

Abstract
As there has been no previous information on the consequences of telomerase expression in genetically altered, mortal cells derived from pre-malignant tissue, we sought to determine the effect of hTERT (human catalytic subunit of telomerase reverse transcriptase) transduction of pre-malignant cell strains from Barrett’s esophagus that do not contain telomerase activity and possess a finite lifespan. Primary cultures of Barrett’s esophageal epithelium transduced with a retrovirus containing hTERT were characterized by growth factor requirements, cytogenetics and flow cytometry. Expression of telomerase lengthened telomeres and greatly extended the lifespan of hTERT transduced (hTERT+) Barrett’s esophagus cells. Growth factor dependency of the hTERT+ cultures remained largely similar to the parental cultures, although there was a modest increase in the ability to grow in agar. Chromosomal instability, measured by both karyotypic and FISH (fluorescence in situ hybridization) analyses, was reduced but not abrogated by hTERT transduction, suggesting that telomerase expression can enhance genomic stability. However, the persistence of residual instability gave rise to new clonal and non-clonal genetic variants, and in one hTERT+ culture a new DNA aneuploid population was observed, the only time such a ploidy shift has been seen in Barrett’s cell strains in vitro. These in vitro observations are analogous to the clinical progression to aneuploidy that often precedes cancer in Barrett’s esophagus, and suggest that reactivation of telomerase may be permissive for continued genetic evolution to cancer. Long-lived Barrett’s esophagus epithelial cultures should provide a useful in vitro model for studies of neoplastic evolution and chemopreventive therapies.
Abbreviations: BPE, bovine pituitary extract; CIN, chromosomal instability; EGF, epidermal growth factor; FBS, fetal bovine serum; hMECs, human keratinocytes and mammary epithelial cells; hTERT, human catalytic subunit of telomerase reverse transcriptase; ITS, insulin-transferrin-selenium; NHDF, normal human diploid fibroblasts; PDL, population doubling level; SKY, spectral karyotyping; SVLT, simian virus 40 large T antigen
Introduction
Introduction of the catalytic unit of human telomerase, hTERT (human catalytic subunit of telomerase reverse transcriptase), into a variety of human cell types typically extends lifespan without in general altering growth factor requirements, tumor-igenicity and chromosomal instability. We have previously established pre-malignant Barrett’s esophagus cell strains containing early genetic changes associated with neoplastic progression in vivo, including inactivation of p53 and p16 (1). These cells have a finite lifespan and do not immortalize spontaneously. This finite lifespan, however, limits the number of cells that can be obtained from culture and complicates the more widespread use of these cells as an in vitro model of Barrett’s esophagus. Expression of telomerase by the forced expression of the reverse transcriptase catalytic subunit (hTERT) allows the extension of the in vitro lifespan of other cell types, including NHDF (normal human diploid fibro-blasts), retinal pigment epithelial (RPE) cells (2) and human endothelial cells (3). Expression of hTERT also confers lifespan extension in human keratinocytes and mammary epithelial cells (hMECs); however, this appears to require the concomitant inactivation of the Rb/p16INK4a pathway (4). The expression of telomerase in both NHDF and RPE cells does not appear to change growth factor requirements, cell cycle checkpoint responses or DNA strand break rejoining activity originally found in the cells prior to hTERT expression (5–7). Expression of telomerase also does not appear to change chromosome stability or confer tumorigenic potential in these cells (2,5,6), although it has been reported that immortalization of hMECs by hTERT can lead to over-expression of the c-myc oncogene (8), a finding that may be related to hTERT induced resistance to TGF-beta in hMECs (9). These studies were all conducted in cell culture systems derived from normal tissues, and we hypothesized that evaluation of hTERT expression in pre-malignant Barrett’s epithelial cell strains might illuminate mechanisms by which telomerase promotes neoplastic evolution.
Thus far, there have been no studies examining the effect of forced hTERT expression in pre-malignant, mortal but chromosomally altered human cells in vitro. Previous work examined the effect of expression of telomerase in cells expressing either SVLT (simian virus 40 large T antigen), E6/E7 and/or H-ras (6,10,11) but not in cells derived from pre-malignant tissue. Retroviral transfection of hTERT into late passage mortal SVLT-Rasval12-transformed human pancreatic cell lines and SVLT-transformed human embryonic kidney cells prevented the onset of crisis (10,12). Altered growth and morphology were observed in both of the hTERT expressing cultures, but the effect on the genetic stability in the cells was not examined. Co-expression of E6/E7 or H-ras with hTERT in NHDF did not confer tumorigenic potential as assessed by growth in soft agar (6). However, co-expression of SVLT and H-ras with hTERT in human embryonic kidney cells or human fibroblasts did increase tumorigenic potential, as assessed by anchorage independent growth in soft agar and tumor formation in nude mice (11). In this study, we investigate the effect of hTERT introduction into Barrett’s epithelial cell strains with regard to altered growth factor requirements and anchorage dependency.
The finite number of cell divisions observed during in vitro cell culture, the Hayflick limit, has been hypothesized to be due to the reduction of telomere length (13). Telomere shortening also appears to contribute to chromosomal instability and rearrangement, due to uncovering of normally protected chromosome ends, which then promotes chromosome recombination and end-to-end associations (14,15). Activation of telomerase has been observed frequently in cancer, including Barrett’s adenocarcinoma (16,17), and the lengthening of telomeres by telomerase might reverse chromosome instability caused by shortened telomeres, allowing continued proliferation of more ‘stabilized’ cells (18). Cytogenetic studies have suggested that lengthening of telomeric sequences can stabilize sites of chromosome breakage (19) or reduce the frequency of dicentric chromosomes (20). We hypothesized that hTERT transduction into pre-malignant Barrett’s epithelial cell strains would lengthen telomeres, enhance cellular lifespan and modulate chromosomal instability. We also wished to establish whether expression of telomerase would lead to phenotypic changes associated with a more malignant phenotype, such as reduced growth factor and anchorage dependency. With answers to these questions, it is possible that hTERT transduced pre-malignant BE epithelial cells could be a valuable in vitro model for pre-clinical studies of Barrett’s esophagus.
Materials and methods
Cell strains and cell culture conditions
Cell strains established previously from Barrett’s esophagus biopsies were maintained in MCDB 153 medium with supplementation that included 5% fetal calf serum, 20 ng/ml EGF (epidermal growth factor) (Gibco, Grand Island, NY), 140 μg/ml BPE (bovine pituitary extract) (Sigma, St Louis, MO), 5 μg/ml insulin, 5 μg/ml transferrin and 5 ng/ml selenium (Sigma), as described previously (1). The four strains utilized are designated as follows: KR-42421 (CP-A); CP-52731 (CP-B); CP-94251 (CP-C) and CP-18821 (CP-D). It has been shown previously that strain CP-A contains wild-type p53, but that the three remaining cultures have p53 LOH and mutation; CP-A, B and C have 9pLOH and all four strains have p16 sequence alterations. 82-6 normal human fibroblasts were grown in Dulbecco’s modified Eagle’s medium with 10% FBS (fetal bovine serum). DLD-1 colon carcinoma cell line were maintained in RPMI containing 16% FBS.
Viral construct and selection of transduced cells
Retroviral transduction was performed using a hTERT retroviral construct or the empty control vector (LXSN) as described previously (4). Cell strains were transduced at the following PDLs (population doubling level) for the selection of clones: CP-A (PDL 22); CP-B (PDL 14); CP-C (PDL 17); CP-D (PDL 13) and at the following PDLs for mass cultures: CP-A (PDL 24); CP-B (PDL 18); CP-C (PDL 17); CP-D (PDL 13). For LXSN+ control clones in chromosome instability studies, CP-A (PDL 15) and CP-C (PDL 17) were transduced with LXSN, selected for resistance to G418, and then directly plated on tissue culture slides at sparse density for individual colony formation.
TRAP (telomere repeat amplification protocol) assay
The TRAP assay was performed as described previously (21). Relative telomerase signal intensity was measured by phosphor imaging analysis.
Telomere hybridization
Cells were plated on slides and lightly fixed in 1% paraformaldehyde for 10 min, followed by 70% ethanol and then air-dried. Hybridization of telomeres was performed with FITC labeled PNA (peptide nucleic acid) probes (Applied Biosystems, Framingham, MA) as described (22). Nuclei were counter stained using TOTO-3 (Molecular Probes, Eugene, OR) and slides were analyzed on a Leica TCS SP confocal microscope (Heidelberg, Germany) using excitation at 488 and 633 nm and emission at 530 (FITC) and 670 nm (TOTO-3).
Telomere restriction fragment length analysis
Average telomere length in cell lines was determined as described previously (23). Blots were hybridized using a 32P-labeled TTAGGG probe, washed and exposed to BioMax film (Eastman Kodak Company, Rochester, NY).
Growth factor requirement assay
3 × 104 cells were each seeded in individual wells of a 24 well tissue culture plate in media containing 0, 10, 50 or 100% of the normal concentrations (see above) of BPE and FBS, and media containing 50 and 100% of the normal concentrations of ITS (insulin-transferrin-selenium) and EGF. At the time of the experiment, the PDL of the parental and hTERT mass cultures, respectively, were as follows: CP-A (PDL 22 and 74.7); CP-B (PDL 8.2 and 86.4); CP-C (PDL 17 and 66.3); CP-D (PDL 13 and 64). Each condition was tested in triplicate wells. Cells were grown and harvested before 100% confluence, from 7 to 12 days post-seeding. Cells were suspended in a small volume of media and counted visually using a hemocytometer.
Anchorage-independent growth assay
Approximately 3.4 × 103 cells were suspended in 0.35% SeaKem GTG agarose (FMC Bioproducts, Rockland, ME) in MCDB media and 5% fetal bovine serum over a 0.5% DNA grade agarose base layer in 6 well tissue culture plates and then fed twice weekly. Colonies of at least 20 mm in diameter (volume 4200 mm3, approximately equivalent to a cluster of eight cells; estimated diameter of one cell is 10 mm for a volume of ~523 mm3) were scored visually under phase contrast and PicoGreen (Molecular Probes) DNA fluorescence microscopy. Control plates were examined after plating but before culture, and confirmed that cell clumps were not present at the outset.
Fluorescence in situ hybridization (FISH)
FISH was performed on 2N and 4N sorted cells as described previously (24), using α-centromeric probes to chromosomes 3, 7, 8 and 18 (Vysis, Downers Grove, IL). At least 200 cells were counted for each probe, except for LXSN transduced short-lived clones, where 100 cells were counted. A copy number was considered modal if it was the most frequently observed number in the group of cells counted.
Cytogenetics
Slides were prepared from colcemid arrested cells and G-banded according to standard cytogenetic methods (25). SKY (spectral karyotyping) was used to identify or confirm the G-band karyotype assignments. Metaphase chromosomes spreads were hybridized with the SKY probe mixture and analyzed using SKYView, imaging software, following the manufacturer’s instructions (Applied Spectral Imaging, Carlsbad, CA). A minimum of five metaphase cells from each cell line was analyzed by SKY. Structural chromosome abnormalities and chromosome gains were considered clonal if the alterations were exhibited in at least two cells from a culture. Whole chromosome losses were considered clonal if observed in at least three cells from a culture.
Flow cytometry
DNA content flow cytometric sorting was used to purify different ploidy populations from samples using a Elite ESP cell sorter (Beckman-Coulter, Fullerton, CA), as described previously (21).
Statistics
To analyze CIN (chromosomal instability) by FISH analysis, the data were transformed to rank, as the percent of FISH abnormal cells had a skewed distribution. ANOVA (analysis of variance) was then used to test the statistical significance of the treatment effects, with the dependent variable being the rank of the percent FISH abnormal cells in a culture, and experimental conditions as independent variables.
Results
Extension of lifespan in Barrett’s cell strains by expression of hTERT
Four Barrett’s esophagus cell strains were retrovirally transduced with hTERT or the empty vector (LXSN) (4). As a control, we attempted to transduce normal esophageal squamous cells but were unable to select any transformants that lived longer than the parental cells (data not shown). This was consistent with the observation that immortalization of certain epithelial cell types requires the inactivation of the p16/Rb pathway (4). We were, however, able to generate hTERT transduced normal human diploid fibroblasts that retained active proliferation at PDLs >100, well in excess of parental fibroblasts (maximum PDL 70).
All hTERT+ cultures showed measurable telomerase activity by the TRAP, whereas none of the parental Barrett’s cell strains showed activity prior to transduction (data not shown). To show that telomerase activity correlated with longer telomeres, TRF (terminal restriction fragment) lengths of parental and hTERT+ cells were measured (23). The TRF of parental cells were 5–6 kb, as compared with ~12 kb for hTERT+ cells (data not shown). TRF provides a conservative estimate of telomere length, as it reflects some subtelomeric sequences as well; FISH with PNA telomere probes showed that the telomere signals in parental cells were at least four to five times dimmer than those in the hTERT+ transductants (Figure 1).
Fig. 1.

Telomere PNA FISH of four Barrett’s epithelial cell cultures before (A, C, E and G) and after (B, D, F and H) transduction with hTERT. Cell strains are CP-D (A and B), CP-C (C and D), CP-B (E and F) and CP-A (G and H). Nuclear fluorescence is shown in blue, telomeres are seen as green dots.
The Barrett’s cell strains have a finite proliferative lifespan, rarely proliferating beyond 30 PDLs (1). No clones from LXSN transduced (LXSN+) cultures were able to proliferate beyond 35 PDLs (range 22–35) whereas all individual hTERT transduced clones have attained at least 69 PDLs (range 69–99) (Table I). Under mass culture conditions, LXSN+ cultures were unable to be passaged beyond a total of 30 PDLs (range 13–30) whereas hTERT+ mass cultures have been maintained for at least 66 PDLs (range 66–92) (Table I). In one case (CP-D), no clones were obtained from the LXSN+ culture, presumably because of the relatively late passage level of the parental strain at the time of infection (infected at PDL 13; average PDL at senescence = 17). In another cell strain, CP-A, infected at late passage (PDL 22; average PDL at senescence = 22), three clones were picked but none grew after selection. The morphology of hTERT+ cells was reminiscent of cells at early passage, being smaller in size with a larger nucleus to cytoplasm ratio and shorter doubling times than LXSN+ cells. This hTERT+ cells morphology persisted even at the latest PDL utilized, as did their more rapid growth rate.
Table I.
hTERT+ and LXSN+ Barrett’s cell growth under clonogenic and mass culture conditions
| Parental
|
Clones
|
Mass cultures
|
||||||
|---|---|---|---|---|---|---|---|---|
| LXSN
|
hTERT
|
LXSN
|
hTERT
|
|||||
| Strain | Max PDL | PDL at infection | No. clones picked | Highest PDLb | No. clones picked | Highest PDLb | Highest PDLb | Highest PDLb |
| CP-A | 24 | 22/24a | 3 | 22 | 16 | 95 | 26 | 78 |
| CP-B | 28 | 14/18 | 14 | 30 | 10 | 99 | – | 92 |
| CP-C | 29 | 17 | 20 | 35 | 17 | 69 | 30 | 66 |
| CP-D | 17 | 13 | 0 | – | 19 | 92 | 13 | 70 |
Represents PDL for clonal and mass culture analysis, respectively.
Highest PDL achieved at the time of writing; includes PDL at time of infection but clones do not include seven to eight PDL accrued to reach usable clone size.
Growth factor requirements of hTERT expressing Barrett’s cells
Although the parental Barrett’s cell strains are mortal, the cells contain genetic changes, including p16 and p53 abnormalities that occur early in neoplastic transformation in vivo (1). These cells do not, however, have many other features commonly associated with transformed cell lines, as they retain stringent growth factor requirements and grow poorly, if at all, in agar. We sought to determine whether expression of telomerase induced phenotypic characteristics of malignant transformation in the Barrett’s cell strains.
We compared the fastidiousness of Barrett’s esophagus hTERT+ mass cultures with non-transduced parental mass cultures by growing the cells in media containing reduced concentrations of FBS, BPE, EGF and ITS. hTERT+ and parental cells showed similar dependency on media growth factors with only a few exceptions. Reduced FBS concentrations greatly inhibited cell proliferation in all cultures, but only CP-D showed any difference between parental and hTERT transduced cells: CP-D hTERT cells had only 14% (P = 0.005) and 47% (P = 0.05) as many cells as the parental strain when grown in 0 and 10% normal FBS, respectively. However, hTERT+ CP-B and CP-C cells showed 34–61% greater cell numbers than parental cells when grown in 0 and 10% of normal concentrations of BPE (P = 0.01–0.001). A 50% reduction in ITS and EGF reduced cell growth by 32 and 45%, respectively, but whereas CP-D cells showed 64% greater growth in reduced EGF (P < 0.001), hTERT+ CP-A and CP-C cells showed 34% (P = 0.008) and 55% (P = 0.001) less growth in reduced EGF, respectively. Only CP-C hTERT cells showed altered growth in reduced ITS compared with parental cells (49% poorer, P = 0.002). Overall, expression of telomerase did not lead to consistent changes in growth factor dependency.
Growth of cells in agar
Colony counts and size measurements are shown in Figure 2. Among the non-transduced Barrett’s cell strains, there was a gradation in the number of colonies observed, with CP-A showing almost no growth in agar and CP-C forming the most colonies. The hTERT+ cultures all formed 3–6-fold more colonies than their corresponding parental strains, a statistically significant difference (two-tailed P = 0.001; Wilcoxon signed rank test). The size of hTERT+ colonies was larger than those formed by parental cells (Figure 2). In all cases, both parental or hTERT+ Barrett’s cell colonies were fewer and smaller than the positive control ras-transformed DLD-1 colon carcinoma cell line colonies; indeed the small size of the Barrett’s cell colonies suggests that these cells are not strictly speaking anchorage independent.
Fig. 2.
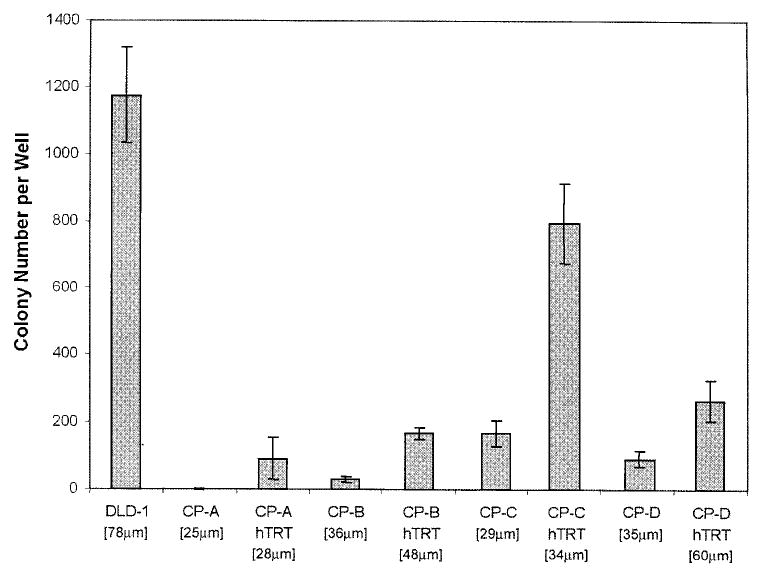
Anchorage independent growth of parental and hTERT transduced Barrett’s cell strains. The number of colonies per well for each strain is shown with error bars representing the standard error of the mean for triplicate wells. The average diameter of colonies for each strain is shown within the brackets. The DLD-1 colon carcinoma cell line served as an anchorage independent positive control.
Effect of hTERT expression on the genetic stability of Barrett’s esophagus cell strains
In Barrett’s esophagus in vivo, aneuploid populations can be detected late in cancer progression (26), on average, 17 months after an elevated G2/4N cell fraction (27). Although DNA aneuploidy is a strong predictor of progression to cancer in Barrett’s esophagus (28), only aneuploid DNA contents above 2.7N are associated with cancer risk (29). Although the cultured Barrett’s esophagus cells all have near-diploid DNA contents with elevated G2/4N fractions (1), we have not observed previously aneuploid populations arising in the parental cultures. Chromosomal instability may, however, precede the detection of aneuploidy, as has been shown in ulcerative colitis and other epithelia (24,30).
The DNA content of parental and hTERT+ cells was analyzed by flow cytometry, with similar ploidy (± 6%) found between hTERT+ and corresponding parental cultures (CP-A, 2.64N; CP-B, 2.27N; CP-C, 2.21N; CP-D clones, 2.43N). However, CP-D hTERT+ cells grown in mass culture exhibited a near-tetraploid aneuploid subpopulation (4.56N) that was not present in the parental culture. The aneuploid population persisted at later passages and therefore appeared to represent a stable population. This is the only example of in vitro generation of a novel DNA aneuploid population that we have encountered in Barrett’s esophagus cells.
As elevated 4N DNA content fractions in vivo are associated with centrosome abnormalities (36), and progression to aneuploidy (27) and cancer (28,29) in Barrett’s esophagus, we measured the proportion of 4N (G2/tetraploid) cells in the parental and hTERT+ cultures (Table II). If shortened telomeres contributed to the accumulation of cells containing a 4N DNA content, expression of telomerase might reduce the proportion of cells in this ploidy fraction. All parental Barrett’s cell strains showed a high 4N fraction, ranging from 22.6 to 33.2% of the total cell population, consistent with previous measurements (1). In three of the four hTERT+ mass cultures the 4N fraction remained elevated at a level similar to that of the corresponding parental culture. The 4N fraction was reduced in all of the hTERT+ clones when compared with the corresponding parental cultures. To distinguish between diploid G2 and tetraploid G1 cells (both have 4N DNA content), we sorted 4N cells from the parental and hTERT+ cultures and used FISH to determine the percentage of tetraploid cells; hTERT transduced cultures retained similar proportions of tetraploid cells as the parental strains (Table II). We conclude that expression of hTERT does not alter the elevated G2/tetraploid phenotype of the BE cell cultures [which is also a phenotype associated with cancer risk in BE patients (28,29)], with the exception of clones, in which we speculate that the reduction of G2/4N cells is due to clonal selection.
Table II.
4N (and tetraploid) fractions of non-transduced parental and hTERT transduced Barrett’s cellsa
| Cell strain | Non-transduced mass culture | hTERT mass culture | hTERT clone 1 | hTERT clone 2 |
|---|---|---|---|---|
| CP-A | 33.2% (20.8%) | 28.6% 27.5%) | 24.6% (25.0%) | 32.3% (20.3%) |
| CP-B | 23% (11.5%) | 10.8% (2.8%) | 13.1% (12%) | 8.7% (5.3%) |
| CP-C | 22.6% (24.3%) | 27.3% (21.3%) | 13.5% (29.5%) | 13.5% (12.0%) |
| CP-D | 30.5% (29.0%) | 28.5% (15.8%) | 24.9% (52.8%) | 17.5% (8.8%) |
4N populations were assessed by flow cytometry. The percentage of the 4N population that was tetraploid was determined by FISH analysis of sorted 4N cells, and is shown in parentheses. The PDL of parental cultures were from 13 to 22 and hTERT+ cultures were from 54 to 83.
Conventional and SKY analyses were performed to identify the structural and numerical chromosome alterations that might be present in the parental and hTERT+ cultures (Table III). Neither parental nor high passage hTERT+ 82-6 human diploid fibroblasts (PDL >100) showed any karyotypic abnormalities by SKY or Giemsa band staining, consistent with a previous report (7). Parental Barrett’s cells exhibited variable degrees of CIN, as evidenced by non-clonal aberrant karyotypes present within the cultures (Table III). CP-A contained minimal clonal karyotypic aberrations, whereas CP-D contained numerous clonal and non-clonal structural and numerical abnormalities (CP-A is the only cell strain derived from tissue obtained from Barrett’s metaplasia rather than high grade dysplasia, and it is also the only strain that does not contain 17p loss of heterozygosity or p53 mutation). In general, the hTERT+ clones contained the same clonal abnormalities as the corresponding non-transduced parental cell strain. However, hTERT+ cells also possessed additional clonal and non-clonal abnormalities not present in the parental cultures. Because clones start, by definition, with a single karyotype, in these cells the non-clonal chromosome aberrations are unambiguous evidence of ongoing genetic instability as cells descend from their progenitor.
Table III.
Karyotypes of parental and hTERT+ cellsa
| Cell culture | PDL | Karyotype (ICSN diagnoses) |
|---|---|---|
| 82-6 mass culture | 38 | 46, XY [10] |
| 82-6 mass culture (hTERT+) | 89 | 46, XY [10] |
| CP-A mass culture | 20 | 47–53, XY, +4, +5, +9, +20, +0–3 mar [cp15] |
| CP-A clone #7 (hTERT+) | 82 | 46–48, XY,i(8)(q10), +20 [cp10] |
| CP-A clone #19 (hTERT+) | 70 | 47–48, XY, +der(1)t(1;6)(q10;?), +20 [8] |
| CP-B mass culture | 15 | 43–45, XY,der(6)t(6;9;12) )(12pter →12p?11.2::6p?11.2 →6q?13::9? →9?),der(9)t(9;14)(q10;q10),−12, der(14)t(6;14)(p12;q?11.2), der(17)t(1;17)(?p31;q10), del(21)(q22)[cp10] |
| CP-B clone #1 (hTERT+) | 99 | 44–46, XY, −4,der(6)t(6;9;12)(12pter →12p?11.2::6p?11.2 →6q?13::9? →9?),der(9)t(9;14)(q10;q10),−12,der(14)t(6;14)(p12;q?11.2), der(17)t(1;17)(?p31;q10),del(21)(q22),+ mar [cp15] |
| CP-B clone #8 (hTERT+) | 91 | 44–46, XY,der(6)t(6;9;12) )(12pter →12p?11.2::6p?11.2 →6q?13::9? →9?), der(9)t(9;14)(q10;q10),−12,der(14)t(6;14)(p12;q?11.2),der(17)t(1;17) (?p31;q10),+18, del(21)(q22)[cp7] |
| CP-C mass culture | 21 | 43–46,XY,der(9)t(9;17)(p10;q10), der(10)t(10;17)(p10;q10), 17, −21,+r,+1–2 mar[cp10] |
| CP-C clone #2 (hTERT+) | 56 | 43–47,XY,der(9)t(9;17)(p10;q10),−17,−21,+r,+1–2 mar[cp8] |
| CP-C clone #11 (hTERT+) | 64 | 42–45,XY,−4,−9,−14,der(9)t(9;17)(p10;q10),−17,−21[cp11] |
| CP-D mass culture | 14 | 39–40,X,-Y,der(1)t(1;22)(p?35;q?12),der(17)t(17;20)(p10;?),−14,−21,−21–22,multiple non-clonal structural (frequently complex) abnormalities, numerical abnormalities and telomeric associations [cp14] |
| CP-D clone #10 (hTERT+) | 86 | 43–45,XY,der(1)t(1;22)(p?35;q?13),del(2)(q10), der(10)t(10;15)(q10;q10), −15,der(17)t(17;20)(p10;?),idic(22)(q13) [cp11] |
| CP-D clone #12 (hTERT+) | 52 | 44–46,XY,der(1)t(1;22)(p?35;q?12),der(3)t(3;10)(q?11;p?26),dup(6) (q?), −10,der(14;15)(q10;q10),der(17)t(17;20)(p?11;?),−19, der(21)t(3;21)(q?12;p12), idic(22)(q13) [cp10] |
The karyotype diagnoses represent the contribution of both G-band and SKY analyses. A ‘?’ indicates a complex chromosomal rearrangement, for which exact breakpoints could not be assigned. Numbers within the brackets represent the number of cells containing the listed karyotype. If the karyotype is the composite from a number cells, this is indicated in brackets by the number of cells prefixed with cp. The clonal aberrations that are not present in the parental culture but are found in the corresponding hTERT+ culture are highlighted in bold type.
Interphase FISH permits examination of larger cell numbers than can be karyotyped; in addition, interphase analysis is useful because some cells with aberrant chromosomes may not be able to enter mitosis, and karyotypic analysis may therefore underestimate the frequency of CIN (32). Centromeric FISH probes to chromosomes 3, 7, 8 and 18 were used to evaluate chromosome copy number in parental and hTERT+ cells (Figure 3), preceded by flow sorting to exclude tetraploid cells. The average variation in chromosome copy number (percent of cells with non-modal FISH spot counts) in the parental and hTERT fibroblast cultures was 1.1 and 1.5%, respectively (probably the background due to variability in probe hybridization and spot identification) in contrast to 7.8 to 19% non-modal cells in the Barrett’s parental mass cultures. Although variable, the hTERT+ cultures had, on average, a significantly lower number of cells with non-modal FISH counts than non-hTERT+ cultures (mean percentage non-modal cells of 7.8% for hTERT+ versus 14.5% in non-hTERT, P = 0.002), suggesting that telomerase expression in the Barrett’s esophagus cultures reduced the amount of CIN as assayed by FISH. Mass cultures had marginally significantly higher numbers of non-modal cells than clonally derived cultures (mean 9.3 versus 11.6%, respectively, P = 0.059). Although chromosomes 3 and 18 had overall higher percentages of non-modal cells than chromosomes 7 and 8 (P = 0.0072), ANOVA showed that the telomerase effect on chromosome instability was chromosome non-specific. Using ANOVA analysis, CIN in mass and clonally derived cultures was directly compared; telomerase reduced the FISH instability in both mass (7.4 versus 14.3%, P = 0.036) and clonally derived cultures (8.6 versus 14.7%, P = 0.0096).
Fig. 3.

Chromosome instability in parental, hTERT+ and LXSN+ cells. The average variation (percent non-modal cells) ± standard error of the mean of parental, hTERT+ and LXSN+ clones and mass cultures are shown. Although the parental strains were not clonogenic under any attempted culture conditions, we were able to grow short-lived LXSN+ clones from two of the cell strains (CP-A and CP-C). 82-6 NHDF cells served as chromosomally stable controls. transduction with hTERT, as the clones tested had undergone at least 40 population doublings after transduction, thus allowing for the possible evolution of cells with reduced anchorage dependency for growth.
Conclusions
Replicative senescence has been proposed to be a mechanism that protects against the evolution of cancer. Limiting the number of population doublings a cell is capable of greatly reduces the probability that the combination of genetic changes needed to form a tumor will occur. The Barrett’s esophagus cells cultured in vitro had short telomeres, and transduction with hTERT led to substantial telomere lengthening. Analysis of chromosomal copy number by FISH demonstrated that chromosomal variability could be reduced by telomere lengthening; however, both karyotypic and FISH analyses indicated that continued genetic evolution of the hTERT+ cells occurred during passaging in vitro. This suggests that CIN is a multifactorial process, in which short telomeres are only one part. The absence of p53 function and abnormally increased numbers of centrosomes are (31) likely to be other such factors.
The new aneuploid population that arose in one hTERT+ mass culture was presumably a consequence of continuing chromosome instability during the extended lifespan of these cells. This hTERT+ culture represents the only new aneuploid population that we have observed to arise during culture of Barrett’s esophagus epithelium. It appears that this observation may parallel neoplastic evolution in vivo, in which aneuploidy follows increased 4N fractions by an average of 17 months (27), and usually precedes the development of esophageal adenocarcinoma (26), which is known to express high levels of hTERT (16,17).
Expression of telomerase in Barrett’s esophagus cells did not appear to give rise to appreciably altered growth factor requirements. However, hTERT transduction enhanced the colony forming efficiency of cells on agar. This is in contrast to the behavior of normal human diploid fibroblasts. The only other report where anchorage independent growth was observed following hTERT transduction was in human embryonic kidney cells and fibroblasts co-expressing SVLT and H-ras (11). We cannot determine whether expression of telomerase per se directly caused a change in the ability of the Barrett’s epithelial cells to grow in agar. Alternatively, this phenotype may have been acquired during culture after
Our work suggests that an important effect of activation of telomerase in neoplastic progression is lifespan extension of cells that are already genetically unstable, thus allowing further cell division and continued genetic evolution towards a malignant phenotype. While stabilization of telomere length may be necessary for progression to cancer (18), neoplastic evolution will not occur without genetic instability, as evidenced by the absence of cancer-associated changes in normal ‘immortalized’ human cells expressing telomerase (2,3,6,7). Our data suggest that although expression of telomerase can reduce the amount of genetic instability present in pre-malignant cells, chromosomal instability can persist at levels that allow continued genetic and karyotypic evolution. The ability to observe this evolution in vitro may provide a useful model of neoplastic progression in Barrett’s esophagus. The extended lifespan of these cells, and their close similarities to the primary cells from which they are derived, should make them a useful model for the study of pre-cancerous Barrett’s esophagus epithelium and for pre-clinical investigations of chemopreventive agents.
Acknowledgments
We thank Sop Chong Kim, Julie Hill and Jennifer Koop for technical help. Grant Support: NIH Grants P01 CA91955, R01 CA61202, T32 AG00057 and R01 CA78855.
References
- 1.Palanca-Wessels MC, Barrett MT, Galipeau PC, Rohrer KL, Reid BJ, Rabinovitch PS. Genetic analysis of long-term Barrett’s esophagus epithelial cultures exhibiting cytogenetic and ploidy abnormalities. Gastroenterology. 1998;114:295–304. doi: 10.1016/s0016-5085(98)70480-9. [DOI] [PubMed] [Google Scholar]
- 2.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 3.Yang J, Chang E, Cherry AM, Bangs CD, Oei Y, Bodnar A, Bronstein A, Chiu CP, Herron GS. Human endothelial cell life extension by telomerase expression. J Biol Chem. 1999;274:26141–26148. doi: 10.1074/jbc.274.37.26141. [DOI] [PubMed] [Google Scholar]
- 4.Kiyono T, Foster SA, Koop JI, McDougall JK, Galloway DA, Klingelhutz AJ. Both Rb/p16INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 5.Jiang XR, Jimenez G, Chang E, Frolkis M, Kusler B, Sage M, Beeche M, Bodnar AG, Wahl GM, Tlsty TD, Chiu CP. Telomerase expression in somatic cells does not induce changes associated with a transformed phenotype. Nature Genet. 1999;21:111–114. doi: 10.1038/5056. [DOI] [PubMed] [Google Scholar]
- 6.Morales CP, Holt SE, Ouellette M, Kaur KJ, Yan Y, Wilson KS, White MA, Wright WE, Shay JW. Absence of cancer-associated changes in human fibroblasts immortalized with telomerase. Nature Genet. 1999;21:115–118. doi: 10.1038/5063. [DOI] [PubMed] [Google Scholar]
- 7.Vaziri H, Squire JA, Pandita TK, Bradley G, Kuba RM, Zhang H, Gulyas S, Hill RP, Nolan GP, Benchimol S. Analysis of genomic integrity and p53-dependent G1 checkpoint in telomerase-induced extended-life-span human fibroblasts. Mol Cell Biol. 1999;19:2373–2379. doi: 10.1128/mcb.19.3.2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Hannon GJ, Beach D. Risky immortalization by telomerase. Nature. 2000;405:755–756. doi: 10.1038/35015674. [DOI] [PubMed] [Google Scholar]
- 9.Stampfer MR, Garbe J, Levine G, Lichtsteiner S, Vasserot AP, Yaswen P. Expression of the telomerase catalytic subunit, hTERT, induces resistance to transforming growth factor beta growth inhibition in p16INK4A (–) human mammary epithelial cells. Proc Natl Acad Sci USA. 2001;98:4498–4503. doi: 10.1073/pnas.071483998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halvorsen TL, Leibowitz G, Levine F. Telomerase activity is sufficient to allow transformed cells to escape from crisis. Mol Cell Biol. 1999;19:1864–1870. doi: 10.1128/mcb.19.3.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Creation of human tumour cells with defined genetic elements. Nature. 1999;400:464–468. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 12.Counter CM, Hahn WC, Wei W, Caddle SD, Beijersbergen RL, Lansdorp PM, Sedivy JM, Weinberg RA. Dissociation among in vitro telomerase activity, telomere maintenance and cellular immortalization. Proc Natl Acad Sci USA. 1998;95:14723–14728. doi: 10.1073/pnas.95.25.14723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allsopp RC, Harley CB. Evidence for a critical telomere length in senescent human fibroblasts. Exp Cell Res. 1995;219:130–136. doi: 10.1006/excr.1995.1213. [DOI] [PubMed] [Google Scholar]
- 14.Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 15.Hande MP, Samper E, Lansdorp P, Blasco MA. Telomere length dynamics and chromosomal instability in cells derived from telomerase null mice. J Cell Biol. 1999;144:589–601. doi: 10.1083/jcb.144.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bachor C, Bachor OA, Boukamp P. Telomerase is active in normal gastrointestinal mucosa and not up-regulated in precancerous lesions. J Cancer Res Clin Oncol. 1999;125:453–460. doi: 10.1007/s004320050302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lord RV, Salonga D, Danenberg KD, et al. Telomerase reverse transcriptase expression is increased early in the Barrett’s metaplasia, dysplasia, adenocarcinoma sequence. J Gastrointest Surg. 2000;4:135–142. doi: 10.1016/s1091-255x(00)80049-9. [DOI] [PubMed] [Google Scholar]
- 18.Artandi SE, DePinho RA. A critical role for telomeres in suppressing and facilitating carcinogenesis. Curr Opin Genet Dev. 2000;10:39–46. doi: 10.1016/s0959-437x(99)00047-7. [DOI] [PubMed] [Google Scholar]
- 19.Meltzer PS, Guan X, Trent JM. Telomere capture stabilizes chromosome breakage. Nature Genet. 1993;4:252–255. doi: 10.1038/ng0793-252. [DOI] [PubMed] [Google Scholar]
- 20.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1929. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wright WE, Shay JW, Piatyszek MA. Modifications of telomeric repeat amplification protocol (TRAP) result in increased reliability, linearity and sensitivity. Nucleic Acid Res. 1995;23:3794–3795. doi: 10.1093/nar/23.18.3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poon SS, Martens UM, Ward RK, Lansdorp PM. Telomere length measurements using digital fluorescence microscopy. Cytometry. 1999;36:267–278. doi: 10.1002/(sici)1097-0320(19990801)36:4<267::aid-cyto1>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 23.Klingelhutz AJ, Barber SA, Smith PP, Dyer K, McDougall JK. Restoration of telomeres in human papillomavirus-immortalized human anogenital epithelial cells. Mol Cell Biol. 1994;14:961–969. doi: 10.1128/mcb.14.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rabinovitch PS, Dziadon S, Brentnall TA, Emond MJ, Crispin DA, Haggitt RC, Bronner MP. Pancolonic chromosomal instability precedes dysplasia and cancer in ulcerative colitis. Cancer Res. 1999;59:5148–5153. [PubMed] [Google Scholar]
- 25.Barsch,M.J., Knutsen,T. and Spurbeck,J.L. (eds) (1997) The AGT Cytogenetics Laboratory Manual, 3rd Edn. Lippencott-Raven Publishers, Phildelphia, pp. 173–197.
- 26.Rabinovitch PS, Reid BJ, Haggitt RC, Norwood TH, Rubin CE. Progression to cancer in Barrett’s esophagus is associated with genomic instability. Lab Invest. 1988;60:65–71. [PubMed] [Google Scholar]
- 27.Galipeau PC, Cowan DS, Sanchez CA, Barrett MT, Emond MJ, Levine DS, Rabinovitch PS, Reid BJ. 17p (p53) allelic losses, 4N (G2/tetraploid) populations and progression to aneuploidy in Barrett’s esophagus. Proc Natl Acad Sci USA. 1996;93:7081–7084. doi: 10.1073/pnas.93.14.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reid BJ, Levine DS, Longton G, Blount PL, Rabinovitch PS. Predictors of progression to cancer in Barrett’s esophagus, baseline histology and flow cytometry identify low- and high-risk patient subsets. Am J Gastroenterol. 2000;95:1669–1676. doi: 10.1111/j.1572-0241.2000.02196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabinovitch PS, Longton G, Blount PL, Levine DS, Reid BJ. Predictors of progression in Barrett’s esophagus III, baseline flow cytometric variables. Am J Gastroenterol. 2001;96:3071–3083. doi: 10.1111/j.1572-0241.2001.05261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hittelman WN, Kim HJ, Lee JS, Shin DM, Lippman SM, Kim J, Ro JY, Hong WK. Detection of chromosome instability of tissue fields at risk, in situ hybridization. J Cell Biochem Suppl. 1996;25:57–62. [PubMed] [Google Scholar]
- 31.Levine DS, Sanchez CA, Rabinovitch PS, Reid BJ. Formation of the tetraploid intermediate is associated with the development of cells with more than four centrioles in the elastase-simian virus 40 tumor antigen transgenic mouse model of pancreatic cancer. Proc Natl Acad Sci USA. 1991;88:6427–6431. doi: 10.1073/pnas.88.15.6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dohner H, Pohl S, Bulgay-Morschel M, Stilgenbauer S, Bentz M, Lichter P. Trisomy 12 in chronic lymphoid leukemias—a meta-phase and interphase cytogenetic analysis. Leukemia. 1993;7:516–520. [PubMed] [Google Scholar]