

Abstract
A novel class of antibiotics based on the antimicrobial properties of immune peptides of multicellular organisms is attracting increasing interest as a major weapon against resistant microbes. It has been claimed that cationic antimicrobial peptides exploit fundamental features of the bacterial cell so that resistance is much less likely to evolve than in the case of conventional antibiotics. Population models of the evolutionary genetics of resistance have cast doubt on this claim. We document the experimental evolution of resistance to a cationic antimicrobial peptide through continued selection in the laboratory. In this selection experiment, 22/24 lineages of Escherichia coli and Pseudomonas fluorescens independently evolved heritable mechanisms of resistance to pexiganan, an analogue of magainin, when propagated in medium supplemented with this antimicrobial peptide for 600–700 generations.
Keywords: antibiotic resistance, antimicrobial peptides, Escherichia coli, Pseudomonas fluorescens, experimental evolution, selection experiment
1. Introduction
The evolution of resistance to every widely used commercial antibiotic has gone a long way toward annulling recent advances in antibacterial chemotherapy. In the past 50 years, resistance to every new antibiotic has appeared in microbial populations within a few years of its introduction (Palumbi 2001). The decline in the effectiveness of current therapies has led to a search for new kinds of agent, including antibiotics based on cationic antimicrobial peptides, which are part of the innate immune system of all multicellular organisms (Hancock et al. 1995; Andreu & Rivas 1998; Zasloff 2002; Reddy et al. 2004; Toke 2005).
Cationic antimicrobial peptides are translated using the ribosomes, in the usual fashion of protein synthesis, and are, therefore, referred to as RAMPs, for ribosomally synthesized antimicrobial peptides (Hancock & Chapple 1999). The RAMPs are gene-encoded polypeptides composed of fewer than 100 amino acids (Huang 2000) and have a net positive charge conferred by lysine and arginine residues (Hancock 2001). The selective toxicity of RAMPs toward bacterial cells is primarily due to an initial electrostatic interaction between the peptide and the anionic phospholipid headgroups in the outer leaflet of the bacterial cytoplasmic membrane (Zasloff 2002; Yeaman & Yount 2003). Although RAMPs are generally thought to act by disrupting membrane integrity, their detailed modes of actions are varied and have been extensively reviewed in Yeaman & Yount (2003), Brogden (2005) and Lohner & Blondelle (2005).
Molecular mechanisms of resistance to RAMPs have been identified in several bacterial groups (Devine & Hancock 2002; Bell & Gouyon 2003; Yeaman & Yount 2003) but may be exceptional because of the fundamental changes in membrane structure that would be needed to confer effective resistance (Zasloff 2002). Host defence peptides have remained effective against bacterial infections for at least 108 years, despite the continual presence of these peptides in bacterial environments, and we may infer that resistance is very unlikely to evolve in the short term (Zasloff 2002). The great diversity of RAMPs, the presence of several different kinds at the infection site and their different modes of action might have impeded the evolution of resistance in natural bacterial populations. Evolutionary biologists have argued, however, that the therapeutic use of particular RAMPs would alter natural environments by creating a source of specific and continued selection that might readily lead to the evolution of resistance (Bell & Gouyon 2003). This is of particular concern because of the possibility of cross-resistance to human defence peptides.
Resistance to RAMPs has been identified as a necessary component of pathogenesis in certain bacteria (Groisman et al. 1992; Peschel & Collins 2001; Fedtke et al. 2004) and has been constructed in the laboratory (Guo et al. 1998; Tamayo et al. 2005). There is no adequate experimental investigation, however, of the evolutionary response of populations to prolonged exposure to a therapeutic dose. Previous selection experiments (Hancock 1997; Ge et al. 1999b) have considered only brief episodes of a few generations of selection; resistance to conventional antibiotics may appear on this time scale, but the evolution of resistance through cumulative change involving several loci cannot be excluded.
The objective of this study was to determine whether the resistance to a RAMP would evolve under continued selection. We designed a selection protocol, where clonal lineages of Pseudomonas fluorescens and Escherichia coli were grown for several hundred generations in media containing increasing concentrations of a RAMP. Our experiment used isogenic lines where resistance can evolve only through the appearance, spread and accumulation of novel chromosomal mutations.
2. Material and methods
(a) Organisms and establishment of populations
We used the gram-negative bacteria P. fluorescens and E. coli to establish clonal lineages of isogenic non-mutator and mutator strains. We employed mutator genotypes because bacteria with abnormally high mutation rates are frequently isolated from clinical infections and this may increase the rate of adaptation to antimicrobial agents (Miller et al. 2002; Chopra et al. 2003). Paul Rainey (University of Auckland) provided P. fluorescens non-mutator strain SBW25 (Lilley & Bailey 1997) and Craig Maclean (McGill University) constructed the otherwise isogenic mutator strain SBW25 Δmuts (Craig Maclean, unpublished). E. coli non-mutator strain PS8, a clonal isolate of REL606 (Sniegowski et al. 1997), and mutator strain PS2350, a mutL genotype (Shaver & Sniegowski 2003), were provided by Paul D. Sniegowski (University of Pennsylvania). In both cases, a defect in the methyl-directed mismatch repair mechanism confers an elevated mutation rate by increasing the occurrence of point mutations after replication by a factor of about 100.
(b) Antimicrobial agent
The selective agent we used is the cationic antimicrobial peptide pexiganan (Gly–Ile–Gly–Lys–Phe–Leu–Lys–Lys–Ala–Lys–Lys–Phe–Gly–Lys–Ala–Phe–Val–Lys–Ile–Leu–Lys–Lys–NH2) as described by Ge et al. (1999a). Pexiganan is an analogue of magainin, an antimicrobial peptide isolated from Xenopus laevis (Zasloff 1987), that is synthetically modified to be used in clinical chemotherapy (Ge et al. 1999a). Stock solutions were filter-sterilized before use. Pexiganan was synthesized and purified as described (Ge et al. 1999a).
(c) Selection experiment
We established for each species six non-mutator and six mutator lines growing in the presence of pexiganan, hereafter referred to as positive selection lines. In addition, we established in both species two non-mutator and two mutator lineages growing in an environment devoid of pexiganan for the entire duration of the experiment, here referred to as control selection lines. Cultures were grown in Mueller–Hinton Broth (MHB; BD Microbiology Systems, Cockeysville, MD) in shaking incubators at 28 °C (P. fluorescens) and 37 °C (E. coli). We transferred lines daily by inoculating 50 μl of stationary phase culture to 5 ml MHB, to give a daily dilution of 100, or about 6–7 doublings. We preserved the ancestral clones frozen at −80 °C in 25% (v/v) glycerol. All lines were initially grown in a pristine environment free of pexiganan for 20 transfers. At transfer 21, we supplemented the medium of the positive selection lines with a non-inhibitory concentration of pexiganan (0.5 μg ml−1) determined by Ge et al. (1999b). Afterwards, we doubled the concentration of pexiganan every 10 transfers, or fewer if the selection lines showed vigorous growth. Every time we increased the concentration of pexiganan, a sample of each line was inoculated to 25% (v/v) glycerol and stored at −80 °C. The experiment was conducted for 100 serial transfers, constituting 600–700 bacterial generations.
We measured the optical density (OD660) of each selection line daily from 200 μl inocula for E. coli or 50 μl aliquots diluted in 150 μl of MHB for P. fluorescens in 96 well microtitre plates using the narrow-beam Universal Microplate Reader (BioTek Instrument, Winooski, VT, USA). The scores for each well were corrected to that of a blank and were considered as a measure of growth of the bacterial population.
(d) In vitro susceptibility testing
We assayed the resistance evolved by measuring the minimal inhibitory concentration (MIC) of pexiganan for each selection line, using samples of the whole population rather than isolated colonies. The method was modified from the broth macrodilution guidelines published by the National Committee for Clinical Laboratory Standards (NCCLS 2003). We measured the growth of each selection line in a serial dilution of twelve pexiganan concentrations. For this, we prepared 5 ml dilutions of pexiganan in unsupplemented MHB and inoculated the vials with 50 μl of stationary phase culture of each line. The growth of these cultures was then measured as previously described after incubation for 24 h. The MIC50 of each culture was estimated as the lowest concentration of pexiganan that reduced growth by 50% or more.
We measured the initial MIC50 values (before selection) for all 32 selection lines from cultures of transfer 20 tested on a dilution series ranging from 0 to 512 μg of pexiganan per ml. Cultures were first reconditioned by two growth cycles in unsupplemented medium. The final MIC50 values (after selection) were measured from fresh stationary phase cultures on a dilution series ranging from 0 to 1024 μg of pexiganan per ml. Since NCCLS does not define standard breakpoints for isolates resistant to RAMPs, we identified the evolution of resistance as a significant increase of MIC50 in positive selection lines compared to that of control selection lines (see Martinez & Baquero 2000).
(e) Stability of resistance
To verify the heritable nature of the evolved resistance, we reconditioned all control and selection lines from frozen stocks and took each through four transfers (about 25–30 generations) in unsupplemented MHB. We then measured growth of two replicates in unsupplemented medium, and in medium containing concentrations of pexiganan near the lower and upper limits of tolerance as established in previous assays.
(f) Cost of resistance
To evaluate the indirect response to selection, we estimated the limiting density, the rate of increase and the lag phase of two replicates of each line of Pseudomonas in unsupplemented medium and in medium containing pexiganan at the MIC.
3. Results
(a) Initial levels of resistance
The level of resistance in the ancestral clones, measured at transfer 20 before any selection, was concordant with previously published MICs of clinical isolates. We estimated MIC50 for the 16 P. fluorescens ancestral lines to range from 2 to 8 μg ml−1 in the non-mutator strain and from 16 to 32 μg ml−1 in the mutator strain. Ge et al. (1999b) reported MIC values of pexiganan from 35 clinical Pseudomonas spp. isolates to range from 4 to 16 μg ml−1. The 16 E. coli ancestral lines had MIC50 between 32 and 128 μg −1 in the non-mutator strain and between 16 and 256 μg ml−1 in the mutator strain. Ge et al. (1999a) first reported E. coli MICs from 53 clinical isolates to range from 4 μg ml−1 to values in excess of 256 μg ml−1. In a second publication, Ge et al. (1999b) reported MIC values from 137 E. coli clinical isolates to range from 2 to 64 μg ml−1.
The positive selection lines and control selection lines had statistically indistinguishable MIC50 at transfer 20 in P. fluorescens (figure 1) non-mutator (variance-ratio test F=3.0; degrees of freedom d.f.=1, 6; p>0.50) and mutator strains (F<1; d.f.=1, 6; p>0.5) and in E. coli (figure 2) non-mutator (F=1.035, d.f.=1, 6, p>0.50) and mutator strains (F<1; d.f.=1, 6; p>0.50).
Figure 1.
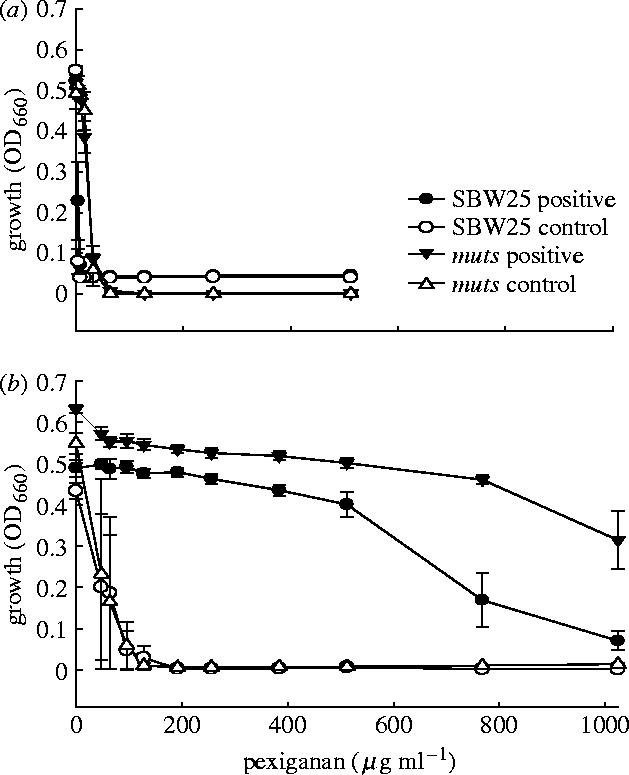
Growth of Pseudomonas fluorescens in relation to concentration of pexiganan. Mean (±s.e.) growth is measured as (a) absorbance before selection and (b) after selection and presents data from positive selection lines and control selection lines for wild-type (SBW25) and mutator (SBW25 Δmuts) genotypes. Note that lineages were not assayed at concentrations above 512 μg ml−1 before selection, since growth was totally inhibited.
Figure 2.
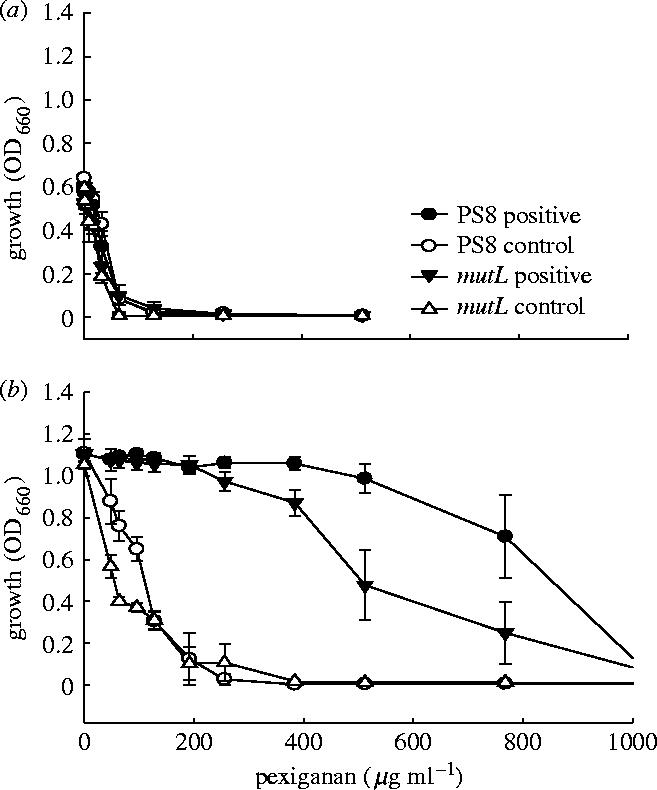
Growth of Escherichia coli in relation to concentration of pexiganan. Mean (±s.e.) growth is measured as absorbance (a) before selection and (b) after selection and presents data from positive selection lines and control selection lines for wild-type (PS8) and mutator (PS8 mutL) genotypes. Note that lineages were not assayed at concentrations above 512 μg ml−1 before selection, since growth was totally inhibited.
(b) Resistance after selection
All P. fluorescens selection lines showed a highly significant increase in growth at high concentration of pexiganan relative to control selection lines (table 1). The six non-mutator positive selection lines evolved MIC50 values above that of the two non-mutator control selection lines (F=44.0; d.f.=1, 6; p<0.001; figure 1b). The six mutator positive selection lines also evolved higher tolerance to pexiganan than the two mutator control selection lines (F=40.4; d.f.=1, 6; p<0.001; figure 1b). All E. coli selection lines also increased in MIC50 values (table 1). The six non-mutator positive selection lines had higher MIC50 values after selection than the control selection lines (F=24.4; d.f.=1, 6; p<0.02; figure 2b). Two of the non-mutator positive selection lines could not be propagated further at a pexiganan concentration of 200 μg ml−1. Although we subsequently selected the two lines for ten additional transfers at 160 μg ml−1, resistance to pexiganan never exceeded 200 μg ml−1. The MIC50 of the six mutator positive selection lines were higher than the corresponding control selection lines values (F=16.7; d.f.=1, 6; p<0.01; figure 2b).
Table 1.
Response to selection for resistance to pexiganan. (Positive lines were cultured in medium supplemented with pexiganan, whereas control lines were cultured in unsupplemented medium. Minimum inhibitory concentration (MIC) was measured using serial macrodilution of pexiganan. Each determination was made on two independent replicates from each line.)
| organism | lineages | selection treatments | no. of lines | MIC ranges | |
|---|---|---|---|---|---|
| before selection | after selection | ||||
| Pseudomonas fluorescens | SBW25 | positive | 6 | 2–8 | 768–1024 |
| control | 2 | 4 | 48–96 | ||
| SBW25 Δmuts | positive | 6 | 16–32 | 1024+ | |
| control | 2 | 32 | 48–96 | ||
| Escherichia coli | PS8 | positive | 4 | 32–128 | 768–1024 |
| control | 2 | 64 | 128 | ||
| PS8 mutL | positive | 6 | 16–256 | 512–1024 | |
| control | 2 | 32–64 | 48–64 | ||
(c) Stability of resistance
After four transfers in unsupplemented medium, all selection lines of both species continued to be capable of undiminished growth at the MIC, whereas all control lines failed to grow.
(d) Cost of resistance
The growth properties of several strains that developed resistance to pexiganan were studied in media alone and in the presence of sublethal concentrations of pexiganan, and compared with the growth properties of control strains. The acquisition of pexiganan resistance did not appear to alter either growth rate or the maximal population density reached in vitro, either in the presence or absence of pexiganan. However, pexiganan resistance was associated with a longer lag phase in the absence of pexiganan, and a shorter lag phase in the presence of pexiganan, compared with the original strain (table 2).
Table 2.
Growth of control and selection lines in unsupplemented medium (MHB) and in medium containing pexiganan at the MIC. (Units of kinetic parameters are: limiting density K, absorbance (known to be highly correlated with cell density, data not shown); rate of increase Vmax, absorbance h−1; lag, h. Values given are overall means for two replicates of all lines. Significance of treatments (Selection, environment of election; Assay, environment of assay) was evaluated from a fixed-effects two-level ANOVA using the among-line variance as denominator in F for the Selection×Assay interaction, and this interaction as denominator for the main effects, with d.f.=1, 12 for all tests. Test-wise significance indicated by asterisk: *p<0.01, **p<0.001.)
| Pseudomonas | arithmetic means | ANOVAs (F) | ||||||
|---|---|---|---|---|---|---|---|---|
| MH broth | pexiganan | treatment | assay | sel×assay | ||||
| control | positive | control | positive | |||||
| SBW25 | K | 0.618 | 0.787 | 0.036 | 0.34 | 13.4* | 46.1** | <1 |
| Vmax | 0.867 | 1.856 | 0.033 | 0.41 | 6.9 | 27.7** | 1.4 | |
| lag | 3.6 | 12.1 | 19.4 | 10.4 | <1 | <1 | 68.7** | |
| Δmuts | K | 0.714 | 0.777 | 0.092 | 0.338 | 7.5 | 57.2** | <1 |
| Vmax | 1.033 | 1.911 | 0.267 | 0.444 | 2.3 | 18.1* | 2.1 | |
| lag | 6.4 | 12.1 | 18.4 | 9.1 | <1 | <1 | 40.1** | |
4. Discussion
Our results show unequivocally that high levels of heritable resistance to a cationic antimicrobial peptide evolved repeatedly in experimental lines of P. fluorescens and E. coli. Since both non-mutator and mutator strains evolved resistance to pexiganan, we conclude that naturally occurring mutation rates are sufficient to create selectable variation in the populations (LeClerc et al. 1996; Oliver et al. 2000). In addition, as microbial infections often involve a population size comparable to that within a 5 ml vial (Lorian 1996), we think that the population parameters of our experiment were reasonably realistic. Whether resistance evolves in any particular clinical situation may depend on the details of how microbes interact with RAMPs near the epithelial surface at the site of infection. Nevertheless, our results show that, as a general rule, population processes can lead to the evolution of mechanisms of resistance to RAMPs in bacterial populations through continued selection, as predicted by Bell & Gouyon (2003).
This experiment contradicts previous reports that resistance is very difficult to obtain in the laboratory (Zasloff 2002). Our experiments, however, incorporated two features lacking in previous investigations. The first was a gradual increase in the concentration of the peptide throughout the experiment. This procedure facilitates the spread of mutations with mildly beneficial effects, which can subsequently be supplemented by further mutations of similar effect, or supplanted by mutations of large effect (see Barbosa & Levy 2000). Screens or short-term experiments at high levels of peptide are likely to be ineffective when single mutants of major effect are rare. The second was the extension of the experiment to 100 serial transfers, allowing time for resistant mutants to appear and spread. The time required for a beneficial mutation with selection coefficient s to spread from mutation–selection equilibrium at an initial frequency m to quasi-fixation in an asexual haploid population is −2(ln m)/s (see Hartl & Clark 1997) and where adaptation requires the accumulation of several mutants of moderate effect a screen lasting a few cycles will not effectively amplify their frequency.
The readiness with which resistance to RAMPs evolves in the laboratory is clearly inconsistent with the view that it will rarely, if ever, evolve in natural communities. On the contrary, it shows that resistance can be expected to evolve rapidly whenever bacterial populations are consistently exposed to elevated levels of RAMPs. At first sight, this conclusion seems inconsistent with the observation that most populations, in fact, lack resistance and are killed by low concentrations of RAMPs. The conditions that bacteria experience in nature are quite different, however, from those that they experience in a laboratory selection experiment. In the laboratory, a lineage can be continually exposed, generation after generation, to a particular stress that limits its growth. This will create intense selection that is very likely to cause specific adaptation. The same situation will often arise when chemical agents are used therapeutically in such a way as to create chronic and widespread contamination. In more natural conditions, a lineage is exposed to a kaleidoscopic array of stresses that change from generation to generation. Even a lineage that is usually associated with animals will encounter a different range of RAMPs in each host species, and, within a single species, very often a different RAMP in different host tissues. Selection will seldom act consistently in any given direction, and is, therefore, unlikely to cause specific adaptation. For similar reasons, bacterial populations expressed only low levels of resistance to conventional antibiotics such as penicillin and streptomycin when they were first used, despite having been exposed to them in the soil for millions of years. Moreover, our selection lines express a substantial cost of resistance in the form of a much longer lag phase in the absence of antibiotic.
The very diversity of RAMPs, indeed, suggests that they have evolved in response to pathogen coevolution. The simplest hypothesis is that each peptide is highly specific, such that the spectrum of RAMPs produced by a given species matches the spectrum of microbes that attack it, and is liable to change as this microbial community changes. Adaptation within each pathogen species provides a further agent of change, implying that each variant is effective only so long as it is rare (in the experience of the pathogen), and loses its effectiveness, through the evolution of resistance by pathogens, should it become more frequent (see Vanhoye et al. 2003 for a discussion of hylid frogs). This interpretation of RAMPs as highly dynamic systems driven by continual host-pathogen coevolution is consistent with the outcome of our experiment.
This study suggests that, like conventional anti-infective agents, the therapeutic use of RAMPS could result in the spread of resistant organisms. These therapeutics should be carefully and appropriately regulated to minimize emergence of resistant organisms from treated individuals and from environments in which large amounts of an anti-infective would be distributed, such as hospitals and stockyards (see O'Brien 2002). It is not our intention to discourage or retard the development of potentially useful antimicrobial agents. What we wish to suggest is that as we develop RAMPs for use as human and veterinary anti-infectives we also seriously consider the consequences of the emergence of resistant organisms. Will organisms that emerge resistant to a synthetic RAMP such as pexiganan also exhibit resistance to endogenous, natural antimicrobial peptides? Would these resistant organisms be more pathogenic than non-resistant strains? Could resistance against a synthetic peptide evolve in vivo, for example, in the setting of the long-term exposure of an animal? How should these concerns be translated into practice by the governmental regulatory bodies that ultimately approve new anti-infectives? Thoughtful analysis of the potential emergence of resistance against these novel anti-infective molecules before they are in widespread use will, in our opinion, help maximize their ultimate benefit to society.
Acknowledgments
We are grateful to Craig MacLean and Paul D. Sniegowski for providing us with the bacterial strains. G. B. is funded by the Natural Sciences and Engineering Research Council of Canada.
Footnotes
As this paper exceeds the maximum length normally permitted, the authors have agreed to contribute to production costs.
References
- Andreu D.J, Rivas L. Animal antimicrobial peptides: an overview. Biopolymer. 1998;47:415–433. doi: 10.1002/(SICI)1097-0282(1998)47:6<415::AID-BIP2>3.0.CO;2-D. 10.1002/(SICI)1097-0282(1998)47:6%3C415::AID-BIP2%3E3.0.CO;2-D [DOI] [PubMed] [Google Scholar]
- Barbosa T.M, Levy S.B. The impact of antibiotic use on resistance development and persistence. Drug Resist. Update. 2000;3:303–311. doi: 10.1054/drup.2000.0167. 10.1054/drup.2000.0167 [DOI] [PubMed] [Google Scholar]
- Bell G, Gouyon P.H. Arming the enemy: the evolution of resistance to self-proteins. Microbiology. 2003;149:1367–1375. doi: 10.1099/mic.0.26265-0. 10.1099/mic.0.C0122-0 [DOI] [PubMed] [Google Scholar]
- Brogden K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005;3:238–250. doi: 10.1038/nrmicro1098. 10.1038/nrmicro1098 [DOI] [PubMed] [Google Scholar]
- Chopra I, O'Neill A.J, Miller K. The role of mutators in the emergence of antibiotic-resistant bacteria. Drug Resist. Update. 2003;6:137–145. doi: 10.1016/s1368-7646(03)00041-4. 10.1016/S1368-7646(03)00041-4 [DOI] [PubMed] [Google Scholar]
- Devine D.A, Hancock R.E.W. Cationic peptides: distribution and mechanisms of resistance. Curr. Pharm. Design. 2002;8:703–714. doi: 10.2174/1381612023395501. 10.2174/1381612023395501 [DOI] [PubMed] [Google Scholar]
- Fedtke I, Gotz F, Peschel A. Bacterial evasion of innate host defenses—the Staphylococcus aureus lesson. Int. J. Med. Microbiol. 2004;294:189–194. doi: 10.1016/j.ijmm.2004.06.016. 10.1016/j.ijmm.2004.06.016 [DOI] [PubMed] [Google Scholar]
- Ge Y, MacDonald D.L, Holroyd K.J, Thornsberry C, Wexler H, Zasloff M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999a;43:782–788. doi: 10.1128/aac.43.4.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, MacDonald D, Henry M.H, Hait H.I, Nelson K.A, Lipsky B.A, Zasloff M, Holroyd K.J. In vitro susceptibility to pexiganan of bacteria isolated from infected diabetic foot ulcer. Diagn. Microbiol. Infect. Dis. 1999b;35:45–53. doi: 10.1016/s0732-8893(99)00056-5. 10.1016/S0732(99)00053-X [DOI] [PubMed] [Google Scholar]
- Groisman E.A, Parra-Lopez C, Salcedo M, Lipps C.J, Heffron F. Resistance to host peptides is necessary for Salmonella virulence. Proc. Natl Acad. Sci. USA. 1992;89:11 939–11 943. doi: 10.1073/pnas.89.24.11939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Lim K.B, Poduje C.M, Daniel M, Gunn J.S, Haclett M, Miller S.I. Lipid A acetylation and bacterial reistance against vertebrate antimicrobial peptides. Cell. 1998;95:189–198. doi: 10.1016/s0092-8674(00)81750-x. 10.1016/S0092-8674(00)81750-X [DOI] [PubMed] [Google Scholar]
- Hancock R.E.M. Peptides antibiotics. Lancet. 1997;349:418–442. doi: 10.1016/S0140-6736(97)80051-7. 10.1016/S0140-6736(97)80051-7 [DOI] [PubMed] [Google Scholar]
- Hancock R.E.M. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 2001;1:156–164. doi: 10.1016/S1473-3099(01)00092-5. 10.1016/S1473-3099(01)00092-5 [DOI] [PubMed] [Google Scholar]
- Hancock R.E.M, Chapple D.S. Peptide antibiotics. Antimicrob. Agents Chemother. 1999;43:1317–1323. doi: 10.1128/aac.43.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock R.E.W, Falla T, Brown M.M. Cationic bactericidal peptides. Adv. Microb. Physiol. 1995;13:135–175. doi: 10.1016/s0065-2911(08)60145-9. [DOI] [PubMed] [Google Scholar]
- Hartl D.L, Clark A.G. Sinauer; New York: 1997. Principles of population genetics. [Google Scholar]
- Huang H.W. Action of antimicrobial peptides: two-state model. Biochemistry. 2000;39:8347–8352. doi: 10.1021/bi000946l. 10.1021/bi000946l [DOI] [PubMed] [Google Scholar]
- LeClerc J.E, Li B, Payne W.L, Cebula T.A. High mutation frequencies among Escherichia coli and Salmonella pathogens. Science. 1996;274:1208–1211. doi: 10.1126/science.274.5290.1208. 10.1126/science.274.5290.1208 [DOI] [PubMed] [Google Scholar]
- Lilley A.K, Bailey M.J. The acquisition of indigenous plasmids by a genetically marked pseudomonad population colonising the phytosphere of sugar beet is related to local environmental conditions. Appl. Environ. Microbiol. 1997;63:1577–1583. doi: 10.1128/aem.63.4.1577-1583.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorian V. Williams & Wilkins laboratory; Baltimore, MD: 1996. Antibiotics in laboratory medicine. [Google Scholar]
- Lohner K, Blondelle S.E. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen. 2005;8:241–256. doi: 10.2174/1386207053764576. 10.2174/1386207053764576 [DOI] [PubMed] [Google Scholar]
- Martinez J.L, Baquero F. Mutation frequencies and antibiotics resistance. Antimicrob. Agents Chemother. 2000;33:1771–1777. doi: 10.1128/aac.44.7.1771-1777.2000. 10.1128/AAC.44.7.1771-1777.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller K, O'Neill A.J, Chopra I. Response of Escherichia coli hypermutators to selection pressure with antimicrobial agents from different classes. J. Antimocrob. Chem. 2002;49:925–934. doi: 10.1093/jac/dkf044. 10.1093/jac/dkf044 [DOI] [PubMed] [Google Scholar]
- NCCLS, 2003 Approved Standards (M7-A4): methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 6th edn. Villanova, PA.
- O'Brien T.F. Emergence, spread, and environmental effect of antimicrobial resistance: how use of an antimicrobial anywhere can increase resistance to any antimicrobial anywhere else. Clin. Infect. Dis. 2002;34(3):S78–S84. doi: 10.1086/340244. 10.1086/340244 [DOI] [PubMed] [Google Scholar]
- Oliver A, Canton R, Campo P, Baquero F, Blazquez J. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science. 2000;288:1251–1254. doi: 10.1126/science.288.5469.1251. 10.1126/science.288.5469.1251 [DOI] [PubMed] [Google Scholar]
- Palumbi S.R. Humans as the World's greatest evolutionary force. Science. 2001;293:1786–1790. doi: 10.1126/science.293.5536.1786. 10.1126/science.293.5536.1786 [DOI] [PubMed] [Google Scholar]
- Peschel A, Collins L.V. Staphylococcal resistance to antimicrobial peptides of mammalian and bacterial origin. Peptides. 2001;22:1651–1659. doi: 10.1016/s0196-9781(01)00500-9. 10.1016/S0196-9781(01)00500-9 [DOI] [PubMed] [Google Scholar]
- Reddy K.V.R, Yedery R.D, Aranha C. Antimicrobial peptides: premises and promises. Int. J. Antimicrob. Agents. 2004;24:536–547. doi: 10.1016/j.ijantimicag.2004.09.005. 10.1016/j.ijantimicag.2004.09.005 [DOI] [PubMed] [Google Scholar]
- Shaver A.C, Sniegowski P.D. Spontaneously arising mutL mutators in evolving Esherishia coli populations are the results of changes in repeat length. J. Bacteriol. 2003;185:6076–6082. doi: 10.1128/JB.185.20.6076-6082.2003. 10.1128/JB.185.20.6076-6082.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski P.D, Gerrish P.J, Lenski R.E. Evolution of high mutation rates in experimental populations of E. coli. Nature. 1997;387:703–705. doi: 10.1038/42701. 10.1038/42701 [DOI] [PubMed] [Google Scholar]
- Tamayo R, Choudhury B, Septer A, Merighi M, Carlson R, Gunn J.S. Identification of cptA, a PmrA-regulated locus required for phosphoethanolamine modification of the Salmonella enterica serovar typhimurium lipopolysaccharide core. J. Bacteriol. 2005;187:3391–3399. doi: 10.1128/JB.187.10.3391-3399.2005. 10.1128/JB.187.10.3391-3399.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toke, O. 2005 Antimicrobial peptides: new candidates in the fight against bacterial infections. Biopolymers (10.1002/bip.20286) [DOI] [PubMed]
- Vanhoye D, Bruston F, Nicolas P, Amichem M. Antimicrobial peptides from hylid and ranin frogs originated from a 150-million-year-old ancestral precursor with a conserved signal peptide but a hypermutable antimicrobial domain. Eur. J. Biochem. 2003;270:2068–2081. doi: 10.1046/j.1432-1033.2003.03584.x. 10.1046/j.1432-1033.2003.03584.x [DOI] [PubMed] [Google Scholar]
- Yeaman M.R, Yount N.Y. Mechanisms of antimicrobial peptides action and resistance. Pharmacol. Rev. 2003;55:27–55. doi: 10.1124/pr.55.1.2. 10.1124/pr.55.1.2 [DOI] [PubMed] [Google Scholar]
- Zasloff M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active, and partial cDNA sequence or a precursor. Proc. Natl Acad. Sci. USA. 1987;84:5449–5453. doi: 10.1073/pnas.84.15.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. 10.1038/415389a [DOI] [PubMed] [Google Scholar]