

Abstract
Transcarboxylase from Propionibacterium shermanii is a 1.2 MDa multienzyme complex that couples two carboxylation reactions, transferring CO2– from methylmalonyl-CoA to pyruvate, yielding propionyl-CoA and oxaloacetate. The 1.9 Å resolution crystal structure of the central 12S hexameric core, which catalyzes the first carboxylation reaction, has been solved bound to its substrate methylmalonyl-CoA. Overall, the structure reveals two stacked trimers related by 2-fold symmetry, and a domain duplication in the monomer. In the active site, the labile carboxylate group of methylmalonyl-CoA is stabilized by interaction with the N-termini of two α-helices. The 12S domains are structurally similar to the crotonase/isomerase superfamily, although only domain 1 of each 12S monomer binds ligand. The 12S reaction is similar to that of human propionyl-CoA carboxylase, whose β-subunit has 50% sequence identity with 12S. A homology model of the propionyl-CoA carboxylase β-subunit, based on this 12S crystal structure, provides new insight into the propionyl-CoA carboxylase mechanism, its oligomeric structure and the molecular basis of mutations responsible for enzyme deficiency in propionic acidemia.
Keywords: carboxyl transferase/crystal structure/domain duplication/multienzyme complex/transcarboxylase
Introduction
Human biotin-dependent carboxylases play central roles in such metabolic pathways as oxidation of odd-chain fatty acids, catabolism of branched amino acids, fatty acid synthesis and gluconeogenesis. Biotin-dependent carboxylases fall into three groups: class I enzymes require ATP, Mg2+ and HCO3–; class II enzymes couple substrate decarboxylation with sodium transport in anaerobes; and class III enzymes, of which Propionibacterium shermanii transcarboxylase (TC) is the only member, couples two carboxylation reactions (Wood and Barden, 1977; Samols et al., 1988; Knowles, 1989). Deficiency of two class I enzymes, pyruvate carboxylase (PC) (Wexler et al., 1994) and propionyl-CoA carboxylase (PCC) (Lamhonwah et al., 1994), can be devastating. Deficiencies of PC, which is the first enzyme in the gluconeogenic pathway and catalyzes the ATP-driven conversion of pyruvate to oxaloacetate, may present as mild lactic acidemia, developmental delay, severe mental retardation or death by 3 months of age (Robinson, 1995). Deficiencies of PCC, which utilizes the product of the final round of β-oxidation of fatty acids, propionyl-coenzyme A (PCoA), to produce methylmalonyl-CoA (MMCoA), may present neonatally as developmental retardation or electroencephalographic (EEG) abnormalities (Fenton and Rosenberg, 1995; Rodríguez-Pombo et al., 1998).
TC has long functioned as a powerful model system for the study of biotin-dependent carboxylases because its subunits share significant sequence homology with the important related human enzymes, are easily isolated and form stable substrate complexes since they have low activity in the absence of the other subunits. TC is a 1.2 MDa multienzyme complex containing 30 polypeptide chains: a catalytic 336 kDa 12S hexameric core; six catalytic 116 kDa 5S dimers; and twelve 12 kDa 1.3S biotinylated linkers (Figure 1A) (Wood and Zwolinski, 1976). The overall TC transcarboxylation reaction consists of two half reactions (Wood and Zwolinski, 1976; Wood, 1979; Wood and Kumar, 1985) (Figure 1B). In the first half reaction, 12S catalyzes COO– transfer from MMCoA to biotin on 1.3S. The second half reaction, catalyzed by 5S, transfers the COO– from the 1.3S biotin to pyruvate. The multienzyme nature of TC, with shuttling of intermediates between different catalytic subunits by a flexible carrier, provides a complex and intriguing target for mechanistic and structural studies. TC thus falls into the general group of multienzyme complexes such as pyruvate dehydrogenase (Coppel et al., 1988; Ho and Patel, 1990; Koike et al., 1990) and glycine decarboxylase (Kume et al., 1991), in which high resolution structures of individual subunits may provide some insight into holo enzyme organization as ambitious efforts continue to crystallize complete multienzyme complexes.
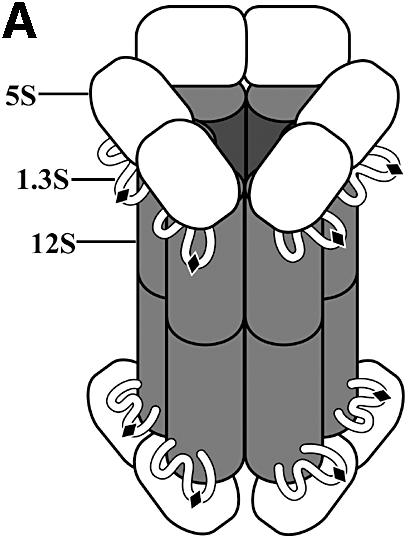

Fig. 1. Transcarboxylase. (A) Electron microscopy-based model (adapted from Wrigley et al., 1977). (B) Half- and full reactions.
TC 12S is especially valuable as a model of the class I human enzymes since their similarity extends beyond biochemical function to multienzyme structure. PCC functions as a dodecamer containing six biotinylated α- and six β-subunits (Fenton and Rosenberg, 1995), and carries out a reaction similar to that of 12S. The human PCC β-chain (PCCβ) shares 50% sequence homology with 12S (Thorton et al., 1993a; Lamhonwah et al., 1994). The carboxyl transferase portion of acetyl-CoA carboxylase, which catalyzes the rate-limiting step in fatty acid synthesis, shares 18% sequence identity with 12S (Abu-Elheiga et al., 1995) and is also predicted to have the same monomer fold. Likewise, TC 5S is functionally and sequentially homologous (27% identity) to the carboxyltransferase portion of human PC, which functions as a homotetramer (Thorton et al., 1993b; Wexler et al., 1994). No high resolution three-dimensional structures are available currently for these related human enzymes.
We have solved the crystal structure of TC 12S bound to its MMCoA substrate at 1.9 Å resolution. This structure identifies active site features important for substrate binding and catalysis, and surfaces which may play a role in holo enzyme assembly. The 12S structure serves as a scaffold for the construction of a molecular model for the homologous human PCCβ, which supports the expectation of conserved active site and mechanism, protein fold and most or all of the oligomerization surfaces. Finally, the homology model allows structural interpretation of PCCβ deficiency disease mutations, reinforcing the value of TC as a model for biotin-dependent carboxylases.
Results and discussion
We have determined two crystal structures of the 336 kDa 12S core subunit of TC. The first, solved by heavy atom phasing methods, showed poor ligand electron density due to substrate degradation. The second, of 12S bound to intact MMCoA, was solved with data from co-crystals re-soaked in artificial mother liquor. This second and more complete 12S–MMCoA–Cd structure is described in the following sections unless the hydrolyzed 12S–CoA–Cd structure is mentioned explicitly.
Overall structure
The crystal’s asymmetric unit contains a ring-shaped 12S–MMCoA–Cd hexamer with overall dimensions of ∼145 × 110 × 50 Å, and with an internal pore diameter of 20 Å (Figure 2A). The 12S subunit has 32 symmetry and contains two stacked trimers, consisting of monomers A–B–C and D–E–F, which are related by 2-fold symmetry. Each monomer folds into two sequential domains: N-terminal domain 1 and C-terminal domain 2. Three cadmium ions and six MMCoA molecules are located between trimers and at the periphery of the ring.

Fig. 2. 12S hexamer. (A) Ribbon diagram viewed down (left) and perpendicular to (right) the 3-fold rotation axis. Monomers are in different colors, MMCoA molecules are in purple space-filling representation, and cadmium ions are yellow spheres. (B) Electrostatic surface of the trimer stacking face (left) and solvent-exposed surface (right).
Hexamer organization
The 12S hexamer can be described as a ring of three dimers, or as two stacked trimeric rings (Figure 2A). Contacts between monomers A and D, B and E or C and F are inter-trimer or opposing monomer interactions. Intra-trimer or adjacent monomer refer to monomers within each trimer (A–B–C or D–E–F). Hexamer stabilization results from a combination of inter- and intra-trimer interactions. Conserved, specific interactions occur at the intersection of dimer pairs (A–D with B–E, B–E with C–F, and C–F with A–D). For example, the contact point of the A–D dimer to the B–E dimer includes four side chain salt bridges: ArgA484 to AspD150, ArgB70 to GluD114, ArgD70 to GluB114 and ArgE484 to AspB150.
The average total buried accessible surface areas between monomers within a ring, and across rings, are 2700 and 9700 Å2, respectively. The interface between the two trimeric rings is very extensive, burying a total of 30 000 Å2 accessible surface area, and gives a high shape complementarity index of 0.72 (Lawrence and Colman, 1993). This inter-trimer interface is composed predominantly of α-helices, and is 65% hydrophobic (Figure 2B). In contrast, the exposed surface of the 12S hexamer is only 47% hydrophobic, and highly negatively charged (Figure 2B), consistent with its calculated net charge at pH 7 of –64. The only prominent exception is the pore through the center of the hexamer, which displays a mixture of hydrophobic and positively charged regions.
Implications for holo enzyme assembly
The striking distribution of surface charges on the 12S hexamer, in combination with calculated net charges at pH 7 of –22 for 5S and –2 for 1.3S, are consistent with the notion that the relatively neutral 1.3S may act in part as a molecular bridge between the very negatively charged 12S and 5S. The first 26 residues of 1.3S are sufficient for holo enzyme assembly (Kumar and Wood, 1982): a peptide consisting of 1.3S residues 1–14 interacts only with 12S, while peptides containing residues 2–26 interact with both 5S and 12S (Kumar et al., 1982). 1.3S residues 1–19 have an intriguing alternating hydrophobicity pattern (MKLKVTVNGTAYDVDVDVD), while residues 20–26 are predominantly hydrophilic (KSHENPM). All the charged amino acids are positively charged in residues 1–12 and negatively charged in residues 13–19; thus the former may interact with some part of the large negatively charged 12S surface while the latter would be more likely to interact with the 12S pore. Residues 20–26 do not have a striking net charge and may interact with some part of 5S. Finally, 1.3S residues 59–78, identified as being transiently associated with both 5S and 12S (Shenoy et al., 1993), contain the three basic residues Lys67, Lys71 and Lys77, which are solvent exposed on the same face of the 1.3S solution NMR structure (Reddy et al., 2000). These positively charged residues would be attracted to both highly negatively charged 12S and 5S surfaces, thus modulating the transient interactions necessary to shuttle between the 12S and 5S active sites.
12S assembly and activity are unaffected by truncation to residue 516, but truncation to residue 507 results in increased lability and decreased rate of assembly into holo TC (Woo et al., 1993). Once assembled, however, 12S(1–507) holo TC has activity and stability similar to those of the wild-type enzyme. In the crystal structure, Tyr507 is located at the C-terminus of an α-helix packing against the adjacent monomer (Figure 3). Intra-chain hydrogen bonding between Tyr507 and Lys510 forms a β-turn that causes the subsequent C-terminus to fold back, leaving residues 509–517 to line the central pore. Residues 518–524 are at the inter-trimer interface and contribute at least eight hydrogen bonds to trimer–trimer stabilization. Truncation to residue 516 would be tolerated since it preserves the substantial hydrophobic interactions stabilizing the inter-trimer interface. In contrast, truncation to residue 507 results in a loss of at least eight intra-trimer hydrogen bonds involving residues 507–514. We speculate that loss of these bonds would lead to a reduced rate of hexamer formation, but for hexamers which do form, the remaining extensive inter-trimer surface would stabilize the complex.

Fig. 3. Contributions of C-terminal residues to intra- and inter-trimer stabilization. (A) Stereo view of interactions involving residues 507–514, with adjacent monomers in blue and green. (B) Stereo view of the interactions involving residues 516–524, with opposing monomers in blue and yellow.
Ion binding
12S–MMCoA crystals are more stable and diffract to higher resolution when grown in the presence of cadmium ions, although 12S is not known to be metal dependent (Ahmad et al., 1972; Wang et al., 2001). In the crystal structure, each Cd2+ is bound by two monomers, and appears to stabilize the trimer–trimer interface. Octahedral coordination is provided by His388 from two opposing monomers, a water-bridging Asp349/Lys391 of the primary monomer and Arg387/Cys524 of the secondary monomer, a second water-bridging Cys524 of the primary monomer and Asp349 of the secondary monomer, as well as two non-bridging waters.
Monomer fold
The six 12S monomers are conformationally very similar, and can be superimposed with an average r.m.s.d. of 0.4 Å for all common Cα atoms. Each monomer is composed of two sequentially folded domains: residues 1–262 form domain 1, while residues 263–524 form domain 2. Each domain has a central seven-stranded β-sheet, with the four middle strands continuing at roughly right angles to form a second β-sheet, which serves as the ligand-binding platform (Figure 4A). All β-strands run parallel, with the exception of strand two in the larger sheet. This central β-sheet region is bracketed by five α-helices. Domain 1 has two additional α-helices at its N-terminus, while domain 2 has an additional α-helix and β-strand at the N-terminus and an additional β-strand at the C-terminus. A flexible loop extending from residues 440 to 470 is disordered in monomers C and D, and stabilized by crystal packing in the other monomers.


Fig. 4. 12S domains. (A) Stereo view superposition of 12S domains: N-terminal domain 1 in blue and C-terminal domain 2 in red. (B) Structure-based sequence alignment of 12S domains 1 (Dom1) and 2 (Dom2), 4-chlorobenzoyl-CoA dehalogenase (PDBid 1NZY), MMCoA decarboxylase from E.coli (PDBid 1EF8 and 1EF9), rat enoyl-CoA hydratase (PDBid 2DUB) and rat dienoyl-CoA isomerase (PDBid 1DCI), with 12S sequence numbers and secondary structure. Residues in the other proteins in structural elements conserved in 12S are in green; amino acids conserved with 12S are underlined. Adenine- and CoA-binding residues are boxed in green and purple, respectively. Amino acids that are reversed relative to the standard CoA-binding motif are boxed in brown. (C) Stereo view superposition of 12S domain 1 (blue) with MMCoA decarboxylase from E.coli (PDB code 1EF9) (black). 12S-bound MMCoA is in space-filling representation.
Domain duplication
Preliminary biochemical studies suggested a 6:12 monomer:MMCoA stoichiometry for 12S (Poto et al., 1978), raising speculations about possible domain duplication. However, these speculations were not pursued due to data reinterpretation in favor of six high affinity and six low affinity sites (G.Kumar, personal communication) along with lack of detectable sequence identity between the N- and C-terminal halves of the 12S sequence. Thus, the revelation of a domain duplication within the 12S monomer is still unexpected. The two 12S domains are structurally very similar, with an r.m.s. fit of 1.6 Å for 175 equivalent Cα atoms (Figure 4A), and provide the basis for a structure-based sequence alignment generating a low 13% sequence identity (Figure 4B). The majority of the residues conserved in domains 1 and 2 are buried and/or participate in conserved intra-monomer hydrogen-bonding interactions; several are found at intra-monomer domain– domain and inter-monomer interfaces. Four pairs of conserved glycines are found at the substrate-binding site (142/373, 181/413, 182/414 and 205/441). The only conserved residue without an obvious important functional or structural role in both domains is Val212/448, which in domain 1 is solvent exposed but in domain 2 packs against MMCoA.
The MMCoA substrate binds predominantly to domain 1. Binding at the corresponding site in domain 2 appears to be unfavorable due to potential steric hindrances, the absence of the CoA-binding motif (discussed below) and the presence of prolines at two positions that in domain 1 provide specific ligand interactions through backbone amide nitrogen atoms. Finally, side chains of domain 2 residues Arg409, Lys410, Tyr412 and Glu436 protrude into the active site and would interfere with ligand binding.
Structural similarity to the crotonase family
A DALI search of known crystal structures (Holm and Sander, 1998) matched the 12S domain fold with that of several CoA-binding proteins. In contrast to the domain-duplicated 12S, these CoA-binding proteins contain a single domain. Superpositions of 12S domain 1 onto the structures of 4-chlorobenzoyl-CoA dehalogenase (Benning et al., 1996), MMCoA decarboxylase (Benning et al., 2000; Figure 4C), enoyl-CoA hydratase (also known as crotonase) (Engel et al., 1998) and dienoyl-CoA isomerase (Modis et al., 1998) give low r.m.s. fit values of 1.7 Å (for 129 Cα atoms), 1.5 Å (124 Cα atoms), 1.6 Å (121 Cα atoms) and 1.4 Å (108 Cα atoms), respectively. From a structure-based sequence alignment, the 12S domain 1 sequence has a low 7–11% identity with the other four CoA-binding enzymes (Figure 4B). The four previously reported CoA-binding protein structures possess the 20 residue enoyl-CoA hydratase/isomerase sequence motif (PROSITE entry PS00166, [LIVM]-[STA]-x-[LIVM]-[DENQRHSTA]-G-x(3)-[AG] (3)-x(4)-[LIVMST]-x-[CSTA]-[DQHP]-[LIVMFY]). The 12S domain 1 contains a similar sequence motif (IAIIAGPCAGGASYSPALTDF, but with the Ala188 and Leu189 sequences in reverse order; this motif is absent in domain 2 (ITVVLRKAYGGSYLAMCNRDL). Thus the 12S domain has a structure and sequence motif very similar to those of the crotonase enzyme family, despite low sequence identity.
Another common feature of 12S and these CoA-binding proteins is that all five form hexamers in solution, and all are crystallized as hexamers with 32 symmetry. Since the crotonase family enzymes have monomers with only one domain, their hexamers are smaller and do not contain the central pore observed in 12S. Three of the four crotonase family structures (4-chlorbenzoyl-CoA dehalogenase, enoyl-CoA hydratase and dienoyl-CoA isomerase) bind substrates at the interface between monomers within the same trimer; MMCoA decarboxylase is the exception, with its substrate-binding site completely defined within each monomer. In contrast, 12S binds its substrate at the interface between monomers in opposing trimers. The four crotonase family hexamers are formed such that the six ligand-binding sites are at the outer faces of the trimers, i.e. away from the trimer–trimer interface. The 12S hexamer is strikingly different since its six substrate-binding sites are located at the trimer–trimer interface (Figure 2A); in this hexamer organization, all 12 potential substrate-binding sites in the 12 domains cannot be occupied simultaneously without substantial steric clashes between pairs of MMCoA molecules.
Active site
The structural similarity between TC 12S and the crotonase family extends beyond the domain fold to the substrate-binding site. In all five proteins, the active site is found near the smaller four-stranded β-sheet. Except for MMCoA decarboxylase, the proteins bind substrate at the interface between monomers (Holden et al., 2001). In 12S, the active site is found where domain 1 of one 12S monomer packs against domain 2 of the opposing monomer from the second trimer (Figure 5A). Approximate 2-fold symmetry brings the two four- stranded β-sheets from these domains 1 and 2 together to form an extended platform; MMCoA binds predominantly to domain 1 of the primary monomer, with its carboxylate leaving group positioned at the interface of the adjacent β-sheets. The flexible (439–475) loop of the secondary monomer’s domain 2, and to a lesser extent the corresponding (204–219) loop of the primary monomer’s domain 1, serve as a canopy for bound substrate. The primary features of the active site include the adenine-binding site (Denessiouk et al., 2001), the oxyanion hole for the MMCoA thioester carbonyl (Babbitt and Gerlt, 1997; Holden et al., 2001) and the binding of the carboxylate leaving group by main chain amides at the N-termini of α-helices.

Fig. 5. MMCoA binding to 12S. (A) Two molecules of MMCoA (purple space-filling) and one cadmium ion (yellow sphere) are at the interface between opposing monomers (yellow and blue). (B) MMCoA (bold) binding in the active site of 12S, with hydrogen bonding interactions (dashed lines) based on distances of 2.4–3.2 Å. (C) MMCoA carboxylate is stabilized by helices starting at residues 414 and 182.
CoA binding
MMCoA is bound to 12S in a U-shape (Figure 5B). Binding of the MMCoA adenine moiety by 12S shows a slight variation of the typical adenine recognition motif (Denessiouk et al., 2001), in that the NH2 group at N(6) can hydrogen-bond to either backbone carbonyls of Gly141 (site III) or Ala143 (site I) (3.0 and 2.9 Å, respectively). In addition, the adenine N(1) hydrogen-bonds to the backbone amide group of Ile145, and a water bridges N(3) and Gln146. Very limited interactions are seen between 12S and the MMCoA phosphates: Arg35 hydrogen-bonds to a P(3) phosphate oxygen and Gln42 is bridged by a water to a P(2) phosphate oxygen.
Oxyanion hole
As has been noted for the four crystallized crotonase superfamily members, the active site contains an oxyanion hole for the thioester carbonyl of the substrate which is composed of main chain nitrogen atoms from His66 and Gly110 in MMCoA decarboxylase (Benning et al., 2000), from Ile117 and Gly173 in dienoyl-CoA isomerase (Modis et al., 1998) and from Ala98 and Gly141 in enoyl-CoA hydratase (Engel et al., 1998). In 12S, the thioester carbonyl group of MMCoA [O(S1)] interacts with an oxyanion hole formed by the main chain nitrogen of Ala143 and a water molecule, which in turn is hydrogen-bonded to the nitrogen atom of Ala183 and the carbonyl group of Ala180 (Figure 5B). This bridging water is held tightly: the six found in the hexamer have an average B-factor of 19 Å2, compared with 38 Å2 for other active site ligand-bound waters, and with 34 Å2 for all 3363 waters. Ala143 is very important in ligand binding: its carbonyl group interacts with the adenine N6 atom, while its nitrogen atom interacts with the MMCoA thioester carbonyl group. Residues Gly182 and Ala183 of the oxyanion hole are at the N-terminus of a conserved α-helix whose dipole moment is expected to play a role in polarizing the thioester carbonyl of the substrate (Benning et al., 1996, 2000; Holden et al., 2001).
Mechanism of carboxyl transferase
In addition to polarization of the MMCoA thioester carbonyl by the 12S oxyanion hole, two helical dipole moments are of interest. MMCoA O(S5) hydrogen-bonds to the backbone nitrogen of Gly182 from the primary monomer, while O(S4) interacts with the backbone nitrogen of Gly414 from the secondary monomer (Figures 5B and C). Interestingly, Gly182 and Gly414 are at the N-termini of helices, which are structurally equivalent when domains 1 and 2 are superimposed, providing a further explanation of why substrate cannot bind to both domains 1 and 2 simultaneously. The glycine interactions orient the carboxylate leaving group approximately perpendicular to the thioester carbonyl (Figures 5C and 6A). These hydrogen bonds, combined with helical dipole moments, may stabilize the negatively charged carboxylate. Conversely, any conformational change that repositions the α-helices will decrease the stability of the carboxylate and may activate it in preparation for transfer to the biotin carrier.
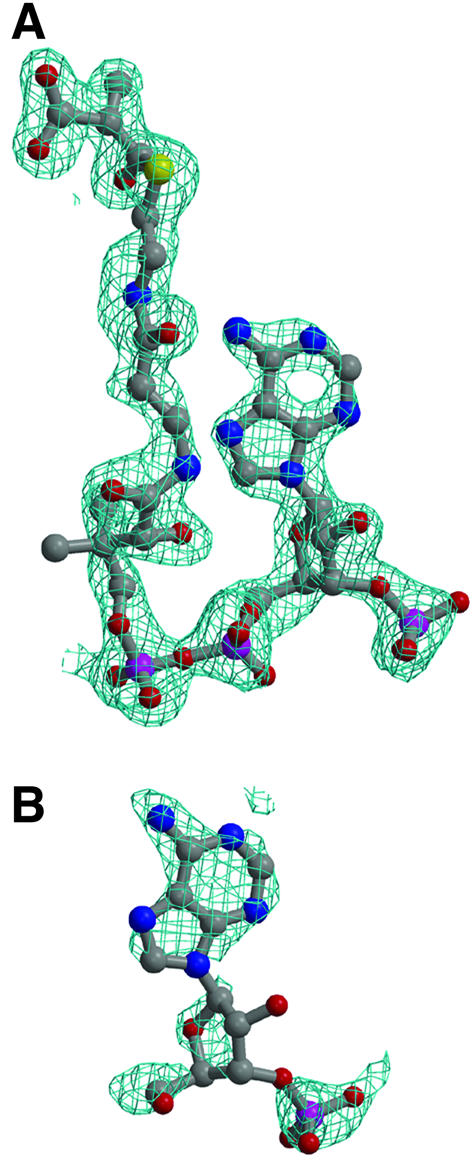
Fig. 6. Simulated annealing omit density (2|Fo| – |Fc| contoured at 1σ) for bound substrate. (A) Intact MMCoA in the 12S–MMCoA–Cd active site. (B) Hydrolyzed MMCoA in the 12S–CoA–Cd crystal.
Two additional waters, bound to MMCoA O(S4) and O(S5), may play a role in catalysis. These waters are observed consistently in the six active sites, and are separated from each other by an average distance of 2.5 Å. The Ile203 carbonyl also interacts with the O(S5)-bound water, and the hydroxyl group of Tyr185 can be swung in to hydrogen-bond with one or both waters with a simple side chain torsional rotation. Activation of a water to abstract the proton from the biotin N1 prior to carboxyl transfer may thus involve either Ile203 or Tyr185. In the 1.3S solution NMR structure, the Lys89 which is biotinylated is located in a surface β-turn (Reddy et al., 2000). Based on considerations for positioning the 1.3S biotin N1 favorably for carboxyl transfer, such as expected proximity to the leaving group, steric accessibility and surface charges (Wood et al., 1963; Knowles, 1989), we have identified access from domain 2 of the active site as a possible direction of biotin approach. An alternative but more restricted approach is via a narrow groove in domain 1 adjacent to the tail of MMCoA.
Comparison of intact and hydrolyzed MMCoA structures
Electron density was clear for the adenine ring, the ribose group and the P(3) phosphate in 12S–CoA–Cd (Figure 6B). No traceable density is clear beyond the P(2) phosphate. Of the adenine N6 interactions with Gly141 and Ala143 observed for the intact MMCoA (Figure 6A), only the latter is also seen for the hydrolyzed MMCoA. In intact MMCoA, N(1) is bound directly to the Ile145 amide, while in hydrolyzed MMCoA an inserted bridging water, which also interacts with the Glu146 side chain, is observed.
In the intact MMCoA structure, residues Ala143, Ala180 and Ala183 are involved in MMCoA thioester interactions. In the hydrolyzed MMCoA structure, no density for the thioester is observed, and these three amino acids interact with the Tyr155 side chain instead, either directly or through a bridging water, which in the intact structure is bound to MMCoA O(S1). Finally, in the absence of the carboxylate leaving group in the hydrolyzed MMCoA structure, the gap between the Gly182 and Gly414 located at the N-termini of domain 1 and domain 2 helices is filled with water molecules. These solvent can interact with Tyr185 upon side chain torsion rotation, as seen for intact MMCoA.
Homology modeling of the propionyl-CoA carboxylase β-subunit (PCCβ)
Human PCC carries out a biotin-dependent carboxylate transfer reaction similar to that catalyzed by 12S (Kaziro and Ochoa, 1964; Mistry and Dakshinamurti, 1964; Northrop, 1969). This nuclear-encoded mitochondrial protein is composed of two subunits, PCCα (72 kDa) and PCCβ (56 kDa), which form an α6β6 heterododecamer (Lamhonwah et al., 1986; Fenton and Rosenberg, 1995). PCCβ contains the CoA-binding site, and its high 50% sequence homology to 12S indicates that the 12S monomeric fold and domain duplication are probably conserved as well (Figure 7A); PCCα has no homology to either of the TC 5S or 1.3S subunits.



Fig. 7. PCCβ homology model. (A) Structure-based sequence alignment of 12S and human PCCβ. Conserved residues are highlighted in black, and sites of PCCβ missense disease mutations are marked with an asterisk (*). The aligned CoA-binding motifs are boxed. (B) Electrostatic surface of the PCCβ homology modeled trimer, for the face involved in 12S inter-trimer packing (left) and for the solvent-exposed surface (right). (C) The PCCβ Arg165Gln, Arg165Trp and Glu168Lys missense mutations. Some or all of the interactions between Arg165 and Glu168, Asp105 would not be possible with Gln165 (cyan), Trp165 (purple) and Lys168 (green). (D) The PCCβ Arg410Trp missense mutation. Interactions with Ser254 in the opposing trimer would not be possible with Trp410 (purple). (E) The PCCβ Thr428Ile missense mutation. The hydrogen bond with Ser443 would not occur with Ile428 (magenta).
Using the 12S fold as a template, we have constructed a homology model of human PCCβ, which is consistent with the expected 6:6 monomer:substrate stoichiometry, and which predicts that substrate will bind primarily to domain 1, as seen in the 12S–MMCoA–Cd crystal structure. The PCCβ domain 1, like that of 12S, contains the 20 residue enoyl-CoA hydratase/isomerase sequence motif with the Ala188 and Leu189 occurring in reverse order, but with a methionine at position five (ISLIMGPCAGGAVYSPALTDF). Since the structural and chemical features of the 12S active site are conserved in the PCCβ homology model, the two enzymes probably have very similar catalytic mechanisms despite TC not sharing the ATP dependence of PCC (whose ATP site is in PCCα). As in 12S, two PCCβ proline residues in the adenine-binding region are predicted to prevent productive substrate binding in domain 2. Also, since two domain 2 basic residues deviate from the isomerase sequence motif (VTVITRKAYGGAYDVMSSKHL), it is unlikely that PCCβ domain 2 will bind any CoA moiety.
Intra-trimer interface residues are mostly conserved in 12S and PCCβ, allowing us to model a similar PCCβ trimer. However, the 12S and PCCβ trimers have dramatically different electrostatic surfaces; thus, it is not obvious from electrostatic considerations which trimer faces pack to form the hexamer. The 12S trimer is very polarized, with a striking negatively charged solvent-exposed surface and a mixed hydrophobic/positively charged inter-trimer packing surface (Figure 2B), while the PCCβ trimer is much less polarized (Figure 7B). The similar 6:6 stoichiometry for PCCβ and 12S would support similar hexamers with active sites at the trimer–trimer interface. However, PCC contains only six biotinylated α-subunits (PCCα), compared with 12 biotinylated 1.3S subunits in TC, and there is no detectable sequence similarity between PCCα and either 1.3S or 5S. It is possible that the PCCβ hexamer may assemble with a ‘tail to tail’ trimer interface rather that the 12S ‘head to head’ interface. The calculated net charges at pH 7 for a PCCα monomer and PCCβ hexamer are –1 and 15, respectively, and very different from those calculated for the three TC subunits, consistent with a potential difference in trimer stacking for the formation of the 12S and PCCβ.
Structural insight into PCCβ disease mutations
PCCβ deficiency results in propionic acidemia, an autosomal recessive disease characterized by accumulation of propionate in the blood, leading to severe metabolic ketoacidosis. Neonatal manifestation of PCCβ deficiency can be life threatening or lead to developmental retardation (Tahara et al., 1990; Fenton and Rosenberg., 1995). Of 20 disease-causing missense mutations resulting in single amino acid changes (Ugarte et al., 1999; http://www.uchsc.edu/sm/cbs/pcc/list_of_pccb_mutations.htm), 14 alter residues conserved in 12S. From the PCCβ homology model, few of the missense disease mutations can be easily predicted to be incompatible with monomer folding, consistent with observations that protein can be detected in patient skin fibroblasts (Chloupkova et al., 2000, 2002; Muro et al., 2001). Instead, most of the mutations are predicted to affect intra-trimer (six mutations) or inter-trimer packing (nine mutations)—thus supporting similar hexamers for 12S and PCCβ—and/or to alter the active site conformation (10 mutations). There does not appear to be any correlation between location or the type of predicted structural consequence and the disease severity observed for the mutation.
Several examples of PCCβ missense mutations that directly or indirectly alter the active site are provided by Arg165Gln/Trp, Glu168Lys, Arg410Trp and Met442Thr. In the PCCβ model, Arg165 and Glu168 form a salt bridge, with Arg165 also interacting with Asp105. The Arg165 backbone atoms line the adenine-binding site, with backbone atoms of the flanking residues directly interacting with adenine N6. The Arg165Gln, Arg165Trp and Glu168Lys mutations would remove some or all of the salt bridge interactions, leading to a change in local conformation (Figure 7C). The larger Arg165Trp side chain is difficult to accommodate and may alter inter-trimer packing as well, consistent with the recombinant mutant showing partial assembly and no defect in specific activity (Chloupkova et al., 2000, 2002). The Arg165Gln side chain can be sterically accommodated, explaining its normal subunit assembly (Muro et al., 2000, 2001); its lack of enzyme activity (Perez-Cerdá et al., 2001) can be explained by the local distortion of active site structure. In the second example, the Arg410Trp mutation also removes an important interaction, in this case with Ser254 in the other trimer. This would alter/disrupt inter-trimer packing and consequently active site formation, since the Arg410 helix is adjacent to the two helices interacting with the MMCoA carboxylate (Figure 7D). In the third case, the Thr428Ile mutation removes an intra-monomer hydrogen bond with Ser443 that would release a restraint on the Ser443-containing helix, whose N-terminus interacts with the MMCoA carboxylate (Figure 7E). Interestingly, the adjacent Met442Thr mutation is expected to have a similar effect: introduction of the branched threonine side chain would be likely to cause a shift in the same helix, thus also altering catalytic activity.
The C-terminal region of PCCβ represents a hot spot of disease mutations since it is altered by the three most common missense mutations (Arg512Cys, Leu519Pro and Asn536Asp), as well as by the Trp531X truncation mutation. Arg512 is located near an intra-trimer interface, forming a salt bridge with Asp325 and two intramolecular hydrogen bonds with main chain oxygen atoms. The Arg512Cys mutation would remove these interactions and alter or interfere with monomer folding, and thus trimer/hexamer formation. Both Leu519Pro and Asn536Asp are also predicted to interfere with hexamer formation: Leu519Pro introduces a kink in a helix at the intra-trimer interface, while Asn536Asp buries a negative charge near Asp212 in the opposing trimer. Finally, the Trp531X truncation removes the (532–539) C-terminus, whose counterpart in 12S wedges between the two domains in the opposing monomer. This truncation removes part of the inter-trimer packing interactions and would compromise hexamer formation. That this PCCβ truncation and most of the missense mutations are predicted to affect either intra-trimer or inter-trimer packing in a PCCβ hexamer with the same trimer stacking as observed for 12S provides some support for conserved hexamer organization for the two proteins.
The TC multienzyme complex has long been a model system for studying the biochemistry of biotin-dependent carboxylation. This 12S–MMCoA–Cd crystal structure reveals an active site and domain fold similarity to the crotonase superfamily, and provides mechanistic implications that extend to human biotin-dependent carboxylases. An analysis of electrostatic surfaces and functional studies provides some understanding of which 12S regions may play a role in holo enzyme assembly; some of these regions, and the 12S domain duplication, appear to be conserved in the human PCCβ enzyme. Structural interpretation of PCCβ deficiency disease mutants is consistent with conserved active site, domain, monomer and trimer structures, and highlights the continued importance of TC as a model biotin-dependent carboxylase.
Materials and methods
Protein expression, purification and complex preparation
TC 12S subunit was expressed and purified as described previously (Woo et al., 1993; Wang et al., 2001). Briefly, Escherichia coli JM109 transformed with a construct encoding the 524 residue 12S polypeptide was grown in YT medium at 37°C overnight before induction with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 6 h. The partially purified lysate from harvested cells was fractionated with 45% ammonium sulfate; the resuspended pellet was subjected to ion exchange and size exclusion columns to obtain pure 12S. The purified protein was concentrated to 10–25 mg/ml in 20 mM potassium phosphate buffer at pH 6.0 containing 1 mM dithiothreitol (DTT) before incubation with 5 mM MMCoA for crystallization. The amino acid sequence of the 12S from the P.shermanii strain used here (Zheng et al., 2002) differs from the sequence originally reported (Woo et al., 1993); the corresponding DNA sequence has been deposited in the EMBL database (ID PS535715).
Crystallization, data collection and processing
Crystals were grown at room temperature by vapor diffusion using equal volumes of the protein sample and well solution containing 19–25% 2-methyl-2,4-pentanediol (MPD), 0.1 M sodium acetate pH 4.5 and with or without 5 mM CdCl2. TC retains catalytic activity at the crystallization pH (1.3 µmol/min/mg at pH 4.5, compared with 14 µmol/min/mg at pH 6.5). The mercury derivative was obtained by adding 0.5 µl of a 100 mM mercury acetate solution to a 6 µl crystallization drop. Crystals were cooled by dunking in liquid nitrogen after stabilizing in a cryoprotectant containing 30% MPD. Diffraction data for crystals of 12S bound to hydrolyzed MMCoA, in the presence and absence of cadmium, were measured on beamline 19-ID at the Advanced Photon Source using 1.0 Å wavelength radiation, while data for a similar crystal derivatized with mercury acetate were measured using an in-house Rigaku R-AXIS IV imaging plate detector mounted on a rotating copper anode source equipped with Yale mirrors. Diffraction data for 12S–MMCoA crystals re-soaked in fresh MMCoA were measured on beamline X9B at the National Synchrotron Light Source, using 1.04 Å wavelength radiation. All data were processed with HKL (Otwinowski and Minor, 1997) and their statistics summarized in Table I.
Table I. Data collection, phasing and refinement statistics.
| 12S–MMCoA–Cda | 12S–CoA–Cda | 12S–CoA–Cd–Hga | 12S–CoA | |
|---|---|---|---|---|
| Data collection |
|
|
|
|
| Space group | C2 | C2 | C2 | C2 |
| Unit cell (Å3) | 114.8 × 200.7 × 146.3 | 113.8 × 200.0 × 145.9 | 115.2 × 201.1 × 146.5 | 115.5 × 201.4 × 146.9 |
| β (°) | 102.4 | 102.97 | 102.4 | 102.7 |
| Wavelength (Å) | 1.04 | 1.00 | 1.54 | 1.00 |
| Resolution (Å) | 20–1.9 (1.97–1.9) | 30–1.9 (1.97–1.9) | 100.0–2.8 (2.9–2.8) | 30–2.1 (2.18–2.1) |
| <I>/<σ(I)> | 15.6 (3.6) | 20.0 (4.9) | 22.3 (5.2) | 16.0 (1.9) |
| Rmerge (%) | 5.7 (26.8) | 5.5 (23.6) | 7.3 (18.7) | 6.5 (34.7) |
| Completeness (%) |
95.4 (97.4) |
92.6 (69.1) |
93.4 (86.7) |
90.2 (58.2) |
| MIR phasing |
|
|
|
|
| Resolution (Å) | 30–2.8 | 30–2.8 | ||
| Number of sites | 3 Cd | 3 Cd | 12 Hg, 3 Cd | |
| Rcullis | 0.86 | 0.871 | ||
| Phasing power | 0.98 | 0.99 | ||
| Mean f.o.m. |
|
|
0.267 (MIRAS) |
0.649 (after DM) |
| Refinement |
|
|
|
|
| Resolution (Å) | 20–1.9 | 30–2.0 | ||
| Rwork, Rfree | 0.168, 0.213 | 0.152, 0.198 | ||
| R.m.s.d. | ||||
| Bond lengths (Å) | 0.0079 | 0.0080 | ||
| Bond angles (°) | 1.41 | 1.50 | ||
| B-factor, mean (Å2) | ||||
| Protein | 21.5 | 19.1 | ||
| Substrate | 36.0 | 77.7 | ||
| Ions | 16.4 | 13.8 | ||
| Other heteroatoms | 42.4 | 25.2 | ||
| Solvent | 33.9 | 31.6 | ||
| R.m.s.d., bonded | 2.4 | 2.3 | ||
| R.m.s.d., angled | 2.9 | 2.8 | ||
| Ramachandran (%) | ||||
| most favored, disallowed | 89.6, 0.0 | 90.0, 0.1 |
Values in parentheses are for the highest resolution shell.
aFor MIRAS phasing, the 12S–CoA–Cd data set was used as native, 12S–CoA as derivative 1 (with negative occupancies) and 12S–CoA–Cd-Hg as derivative 2.
Structure determination and refinement
The positions of three cadmium ions were solved manually from peaks in an isomorphous difference Patterson function; those for 12 mercury ions were determined from cross-difference Fourier calculations. Non-crystallographic 3- and 2-fold axes corresponding to the 32 symmetry within the 12S hexamer in the asymmetric unit were identified by inspection of the cadmium and mercury positions. Heavy atom phasing calculations were with SHARP (de La Fortelle and Bricogne, 1997); density modification, including solvent flattening, histogram matching and non-crystallographic symmetry averaging, were with DM (CCP4, 1994).
Iterative cycles of graphical model building with O (Jones et al., 1991) and refinement calculations with CNS (Brünger et al., 1998) were carried out until convergence. Non-crystallographic symmetry restraints were applied only during initial refinement. Five percent of the reflections were used for Rfree calculations, and DDQ (van den Akker and Hol, 1997) was used to identify local model errors and solvent molecules. During the refinement of the first co-crystal structure, substrate density was clear for only the adenosine moiety; Raman studies of similar crystals showed that the MMCoA substrate had hydrolyzed (Zheng et al., 2002), and thus we designate these crystals 12S–CoA–Cd. For the 12S–MMCoA crystal re-soaked in fresh MMCoA solution, refinement was performed similarly, initially using the protein coordinates for the 12S–CoA–Cd model. Substrate density is complete and well defined for intact MMCoA for this structure, which is designated 12S–MMCoA–Cd and is the focus of the structure description and analysis. For this crystal, residual difference density, unaccounted for by protein, intact MMCoA substrates, cadmium ions or solvent, was interpreted as methylmalonic acid (MM), one of the products of hydrolysis of MMCoA, and MPD. The final refined model for 12S–MMCoA–Cd includes 3093 protein residues: 8–524 for monomer A, 9–524 for monomer B, 9–453 and 460–524 for monomer C, 7–455 and 461–524 for monomer D, 5–524 for monomer E, and 8–524 for monomer F. Also refined were three cadmium ions, six MMCoA, six MM, three MPD and 3363 water molecules. Alternative side chain conformations were refined for 22 amino acids. Refinement statistics are listed in Table I.
Ramachandran analysis using PROCHECK (Laskowski et al., 1993) showed that only one residue is in a disallowed region. The electron density is well defined for this E409Arg residue; its main chain conformation is stabilized by hydrogen bonding interactions between its backbone carbonyl group and the backbone nitrogen of E436Glu, as well as hydrogen bonding between its backbone nitrogen and the backbone carbonyl of E369Val. The six polypeptide chains are very similar in conformation, with an average r.m.s.d. of 0.4 Å calculated for pairwise superposition of their Cα atoms. Since chain E is the most complete in the refined model, and the electron density for its bound MMCoA is intact and well defined, this monomer is used for the detailed description of protein fold and comparison with other structures.
Homology modeling and calculations
A homology model for the β-subunit of human PCC was constructed using an alignment of its sequence with 12S and the InsightII software package (MSI, San Diego). Net molecular charges were calculated with PROTEAN (DNASTAR, Madison), and secondary structure was predicted with SSpro (Baldi et al., 1999). Molecular figures were generated using MOLSCRIPT (Kraulis, 1991), BOBSCRIPT (Esnouf, 1999), Raster3D (Merritt and Bacon, 1997) and GRASP (Nicholls et al., 1991).
Coordinates and structure factors
Coordinates and structure factors for 12S bound to MMCoA and to hydrolyzed MMCoA have been deposited with the RCSB PDB under accession codes 1ON3 and 1ON9, respectively.
Acknowledgments
Acknowledgements
We thank F.van den Akker for many helpful discussions. P.R.H. is partially supported by NIH training grant GM08803. Research in the V.C.Y. and P.R.C. laboratories is funded by NSF MCB0077488 and NIH DK053053, respectively. Diffraction data were measured at NSLS X9B, supported by the NIH and the US Department of Energy, and at APS 19-ID, supported by the US Department of Energy.
References
- Abu-Elheiga L., Jayakumar,A., Baldini,A., Chirala,S.S. and Wakil,S.J. (1995) Human acetyl-CoA carboxylase: characterization, molecular cloning and evidence for two isoforms. Proc. Natl Acad. Sci. USA, 92, 4011–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad F., Lygre,D.G., Jacobson,B.E. and Wood,H.G. (1972) Transcarboxylase: XII. Identification of the metal-containing subunits of transcarboxylase and stability of the binding. J. Biol. Chem., 247, 6299–6305. [PubMed] [Google Scholar]
- Babbitt P.C. and Gerlt,J.A. (1997) Understanding enzyme superfamilies. Chemistry as the fundamental determinant in the evolution of new catalytic activities. J. Biol. Chem., 272, 30591–30594. [DOI] [PubMed] [Google Scholar]
- Baldi P., Brunak,S., Frasconi,P., Pollastri,G. and Soda,G. (1999) Exploiting the past and the future in protein secondary structure prediction. Bioinformatics, 15, 937–946. [DOI] [PubMed] [Google Scholar]
- Benning M.M., Taylor,K.L., Liu,R.-Q., Yang,G., Xiang,H., Wesenberg,G., Dunaway-Mariano,D. and Holden,H.M. (1996) Structure of 4-chlorobenzoyl coenzyme A dehalogenase determined to 1.8 Å resolution: an enzyme catalyst generated via adaptive mutation. Biochemistry, 35, 8103–8109. [DOI] [PubMed] [Google Scholar]
- Benning M.M., Haller,T., Gerlt,J.A. and Holden,H.M. (2000) New reactions in the crotonase superfamily: structure of methylmalonyl-CoA decarboxylase from Esherichia coli. Biochemistry, 39, 4630–4639. [DOI] [PubMed] [Google Scholar]
- Brünger A.T. et al. (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Chloupkova M., Ravn,K., Schwartz,M. and Kraus,J.P. (2000) Changes in the carboxyl terminus of the β subunit of human propionyl-CoA carboxylase affect the oligomer assembly and catalysis: expression and characterization of seven patient-derived mutant forms of PCC in Escherichia coli. Mol. Genet. Metab., 71, 623–632. [DOI] [PubMed] [Google Scholar]
- Chloupkova M. MacLean,K.N., Alkhateeb,A. and Kraus,J.P. (2002) Propionic acidemia: analysis of mutant propionyl-CoA carboxylase enzymes expressed in Escherichia coli. Hum. Mutat., 19, 629–640. [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D, 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Coppel R.L., McNeilage,L.J., Surh,C.D., van de Water,J., Spithill,T.W., Whittingham,S. and Gershwin,M.E. (1988) Primary structure of the human M2 mitochondrial autoantigen of primary biliary cirrhosis: dihydrolipoamide acetyltransferase. Proc. Natl Acad. Sci. USA, 85, 7317–7321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Fortelle E. and Bricogne,G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol., 276, 472–494. [DOI] [PubMed] [Google Scholar]
- Denessiouk K.A., Rantanen,V.V. and Johnson,M.S. (2001) Adenine recognition: a motif present in ATP-, CoA-, NAD-, NADP- and FAD-dependent proteins. Proteins, 44, 282–291. [DOI] [PubMed] [Google Scholar]
- Engel C.K., Kiema,T.R., Hiltunen,J.K. and Wierenga,R.K. (1998) The crystal structure of enoyl-CoA hydratase complexed with octanoyl-CoA reveals the structural adaptations required for binding of a long chain fatty acid-CoA molecule. J. Mol. Biol., 275, 847–859. [DOI] [PubMed] [Google Scholar]
- Esnouf R.M. (1999) Further additions to MolScript version 1.4, including reading and contouring of electron-density maps. Acta Crystallogr. D, 55, 938–940. [DOI] [PubMed] [Google Scholar]
- Fenton W.A. and Rosenberg,L.E. (1995) Disorders of propionate and methylmalonate metabolism. In Scriver,C.R., Beaudet,A., Sly,W.S. and Valle,D. (eds), Metabolic and Molecular Bases of Inherited Disease, 7th edn. McGraw-Hill, New York, NY, pp. 1423–1449.
- Ho L. and Patel,M.S. (1990) Cloning and cDNA sequence of the β-subunit component of human pyruvate dehydrogenase complex. Gene, 86, 297–302. [DOI] [PubMed] [Google Scholar]
- Holden H.M., Benning,M.M., Haller,T. and Gerlt,J.A. (2001) The crotonase superfamily: divergently related enzymes that catalyze different reactions involving acyl coenzyme A thioesters. Acc. Chem Res., 34, 145–157. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander,C. (1998) Touring protein fold space with Dali/FSSP. Nucleic Acids Res., 26, 316–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kaziro Y. and Ochoa,S. (1964) The metabolism of propionic acid. Adv. Enzymol., 26, 283–378. [DOI] [PubMed] [Google Scholar]
- Knowles J.R. (1989) The mechanism of biotin-dependent enzymes. Annu. Rev. Biochem., 58, 195–221. [DOI] [PubMed] [Google Scholar]
- Koike K., Urata,Y., Matsuo,S. and Koike,M. (1990) Characterization and nucleotide sequence of the gene encoding the human pyruvate dehydrogenase α subunit. Gene, 93, 307–311. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr., 24, 946–950. [Google Scholar]
- Kumar G.K. and Wood,H.G. (1982) Intrinsic fluorescence of transcarboxylase during subunit–subunit interactions. Biochem. Int., 4, 605–616. [Google Scholar]
- Kumar G.K., Bahler,C.R., Wood,H.G. and Merrifield,R.B. (1982) The amino acid sequences of the biotinyl subunit essential for the association of transcarboxylase. J. Biol. Chem., 257, 13828–13834. [PubMed] [Google Scholar]
- Kume A., Koyata,H., Sakakibara,T., Ishiguro,Y., Kure,S. and Hiraga,K. (1991) The glycine cleavage system. Molecular cloning of the chicken and human glycine decarboxylase cDNAs and some characteristics involved in the deduced protein structures. J. Biol. Chem., 266, 3323–3329. [PubMed] [Google Scholar]
- Lamhonwah A.-M., Barankiewicz,T.J., Willard,H.F., Mahuran,D.J., Quan,F. and Gravel,R.A. (1986) Isolation of cDNA clones coding for the α and β chains of human propionyl-CoA carboxylase: chromosomal assignments and DNA polymorphisms associated with PCCA and PCCB genes. Proc. Natl Acad. Sci. USA, 83, 4864–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamhonwah A.-M., Leclerc,D., Loyer,M., Clarizio,R. and Gravel,R.A. (1994) Correction of the metabolic defect in propionic acidemia fibroblasts by microinjection of a full-length cDNA or RNA transcript encoding the propionyl-CoA carboxylase β subunit. Genomics, 19, 500–505. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., MacArthur,M.W., Moss,D.S. and Thornton,J.M. (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr., 26, 283–291. [Google Scholar]
- Lawrence M.C. and Colman,P.M. (1993) Shape complementarity at protein–protein interfaces. J. Mol. Biol., 234, 946–950. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Bacon,D.J. (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol., 277, 505–524. [DOI] [PubMed] [Google Scholar]
- Mistry S.P. and Dakshinamurti,K. (1964) Biochemistry of biotin. In Harris,R.S., Wool,I.G., Loraine,J.A., Marrian,G.F. and Thimann,K.V. (eds), Vitamins and Hormones, Vol. 22. Academic Press, New York, NY, pp. 1–55. [DOI] [PubMed]
- Modis Y., Filppula,S.A., Novikov,D.K., Norledge,B., Hiltunen,J.K. and Wierenga,R.K. (1998) The crystal structure of dienoyl-CoA isomerase at 1.5 Å resolution reveals the importance of aspartate and glutamate side chains for catalysis. Structure, 6, 957–970. [DOI] [PubMed] [Google Scholar]
- Muro S., Pérez,B., Rodríguez-Pombo,P., Desviat,L.R., Pérez-Cerda,C. and Ugarte,M. (2000) Mutations affecting the β–β homomeric interaction in propionic acidaemia: an approach to the determination of the β-propionyl-CoA carboxylase functional domains. J. Inherit. Metab. Dis. Dis., 23, 300–304. [DOI] [PubMed] [Google Scholar]
- Muro S., Pérez,B., Desviat,L.R., Rodríguez-Pombo,P., Pérez-Cerda,C., Clavero,S. and Ugarte,M. (2001) Effect of PCCB gene mutations on the heteromeric and homomeric assembly of propionyl-CoA carboxylase. Mol. Genet. Metab., 74, 476–483. [DOI] [PubMed] [Google Scholar]
- Nicholls A., Sharp,K. and Honig,B. (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins, 11, 281–296. [DOI] [PubMed] [Google Scholar]
- Northrop D.B. (1969) Transcarboxylase VI. Kinetic analysis of the reaction mechanism. J. Biol. Chem., 244, 5808–5819. [PubMed] [Google Scholar]
- Otwinowski Z. and Minor,W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Perez-Cerdá C., Clavero,S., Desviat,L.R., Pérez,B., Rodríguez-Pombo,P. and Ugarte,M. (2001) Expression of PCCA and PCCB mutations in PCC-deficient fibroblasts. J. Inherit. Metab. Dis., 24(Suppl. 1), 56. [Google Scholar]
- Poto E.M., Wood,H.G., Barden,R.E. and Lau,E.P. (1978) Photoaffinity labeling and stoichiometry of the coenzyme A ester sites of transcarboxylase. J. Biol. Chem., 253, 2979–2983. [PubMed] [Google Scholar]
- Reddy D.V., Shenoy,B.C., Carey,P.R. and Sonnichsen,F.D. (2000) High resolution solution structure of the 1.3S subunit of transcarboxylase from Propionibacterium shermanii. Biochemistry, 39, 2509–2516. [DOI] [PubMed] [Google Scholar]
- Robinson B.H. (1995) Lactic acidemia (disorders of pyruvate carboxylases, pyruvate dehydrogenase). In Scriver,C.R., Beaudet,A., Sly,W.S. and Valle,D. (eds), Metabolic and Molecular Bases of Inherited Disease, 7th edn. McGraw-Hill, New York, NY, pp. 1479–1499.
- Rodríguez-Pombo P., Hoenicka,J., Muro,S., Pérez,B., Perez-Cerdá,C., Richard,E., Desviat,L.R. and Ugarte,M. (1998) Human propionyl-CoA carboxylase β subunit gene: exon-intron definition and mutation spectrum in Spanish and Latin American propionic acidemia patients. Am. J. Hum. Genet., 63, 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samols D., Thornton,C.G., Murtif,V.L., Kumar,G.K., Hase,F.C. and Wood,H.G. (1988) Evolutionary conservation among biotin enzymes. J. Biol. Chem., 263, 6461–6464. [PubMed] [Google Scholar]
- 4Shenoy B.C., Kumar,G.K. and Samols,D. (1993) Dissection of the biotinyl subunit of transcarboxylase into regions essential for activity and assembly. J. Biol. Chem., 268, 2232–2238. [PubMed] [Google Scholar]
- Tahara T., Kraus,J.P. and Rosenberg,L.E. (1990) An usual insertion/deletion in the gene encoding the β-subunit of propionyl-CoA carboxylase is a frequent mutation in Caucasian propionic acidemia. Proc. Natl Acad. Sci. USA, 87, 1372–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorton C.G. et al (1993a) Primary structure of the monomer of the 12S subunit of transcarboxylase as deduced from DNA and characterization of the product expressed in Escherichia coli.J. Bacteriol., 175, 5301–5308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorton C.G. et al. (1993b) Primary structure of the 5S subunit of transcarboxylase as deduced from the genomic DNA sequence. FEBS Lett., 330, 191–196. [DOI] [PubMed] [Google Scholar]
- Ugarte M. et al. (1999) Overview of mutations in the PCCA and PCCB genes causing propionic acidemia. Hum. Mutat., 14, 275–282. [DOI] [PubMed] [Google Scholar]
- van den Akker F. and Hol,W.G.J. (1999) Difference density quality (DDQ): a method to assess the global and local correctness of macromolecular crystal structures. Acta Crystallogr. D, 55, 206–218. [DOI] [PubMed] [Google Scholar]
- Wang Y.F., Hyatt,D.C., Rivera,R.E., Carey,P.R. and Yee,V.C. (2001) Crystallization and preliminary X-ray analysis of the 12S central subunit of transcarboxylase from Propionibacterium shermanii.Acta Crystallogr. D, 57, 266–268. [DOI] [PubMed] [Google Scholar]
- Wexler I.D. et al. (1994) Primary amino acid sequence and structure of human pyruvate carboxylases. Biochim. Biophys Acta, 1227, 46–52. [DOI] [PubMed] [Google Scholar]
- Woo S.B., Shenoy,B.C., Wood,H.G., Magner,W.J., Kumar,G.K., Beegen,H. and Samols,D. (1993) Effect of deletion from the carboxyl terminus of the 12S subunit on activity of transcarboxylase. J. Biol. Chem., 268, 16413–16419. [PubMed] [Google Scholar]
- Wood H.G. (1979) The anatomy of transcarboxylase and the role of its biotins. CRC Crit. Rev. Biochem., 7, 143–160. [DOI] [PubMed] [Google Scholar]
- Wood H.G. and Barden,R.E. (1977) Biotin enzymes. Annu. Rev. Biochem., 46, 385–413. [DOI] [PubMed] [Google Scholar]
- Wood H.G. and Kumar,G.K. (1985) Transcarboxylase: its quaternary structure and the role of the biotinyl subunit in the assembly of the enzyme and in catalysis. Ann. N. Y. Acad. Sci., 447, 1–21. [DOI] [PubMed] [Google Scholar]
- Wood H.G. and Zwolinski,G.K. (1976) Transcarboxylase: role of biotin, metals and subunits in the reaction and its quaternary structure. CRC Crit. Rev. Biochem., 4, 47–122. [DOI] [PubMed] [Google Scholar]
- Wood H.G., Allen,S.H.G., Stjernholm,R. and Jacobson,B. (1963) Transcarboxylase III. Purification and properties of methylmalonyl-oxaloacetic transcarboxylase containing tritiated biotin. J. Biol. Chem., 238, 547–556. [PubMed] [Google Scholar]
- Wrigley N.G., Chiao,J.-P. and Wood,H.G. (1977) Electron microscopy of the large form of transcarboxylase with six attached subunits. J. Biol. Chem., 252, 1500–1504. [PubMed] [Google Scholar]
- Zheng X., Rivera-Hainaj,R.E., Zheng,Y., Pusztai-Carey,M., Hall,P.R., Yee,V.C. and Carey,P.R. (2002) Substrate binding induces a cooperative conformational change in the 12S subunit of transcarboxylase: Raman crystallographic evidence. Biochemistry, 41, 10741–10746. [DOI] [PubMed] [Google Scholar]