothblum,L.I. and Moss,T. (2001) An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell, 8, 1063–1073. [DOI] [PubMed] [Google Scholar]
- Sutcliffe J.E., Cairns,C.A., McLees,A., Allison,S.J., Tosh,K. and White,R.J. (1999) RNA polymerase III transcription factor IIIB is a target for repression by pocket proteins p107 and p130. Mol. Cell. Biol., 19, 4255–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutcliffe J.E., Brown,T.R.P., Allison,S.J., Scott,P.H. and White,R.J. (2000) Retinoblastoma protein disrupts interactions required for RNA polymerase III transcription. Mol. Cell. Biol., 20, 9192–9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann M., Wang,Z. and Roeder,R.G. (2000) A stable complex of a novel transcription factor IIB-related factor, human TFIIIB50 and associated proteins mediate selective transcription by RNA polymerase III of genes with upstream promoter elements. Proc. Natl Acad. Sci. USA, 97, 14200–14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. (2000) An encore for ribosome biogenesis in the control of cell proliferation. Nat. Cell Biol., 2, E71–E72. [DOI] [PubMed] [Google Scholar]
- Tower J. and Sollner-Webb,B. (1988) Polymerase III transcription factor B activity is reduced in extracts of growth-restricted cells. Mol. Cell. Biol., 8, 1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai F.T. and Sigler,P.B. (2000) Structural basis of preinitiation complex assembly on human pol II promoters. EMBO J., 19, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-D., Yuh,C.-H., Dang,C.V. and Johnson,D. (1995) The hepatitis B virus X protein increases the cellular level of TATA-binding protein, which mediates transactivation of RNA polymerase III genes. Mol. Cell. Biol., 15, 6720–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-D., Trivedi,A. and Johnson,D.L. (1997) Hepatitis B virus X protein induces RNA polymerase III-dependent gene transcription and increases cellular TATA-binding protein by activating the Ras signalling pathway. Mol. Cell. Biol., 17, 6838–6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Stott,D. and Rigby,P.W.J. (1989) Regulation of RNA polymerase III transcription in response to F9 embryonal carcinoma stem cell differentiation. Cell, 59, 1081–1092. [DOI] [PubMed] [Google Scholar]
- White R.J., Gottlieb,T.M., Downes,C.S. and Jackson,S.P. (1995a) Mitotic regulation of a TATA-binding-protein-containing complex. Mol. Cell. Biol., 15, 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Gottlieb,T.M., Downes,C.S. and Jackson,S.P. (1995b) Cell cycle regulation of RNA polymerase III transcription. Mol. Cell. Biol., 15, 6653–6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White R.J., Trouche,D., Martin,K., Jackson,S.P. and Kouzarides,T. (1996) Repression of RNA polymerase III transcription by the retinoblastoma protein. Nature, 382, 88–90. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J. and Davis,R.J. (2000) A central control for cell growth. Nature, 403, 255–256. [DOI] [PubMed] [Google Scholar]
- Winter A.G., Sourvinos,G., Allison,S.J., Tosh,K., Scott,P.H., Spandidos,D.A. and White,R.J. (2000) RNA polymerase III transcription factor TFIIIC2 is overexpressed in ovarian tumours. Proc. Natl Acad. Sci. USA, 97, 12619–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]