

Abstract
Conventional kinesin is a processive, microtubule-based motor protein that drives movements of membranous organelles in neurons. Amino acid Thr291 of Drosophila kinesin heavy chain is identical in all superfamily members and is located in α-helix 5 on the microtubule-binding surface of the catalytic motor domain. Substitution of methionine at Thr291 results in complete loss of function in vivo. In vitro, the T291M mutation disrupts the ATPase cross-bridge cycle of a kinesin motor/neck construct, K401-4 (Brendza, K. M., Rose, D. J., Gilbert, S. P., and Saxton, W. M. (1999) J. Biol. Chem. 274, 31506–31514). The pre-steady-state kinetic analysis presented here shows that ATP binding is weakened significantly, and the rate of ATP hydrolysis is increased. The mutant motor also fails to distinguish ATP from ADP, suggesting that the contacts important for sensing the γ-phosphate have been altered. The results indicate that there is a signaling defect between the motor domains of the T291M dimer. The ATPase cycles of the two motor domains appear to become kinetically uncoupled, causing them to work more independently rather than in the strict, coordinated fashion that is typical of kinesin.
Structural analyses of kinesin (2-5) and family relatives Ncd (6, 7) and Kar3 (8) have revealed that the topography of the nucleotide-binding pocket is quite similar to that of myosins and the G proteins (9, 10). The results suggest further that these proteins may share a common mechanism to sense the state of the nucleotide bound at the active site and to respond through structural transitions to communicate with protein partners. For dimeric kinesin, coordination of the motor domains is controlled in part by the nucleotide state at the active sites (11-13) and is required for the cyclic interaction with the microtubule lattice for unidirectional, processive movement. Through pre-steady-state kinetic analysis, it has been determined that ATP binding by the microtubule-bound motor domain allows the partner motor domain to bind to the microtubule and quickly release its ADP (12-13). Other points of coordination are also evident. Thus, the two motor domains of the kinesin heavy chain (KHC)1 dimer are at different stages in their ATPase cycles at any given time, keeping the heads out of phase and allowing for processive movement (reviewed in Refs. 14 and 15).
Much has been learned about kinesin through structural studies. The first motor domain structural intermediate to be determined was the KHC·ADP intermediate (3-5). Recently, the structures of KHC under conditions that mimic different nucleotide states were solved (16). This work revealed that in the absence of microtubules there is little change in KHC structure regardless of the nucleotide present. Thus, with no existing structures of microtubule·kinesin intermediates, the transition states important for efficient ATP hydrolysis remain enigmatic. Because it is difficult to integrate mechanistic models with the available information, alternative approaches are required to understand the structural requirements for motor coordination and processive movement.
Site-directed mutagenesis has already identified structural elements that are important for certain aspects of KHC function. Studies by Woehlke et al. (17) highlighted charged surface residues necessary for the interaction of KHC with microtubules. Romberg et al. (18) showed that the neck region of KHC is important for efficiency but is not essential for processive movement. They proposed a model in which interactions between the β-sheet region of the neck (β9 and β10) and the catalytic domain are disrupted such that the neck region could adopt a more extended conformation to allow head separation during times when both heads are bound to the microtubule. This hypothesis is supported by the cryo-EM diffraction studies of microtubule·kinesin complexes by Hoenger et al. (19).
Studies of mutant KHCs may help us understand both the ATPase mechanism of kinesin and associated structural rearrangements that allow for efficient utilization of the energy generated through ATP hydrolysis. Moore et al. (20) determined that a point mutation in β6 of Ncd greatly reduces the affinity of the motor for microtubules and reduces the velocity of microtubule-based movement. Song and Endow (21) have shown that a point mutation in α4 of both Kar3 and Ncd uncouple the nucleotide and microtubule-binding sites, perhaps due to a break in Loop11-mediated communication between the two sites. Recent studies by Rice et al. (22) have provided insight into structural changes that may allow for force generation and plus end-directed movement of KHC. They have shown that ATP binding promotes a large conformational change in the kinesin neck linker that is directed toward the plus end of the microtubule. These results with monomeric kinesin K349 emphasize that there are conformational transitions in the microtubule-bound kinesin dimer during ATP turnover that are beyond the view of current crystallography efforts.
To gain insight into the mechanisms of kinesin mechanochemistry in vivo, we screened for recessive lethal mutations in the kinesin heavy chain (Khc) that disrupt axonal transport (1, 23). One of the mutations found, Khc4, causes in vivo phenotypes that suggest a nearly complete loss of function. Sequencing revealed that there is a threonine to methionine substitution at amino acid position 291, an almost completely conserved residue in the kinesin superfamily.2 Thr291 is located in α-helix 5, which lies on the microtubule-binding surface of the motor domain. The steady-state ATPase kinetics showed that this mutation causes defects in both ATP and microtubule binding. The kcat is very similar to wild type, yet there is a 3-fold increase in both Km, ATP and K0.5, Mt (1). In an effort to determine more precisely how this mutation alters the ATPase cycle and consequently how Thr291 contributes to KHC mechanochemistry, we have pursued a mechanistic analysis of K401-4.
The results provide direct evidence that ATP binding is significantly weakened by the T291M mutation and that the binding pocket has lost the ability to distinguish ATP from ADP. These data suggest that structural arrangements important for sensing the γ-phosphate have been disrupted. Furthermore, the kinetics indicate that the two motor domains of the mutant dimer are defective in their head-head communication. Thus, the T291M substitution allows the motor domains to work more independently rather than in the strictly coordinated fashion that is characteristic of kinesin.
EXPERIMENTAL PROCEDURES
Materials
[α-32P]ATP (>3000 Ci/mmol) was from NEN Life Science Products; polyethyleneimine-cellulose TLC plates (EM Science of Merck, 20 × 20 cm, plastic-backed) were from VWR Scientific (Bridgeport, NJ); Taxol (Taxus brevifolia) was from Calbiochem-Novabiochem; ATP, GTP, AMP-PNP, and S-Sepharose were from Amersham Pharmacia Biotech (Uppsala, Sweden); Bio-Rad Protein Assay, ovalbumin, IgG, and DEAE-Sephacel were from Bio-Rad.
Protein Purification
K401-4 was bacterially expressed, purified, and characterized as described previously (1). Six preparations of K401-4 were used in the experiments reported, and each was evaluated to determine the steady state parameters: kcat = 18 ± 0.6 s−1, Km, ATP = 224 ± 4.9 μm, and K0.5, Mt = 3.6 ± 0.01 μm. All experiments reported were performed in ATPase buffer (50 mm HEPES, pH 7.2, with KOH, 5 mm magnesium acetate, 0.1 mm EGTA, 0.1 mm EDTA, 50 mm potassium acetate) at 25 °C.
Analytical Ultracentrifugation
Experiments were conducted at 42,000 rpm in a Beckman Optima XLA analytical ultracentrifuge equipped with absorbance optics and an An60Ti rotor. ATPase buffer was used at 24.7 °C. Velocity data were analyzed using DCDT+, version 1.12 (25). The reported weight average sedimentation coefficient values obtained from DCDT+ are corrected for the solution density and viscosity (26) and are calculated by a weighted integration over the entire range of sedimentation coefficients covered by the g(s) distribution. To verify these results, the data were also analyzed with DCDT (27) and, where appropriate, SVEDBERG, version 6.37 (28).
Microtubule Preparation for the Kinetic Experiments
Purified tubulin was cold-depolymerized and clarified the morning of each experiment. Microtubules were assembled by the addition of taxol to 20 μM. The microtubules were collected by centrifugation, and the microtubule pellet was resuspended in ATPase buffer plus 20 μm taxol to stabilize the microtubules (29). For all of the experiments in which microtubules were present, 20 μm taxol was included to maintain the polymer state.
Stopped-flow Experiments
The pre-steady-state kinetics of mantATP binding, mantADP release, K401 binding to microtubules, and detachment of K401 from microtubules were measured using a KinTek StopFlow Instrument (SF-2001, KinTek Corp., Austin, TX) at 25 °C in ATPase buffer. N-methylanthraniloyl fluorescence (mantATP and mantADP) was excited at 360 nm (mercury arc lamp) and detected after being passed through a 400-nm cut-off filter. mantATP binding data in Fig. 1B were fit to the equation,
| (Eq. 1) |
where kobs is the rate constant obtained from the exponential phase of the fluorescence change, k+1 defines the second order rate constant for mantATP binding, and k−1 corresponds to the observed rate constant of mantATP release as determined by the y intercept. The dissociation kinetics of Mt·K401 complex and association kinetics of K401 with microtubules were determined by the change in turbidity monitored at 340 nm. All concentrations reported are final after mixing. For Fig. 3B, the observed rate constants of microtubule association were fit to the equation,
| (Eq. 2) |
where kobs is the rate constant obtained from the fast, exponential phase, k+5 defines the second order rate constant for microtubule association, and k−5 corresponds to the observed rate constant of motor dissociation as determined by the y intercept.
Fig. 1.
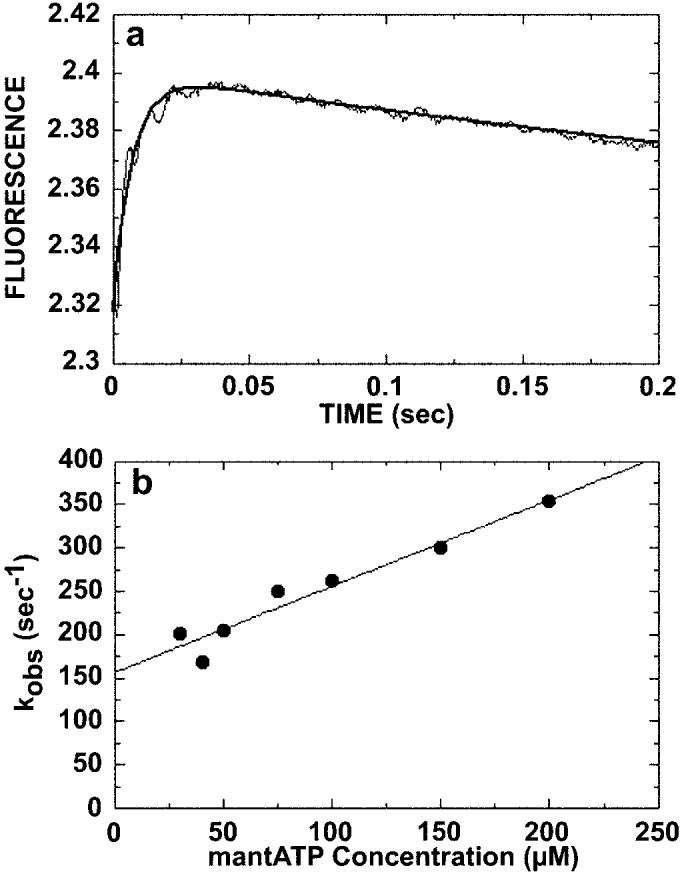
Kinetics of ATP binding. The preformed Mt·K401-4 complex (30 μm tubulin, 10 μm K401-4) was rapidly mixed in the stopped-flow apparatus with varying concentrations of mantATP (20–200 μm). A, a representative stopped-flow record (average of seven individual traces) from an experiment at 20 μm mantATP. An increase in fluorescence was observed, and the smooth line is the fit of the data to a double exponential. The observed rate of the initial fast exponential phase was 135 ± 4.3 s−1. The slow, declining phase at 3.1 ± 0.9 s−1 is too slow to be ATP hydrolysis and is believed to represent an isomerization after initial mantATP binding. B, the observed rate constant of the initial, fast phase increased linearly with increasing mantATP concentration. The data were fit to Equation 1, and the slope provides the apparent second order rate constant for mantATP binding, k+1 = 1 ± 0.1 μm−1 s−1, and the y intercept provides the off-rate, k−1 = 157 ± 11.8 s−1 (Scheme 1).
Fig. 3.
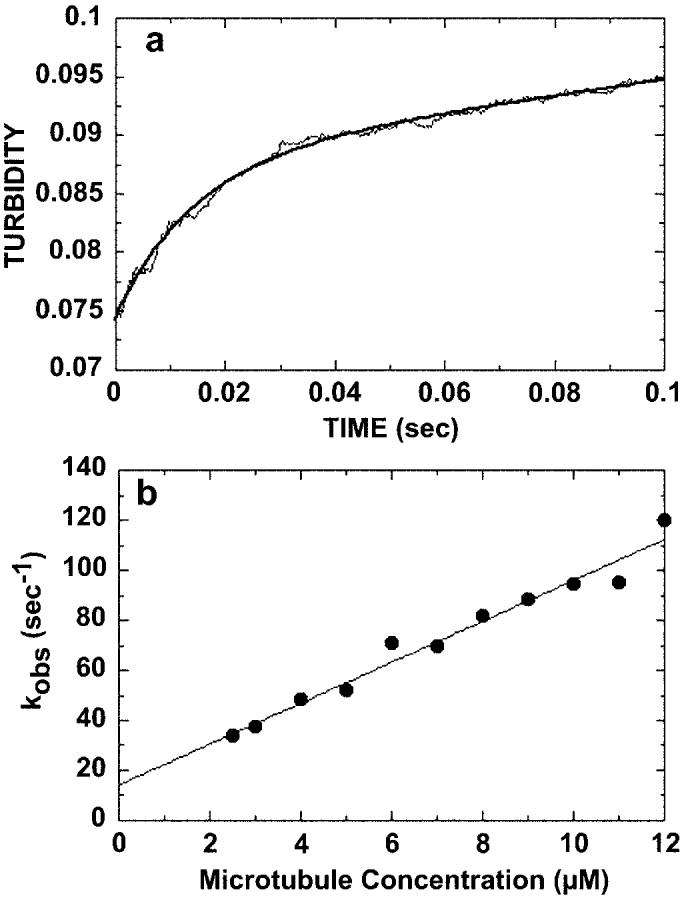
Kinetics of microtubule binding. 4 μm K401-4·ADP was rapidly mixed with varying concentrations of microtubules (2.5–12 μm), and turbidity was monitored in the stopped-flow apparatus. A, a representative stopped-flow record at 7 μm microtubules, which is the average of seven traces. The smooth line is the best fit of the data to a single exponential plus a linear term. The rate constant of the initial exponential phase is 70 ± 1.6 s−1. The second phase at a rate of 0.07 ± 0.001 s−1 is too slow to be considered on the pathway and is attributed to a conformational change after microtubule association. B, the rate constant from the initial fast phase of each transient was plotted as a function of microtubule concentration. The data were fit to Equation 2, the slope providing the apparent second order rate constant for microtubule association, k+5 = 8.2 ± 0.5 μm−1 s−1, and the y intercept providing k−514.3 ± 3.7 s−1.
Rapid Quench Experiments
The pre-steady-state experiments to determine the rate constant of ATP hydrolysis were performed with a rapid chemical quenched-flow instrument (KinTek Corp., Austin, TX) at 25 °C in ATPase buffer as described previously (30). For each time point, a preformed Mt·K401 complex (10 μm K401, 25 μm tubulin, 20 μm taxol; final concentrations after mixing) was reacted with [α-32P]ATP for times ranging from 5 to 400 ms. The reaction mixture was then quenched with 4 n HCl and expelled from the instrument. Chloroform (100 μl) was added immediately to the reaction mix to denature the protein, followed by neutralization (pH 7–7.8) by the addition of 2 m Tris, 3 m NaOH. The acid quench stops the reaction and denatures the protein; therefore, the product formed is the sum of three intermediates: K·[α-32P]ADP·Pi, K·[α-32P]ADP, and [α-32P]ADP released from the active site. The concentration of product ([α-32P]ADP) was plotted as a function of time, and the data were fit to the burst equation,
| (Eq. 3) |
where A is the amplitude of the burst, representing the formation of [α-32P]ADP·Pi at the active site; kb is the rate constant of the pre-steady-state burst; kss is the rate constant of the linear phase, corresponding to steady-state turnover; and t is the time in seconds.
Molecular Modeling
All molecular modeling was performed on a Silicon Graphics workstation using UCSF MidasPlus Molecular Interactive Display and Simulation software (Computer Graphics Laboratory, University of California, San Francisco, CA).
RESULTS
Pre-steady-state Kinetics of mantATP Binding
To explore the apparent weak binding of ATP by K401-4 revealed by the steady-state Km, ATP (1), we began the pre-steady-state kinetic analysis by measuring the kinetics of ATP binding and ATP hydrolysis (Figs. 1 and 2). For the ATP binding studies, the fluorescent ATP analog mantATP was used (Fig. 1). This analog has been used in previous characterizations of kinesin motors and has been shown experimentally to be a good ATP analog because of the fluorescence enhancement and similarity to ATP (31, 32). For K401-wt, the microtubule-activated steady-state kcat reported was 19 s−1 for both ATP and mantATP with the Km, ATP = 62 μm and Km, mantATP = 150 μm (32). The preformed Mt·K401-4 complex (30 μm microtubules, 10 μm K401-4) was rapidly mixed with varying concentrations of mantATP in the stopped-flow apparatus, and the change in fluorescence was monitored. A representative stopped-flow record is shown in Fig. 1A. Binding of mantATP causes a biphasic fluorescence transient with a rapid exponential increase in fluorescence (associated with mantATP binding) followed by a significantly slower exponential decrease in fluorescence. The observed decrease in fluorescence is independent of substrate concentration (kobs = 3.1–3.8 s−1) and is not fast enough to be attributed to ATP hydrolysis. This biphasic fluorescence transient is characteristic of kinesin and has been observed by others (34, 36). The smooth line is the best fit of the data to a double exponential, providing kobs of the initial, fast reaction at 135 s−1. The rate of the fast exponential phase increased as a function of ATP concentration, and the data were fit to Equation 1 (Fig. 1B). The slope of the line provided the second order rate constant for mantATP binding, k+1 = 1 μm−1 s−1, which is similar to the observed rate constant for K401-wt at 2 μm−1 s−1 (30, 34). The y intercept predicts an off rate for mantATP with k−1 = 157 s−1.
Fig. 2.
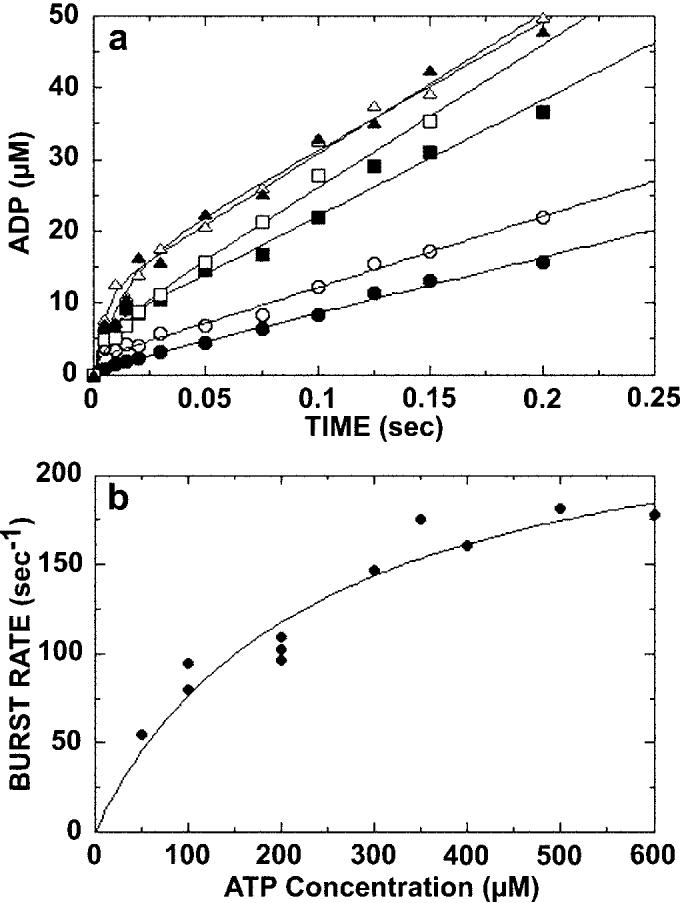
Kinetics of ATP hydrolysis. A preformed Mt·K401-4 complex (10 μm K401-4, 25 μm tubulin) was rapidly mixed with varying concentrations of [α-32P]MgATP in a chemical quenched-flow instrument. The reaction times varied from 5 to 400 ms, and the data were fit to the burst equation (Equation 3). A, the transients for ATP hydrolysis in the presence of 100 μm ([UNK]), 200 μm (○), 300 μm (■), 400 μm (□), 500 μm (△), and 600 μm (▲) [α-32P]MgATP. Only the first 200 ms of each transient are shown to expand the time domain of the initial burst phase. B, the rate constants (kb) of the pre-steady-state burst phase determined from each transient in A were plotted as a function of [α-32P]MgATP concentration. Data from other experiments were included in this plot. The fit of the data to a hyperbola provides the maximum rate constant for the burst phase, kb = 257 ± 30 s−1 and Kd, ATP = 236 ± 62 μm. At high ATP concentrations, ATP binding no longer limits the first order reaction of ATP hydrolysis; therefore, the rate constant for ATP hydrolysis (k+2) = 257 ± 30 s−1 (Scheme 1).
Pre-steady-state Kinetics of ATP Hydrolysis
We next measured the kinetics of ATP hydrolysis for K401-4 through a series of acid quench experiments (Fig. 2). The preformed Mt·K401-4 complex (25 μm microtubules, 10 μm K401-4) was rapidly mixed with [α-32P]MgATP in the chemical quench instrument. Fig. 2A shows the time course of ATP hydrolysis by K401-4 at six different ATP concentrations. Each transient was biphasic with an initial exponential rate of ADP·Pi formation (the burst), followed by a slower rate of product formation (the linear phase) corresponding to steady-state turnover. The exponential burst of product formation at the active site indicates that a step after ATP hydrolysis is rate-limiting for K401-4 as observed for K401-wt (30). At high ATP concentrations, ATP binding becomes faster than ATP hydrolysis. Because substrate binding is no longer limiting, the maximum rate constant for the exponential burst phase is the rate constant of ATP hydrolysis. The rate constant for ATP hydrolysis (k+2 = 257 s−1) was determined by plotting the burst rates as a function of ATP concentration (Fig. 2B) and fitting the data to a hyperbola. Note that the K401-4 rate constant (257 s−1) is significantly faster than the 100 s−1 rate constant observed for dimeric K401-wt ATP hydrolysis (30). It is interesting that a monomeric KHC motor domain (K341) also shows a rapid rate of ATP hydrolysis at >300 s−1 (35). For K401-4, the Kd,ATP is 236 μm (Fig. 2B), which is equivalent to the steady-state Km, ATP at 236 μm (1). Comparison of the Kd, ATP for K401-wt at 60 μm (30) with the Kd, ATP for K401-4 indicates that the mutant motor binds ATP much more weakly. The acid quench experiments provide direct evidence that the T291M mutation significantly weakens ATP binding and increases the rate of ATP hydrolysis.
Association Kinetics of K401-4 and Microtubules
We next looked at the rate of K401-4·ADP binding to the microtubule (Fig. 3) to determine if a microtubule binding defect was indeed contributing to the abnormally high K0.5, Mt measured through steady-state analysis (1). The rate of formation of the Mt·K401-4 complex was monitored by turbidity measurements in the stopped-flow instrument. K401-4·ADP (4 μm) was rapidly mixed with taxol-stabilized microtubules (7 μM), and the change in turbidity was recorded. In this experiment, an increase in turbidity was interpreted as binding of K401 to the microtubule. A representative stopped-flow record is shown in Fig. 3A. The solid line is the best fit of the data to a single exponential function and a linear term, providing the kobs of the fast exponential phase at 70 s−1. Fig. 3B shows that the observed rates of microtubule association increased linearly as a function of microtubule concentration. These data were fit to Equation 2, the slope of which provides the apparent second order rate constant for binding k+5 = 8 μm−1 s−1; this constant is somewhat less than K401-wt measured at 11–15 μm−1 s−1 (15, 32). The y intercept predicts a significant off-rate (k−5 = 14 s−1), which was not seen for K401-wt (32). Therefore, the T291M mutation appears to weaken the binding between the KHC motor domains and the microtubule.
ADP Release Kinetics from Mt·K·ADP
We also measured the kinetics of ADP release from the Mt·K401-4·ADP intermediate (Fig. 4) using mantADP. K401-4 was incubated with mantADP at a ratio of 4:1 to allow for exchange of ADP resident at the active site with mantADP. A preformed K401-4·mantADP complex (3 μm K401-4, 12 μm mantADP) was rapidly mixed with microtubules plus MgATP in the stopped-flow apparatus. As mantADP was released from the more hydrophobic environment of the active site into the aqueous buffer, its fluorescence was quenched. The MgATP (1 mm) present in solution blocked the subsequent rebinding of any mantADP to K401. Fig. 4A shows a representative stopped-flow record. The solid line is the fit of the data to a single exponential function and a linear term, providing the kobs of the fast exponential reaction at 31 s−1. Fig. 4B shows that the exponential rate constant associated with the fluorescence change upon mantADP release increased with increasing microtubule concentration. The fit of the data to a hyperbola provides a maximum rate constant of mantADP release, k+6 = 234 s−1 with a K0.5, Mt at 34 μm. The rate constant of mantADP release is consistent with the fast ADP dissociation kinetics reported for K401-wt at 200–300 s−1 (32), yet the K0.5, Mt for K401-4 is larger than that for K401-wt (15 μm). These results are consistent with the interpretation that the T291M mutant protein requires a higher concentration of microtubules both for half-maximal activation of steady-state turnover at 3.6 versus 1 μmm for K401-wt (1) and for mantADP release (34 versus 15 μm for K401-wt).
Fig. 4.
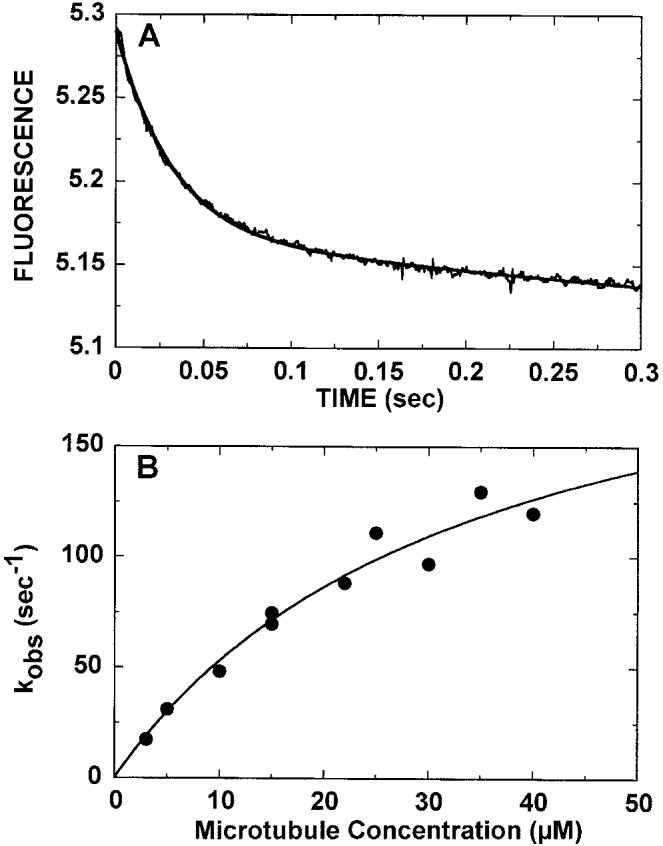
Kinetics of mantADP release. The K401-4·mantADP complex (3 μm K401-4, 12 μm mantADP) was preformed such that K401-4 contained mantADP bound to both heads. The complex was rapidly mixed in the stopped-flow apparatus with varying concentrations of microtubules (3–40 μm)plus 1 mm MgATP. A, a representative stopped-flow record (average of seven traces) at 5 μm microtubules. The smooth line is the fit of the data to a single exponential plus a linear term. The kobs of the fast exponential phase was 31 ± 0.4 s−1. B, the rate constant from the exponential phase increased as a function of microtubule concentration. The fit of the data to a hyperbola provided the maximum rate constant of mantADP release, k+6 = 234 ± 41 s−1 with K 0.5, Mt at 34.2 ± 10μm.
ATP-promoted Dissociation Kinetics of the Mt·K401-4 Complex
The effects of the T291M mutation on ATP-promoted detachment of K401·ADP from the microtubule were examined by following changes in turbidity (Fig. 5). The preformed Mt·K401-4 complex (2.9 μm microtubules, 3 μm K401-4) was rapidly mixed with MgATP plus 100 mm KCl in the stopped-flow apparatus. In this experiment, a decrease in turbidity was interpreted as release of the motor from the microtubule. The addition of 100 mm KCl weakens rebinding of the motor to the microtubule after detachment to allow for accurate measurement of the dissociation kinetics (32). Fig. 5A shows a representative stopped-flow record. The solid line is the best fit of the data to a double exponential, providing a kobs of the initial fast exponential phase at 49 s−1. The kobs increased as a function of ATP concentration, and the fit of the data to a hyperbola provides a maximum rate constant of detachment, k+3 = 60 s−1 with a K0.5, ATP of 280 μm (Fig. 5B). This K0.5, ATP is in agreement with both the Km, ATP at steady-state conditions (1) and the Kd,ATP determined by acid quench experiments (Fig. 2B), reinforcing the interpretation that K401-4 binds ATP weakly.
Fig. 5.
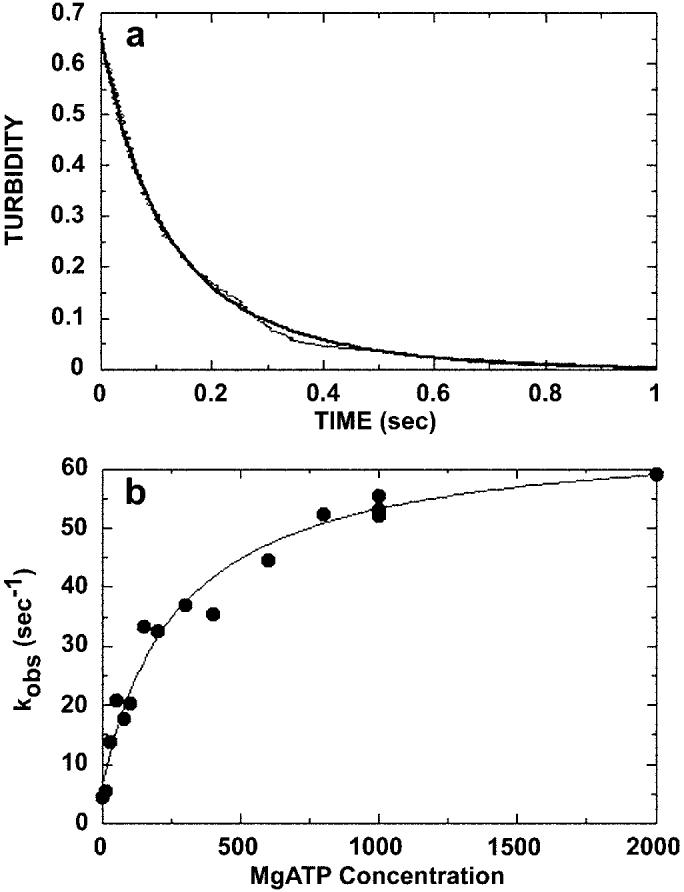
ATP-promoted dissociation of the Mt·K401-4 complex. The preformed Mt·K401-4 complex (3 μm K401-4, 2.9 μm tubulin) was rapidly mixed with varying concentrations of MgATP (10 μm to 2 mm) plus 100 mm KCl, and turbidity was monitored in the stopped-flow apparatus. The salt was added to slow rebinding of K401-4 to microtubules after dissociation. A, a representative stopped-flow record (average of six traces) for 1 mm MgATP. The smooth line is the fit of the data to a double exponential. The kobs of the initial exponential phase was 49.4 ± 0.8 s−1. The second exponential phase was too slow to be considered on the pathway. B, the rate constant of the exponential phase for each transient was plotted as a function of MgATP concentration. The data were fit to a hyperbola, providing the maximum dissociation rate constant k+3 of 59.8 ± 2.9 s−1 and K0.5, ATP = 280 ± 57.1 μm.
It is striking that for both K401-wt and a dimeric human KHC construct K379 (36), this ATP-promoted dissociation step was observed at rates of 12–14 s−1, substantially slower than the 60 s−1 observed for K401-4. The motor domains of wild type dimeric KHC are coupled at this step such that ATP binding by the microtubule-attached motor domain stimulates microtubule binding of the partner motor domain and rapid release of its ADP (11-13). Johnson and colleagues proposed that the intrinsic rate constant for ATP-promoted detachment for a single motor domain is 50 s−1 (13). This constant is faster than the observed rate of 12–14 s−1 measured for the motor domain dimer because the alternating site mechanism only allows one motor domain to be active at a time (13, 15, 32, 34). Thus, the net rate of any step occurring in sequence is influenced by the rates of the other steps. The intrinsic reaction rates for K401-wt have been determined through global fitting of the data to a complete kinetic mechanism using computer simulation (34). The observed rate of detachment for K401-4 (60 s−1) is very different from the observed rate of 12–14 s−1 but is very similar to the intrinsic rate constant of detachment for K401-wt at 50 s−1. These results suggest that if the motor domains of K401-4 were dimerized as expected, they are no longer tightly coupled and are acting more independently.
mantADP Dissociation Kinetics from the Mt·K401-4·mantADP Complex
To evaluate further the possible loss of motor domain cooperativity in K401-4, a series of mantADP dissociation experiments were performed. Previously, it was reported that microtubule-stimulated ADP release from dimeric KHC is biphasic. ADP release from the first head is fast, but release from the second head is delayed until the first head can bind ATP (11-13). If the T291M mutation in K401-4 frees the dimer from the constraint of cooperativity, the biphasic nature of ADP release should be altered. Initially, we examined the rate of mantADP release from both sites of K401-wt in the presence of ATP or ADP (Fig. 6A, inset). As in our previous experiments, ADP at the active site was exchanged with mantADP. The K401·mantADP complex (3 μm K401, 6 μm mantADP) was rapidly mixed with microtubules (10 μm tubulin) plus 2 mm MgATP or 2 mm MgADP. Representative stopped-flow records for K401-wt and K401-4 are shown in Fig. 6A. For K401-wt, ATP promoted rapid release of mantADP from both motor domains, and ADP caused a dramatic biphasic response with rapid release of mantADP from the first motor domain (approximately 50% of the amplitude) and very slow release of mantADP from the second motor domain (approximately 50% of the amplitude). Fig. 6A shows representative stopped-flow records from the same experiment performed with K401-4. In contrast to the K401-wt results, there was rapid release of mantADP regardless of the nucleotide added (ATP or ADP); thus, the two curves superimpose, and the rate constants for mantADP release were fast (116 s−1 with ADP and 108 s−1 with ATP). These results indicate that the T291M mutation completely eliminates the biphasic nature of microtubule-stimulated ADP release from dimeric K401. Similar kinetics of mantADP release have been seen with K341, a monomeric KHC motor domain construct (13).
Fig. 6.
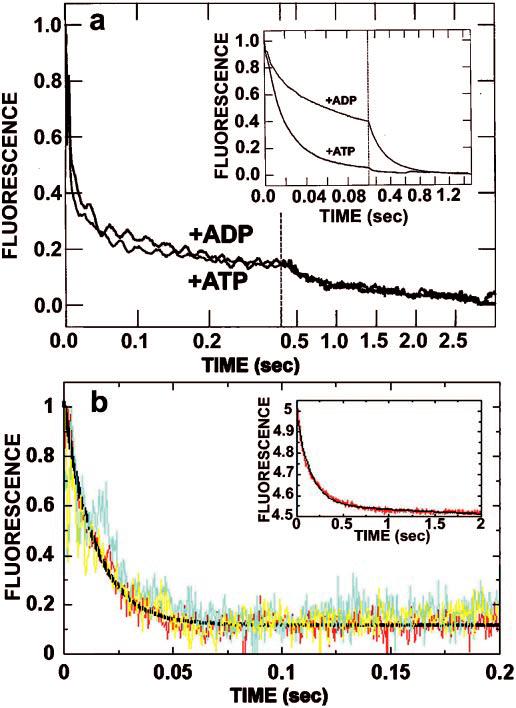
mantADP release in the presence of ATP or ADP. A, kinetics of mantADP release from mutant K401-4 promoted by ATP or ADP. These are representative stopped-flow records (average of six traces) in which the K401-4·mantADP complex (3 μm K401-4, 12 μm mantADP; preformed such that K401-4 contained mantADP bound to both heads) was rapidly mixed with 10 μm microtubules plus either 1 mm MgATP or 1 mm MgADP. It is noteworthy that the mantADP release transients promoted by ADP and ATP are indistinguishable (115.8 ± 4.5 and 107.7 ± 4.0 s−1, respectively). Only 15% of the total amplitude is associated with the second phase. Time domains are 0–400 ms and 400 ms to 3 s. Inset, mantADP release from K401-wt initiated by ATP and ADP. The experiment shown in a was repeated with wild type (3 μm K401 6 μm mantADP). Note that the ADP-promoted kinetic transient shows that approximately 50% of the release amplitude was associated with the initial fast exponential phase (53.6 ± 0.7 s−1), and 50% was associated with the second, much slower exponential phase (4.5 ± 0.02 s−1). Time domains are 0–100 ms and 100 ms to 2 s. B, kinetics of mantADP release from the high affinity nucleotide binding site of K401-4 and K401-wt as a function of ATP or ADP. The Mt·K401-4·mantADP complex was preformed (6 μmm K401-4, 3 μm mantADP, 15 μm microtubules; one mantADP bound per K401-4 dimer) and mixed with either 1 mm MgATP or 1 mm MgADP. Note that the kinetics of mantADP release promoted by ADP and ATP are similar with kobs = 61.1 ± 2.7 s−1 for ATP (yellow) and 54.3 ± 2.5 s−1 for ADP (blue). This experiment was repeated with K401-wt (red), and similar results were observed with 1 mm MgATP, kobs = 73.3 ± 1.7 s−1. Inset, the same experiment repeated with K401-wt with 1 mm MgADP. Note that the rate of ADP release in the presence of ADP is significantly slower for K401-wt; kobs = 6.6 ± 0.1 s−1.
These results and those from ATP-promoted detachment of K401-4·ADP from microtubules suggest that the T291M mutation induces nearly independent, monomeric behavior. K401-4, with ∼60 coiled-coil-forming amino acids of the neck, should be dimeric. To determine the correctness of this supposition, sedimentation velocity studies (26) were performed with monomeric K341, dimeric K401-wt, and K401-4 (Fig. 7). We found that K401-wt and K401-4 had similar values (5.50 and 5.42, respectively), and these were substantially greater than the value for monomeric K341 at 3.32. In the case of K401-wt, there is a shift in the g(s) curves to higher sedimentation coefficient values with increasing protein concentration, which reflects the previously observed 1-2-4 self-association mechanism for K401-wt (26). In Fig. 7, the g(s) curves of K401-4 do not show the same degree of shift in the g(s) curves with increasing protein concentration, indicative that the mutant form of K401 is more stable as a dimer. There is some centrifugal tailing in the g(s) curves for K401-4, suggesting that it may still associate to form a tetramer, but this degree of association is much weaker than that observed for K401-wt. The majority of the K401-4 protein sediments near a peak position of 4.9 S, as determined by a two-species fit by SVEDBERG (version 6.37); therefore, the predominant species of the mutant is the dimer. Because K401-4 is dimeric, the mantADP release kinetics indicate that the two motor domains behave in a more kinetically independent manner. We conclude that the T291M mutation alters structural features that are important for the communication between motor domains and required for the alternating site ATP hydrolysis mechanism.
Fig. 7.
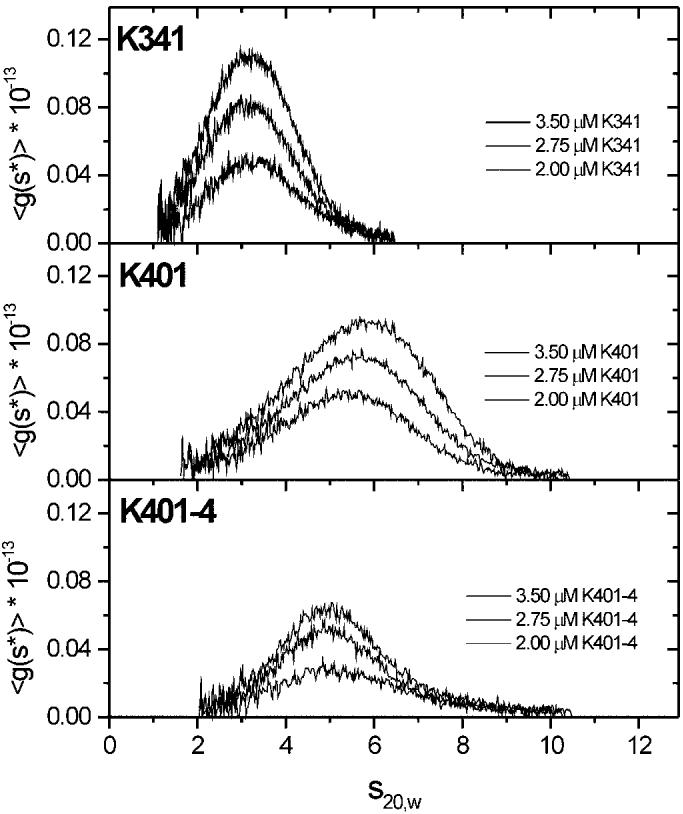
Sedimentation coefficient distribution profiles for K341, K401-wt, and K401-4. The sedimentation velocity data were analyzed for sedimentation distribution g(s) in the presence of 20 μm MgATP with motor concentrations from lowest to highest (bottom to top): 2.0, 2.75, and 3.5 μm. The average value for each construct is reported based on the data obtained at three different protein concentrations for each: .
Scheme1.
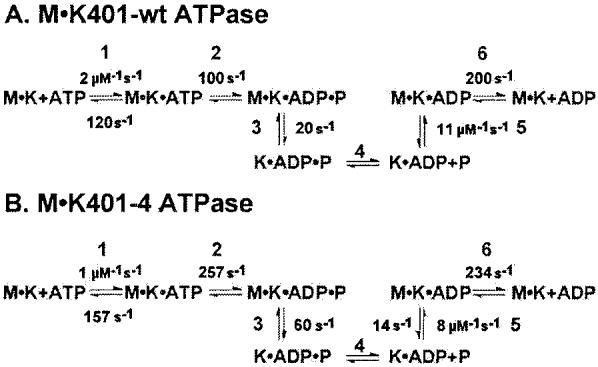
Observed kinetics of K401-wt and K401-4.
mantADP Dissociation Kinetics from Each Site of Dimeric K401-4
The observation that microtubules in the presence of ADP elicit rapid release of mantADP from both motor domains of K401-4 (Fig. 6A) suggests that the active site responds to ADP in the same manner that it responds to ATP. Both seem to serve equally well to stimulate microtubule binding by the second motor domain. To evaluate this hypothesis further, we examined the rate of mantADP release from the second motor domain; the head that keeps ADP more tightly bound in the presence of microtubules and is believed to be detached or weakly bound to the microtubule (Fig. 6B). An equilibrium mixture was prepared in which 6 μm K401-4 (or K401-wt) was incubated with 3 μm mantADP and 15 μm microtubules. At these conditions, only one mantADP is expected to be bound per kinesin dimer (12). mantADP release is then activated by rapidly mixing with either ATP or ADP in the stopped-flow instrument. Under these conditions, the added ATP or ADP should bind the active site of the microtubule-attached head, allowing the second motor domain to bind the microtubule to drive release of its mantADP (12). For K401-wt, mantADP release promoted by ATP was fast at 73 s−1 (Fig. 6B, red trace), yet ADP-promoted release of mantADP was extremely slow at 6.6 s−1 (Fig. 6B, inset). In contrast, for K401-4 there was no appreciable difference in the mantADP dissociation kinetics using either ATP (kobs = 61 s−1, Fig. 6B, yellow trace) or ADP (kobs = 54 s−1, Fig. 6B, blue trace) to promote mantADP release from the high affinity site of K401-4. These data show that K401-4 is able to hold mantADP at the high affinity site despite the fact that this mutant binds nucleotide much more weakly than wild type kinesin. Therefore, there is some degree of cooperativity between the motor domains to establish a difference in nucleotide binding at head 1 versus head 2. More importantly, because ATP and ADP both stimulate rapid release of mantADP from the high affinity site, yet buffer does not (data not shown), the results indicate that the low affinity site (head 1) has lost the ability to distinguish ATP from ADP.
DISCUSSION
To gain a deeper understanding of the mechanisms employed by kinesin to produce processive movement, we examined in detail the effects of a specific amino acid substitution on the mechanochemical cycle (see Schemes 1 and 2). Our steady-state kinetic analysis indicated that the T291M mutation causes defects in both ATP and microtubule binding (1). The pre-steady-state kinetics presented here show that the mutant motor binds ATP and microtubules more weakly than K401-wt. Moreover, our analysis reveals a defect in the signaling mechanism between the motor domains of the mutant dimer, suggesting that the T291M change significantly disrupts the alternating site mechanism of ATP turnover for dimeric kinesin.
In our initial studies, we reported an almost 4-fold increase in the K0.5, Mt and attributed it to weaker microtubule binding by K401-4 (1). Through our mantADP release studies (Fig. 4), the measured K0.5, Mt at 34 μm is much larger than that for K401-wt (15 μm). The microtubule association kinetics (Fig. 3) also reveal an appreciable off-rate for microtubules (k−5 = 14 s−1), which is not seen with K401-wt (32). These results confirm that microtubule binding for K401-4 is much weaker than that seen for K401-wt.
We also reported a 3-fold increase in the Km, ATP and attributed it to weaker ATP binding by K401-4 (1). There is a significant off-rate (k−1 = 157 s−1) observed in the mantATP binding experiment (Fig. 1), which shows weak mantATP binding. This finding is reinforced by our ATP hydrolysis studies, which define the Kd, ATP at 236 μm. For K401-wt, the Kd,ATP at 60 μm (30) indicates significantly tighter ATP binding by wild type kinesin. Thus, the pre-steady-state kinetic analysis confirms our original supposition that ATP binding by K401-4 is significantly weaker than K401-wt.
The most striking difference in the ATPase cycles of mutant and wild type K401 is seen in the kinetics of rate-limiting detachment from microtubules. The rate of detachment for K401-4 (k+3 = 60 ± 3 s−1) is quite fast relative to wild type dimeric kinesin constructs at 12–14 s−1 (32, 36). Furthermore, the observed rate constant at 60 s−1 is very similar to the intrinsic rate constant of detachment for K401-wt at 50 ± 8s−1 (34), and the centrifugation analysis documents that K401-4 is dimeric (Fig. 7). These results show that the mutant motor domains exhibit some degree of kinetic uncoupling.
Scheme 2.
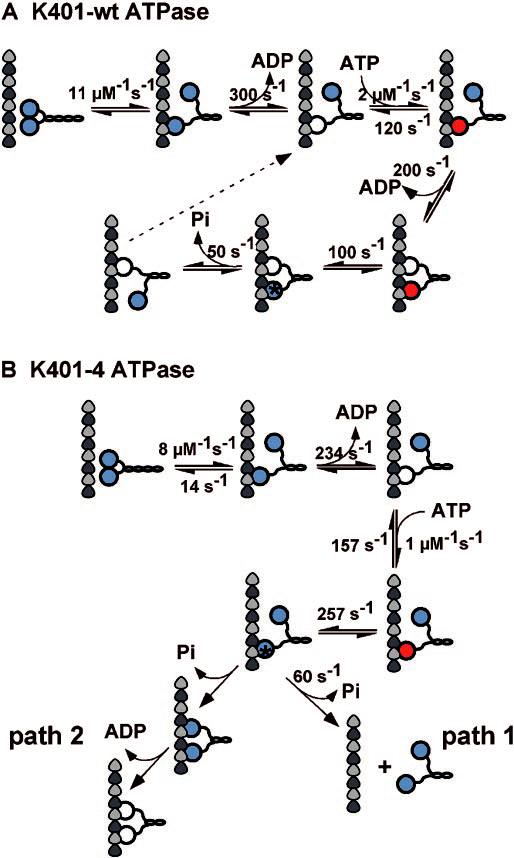
Microtubule·kinesin ATPase mechanism.
The observation of motor domain uncoupling in K401-4 indicates that the alternating site catalysis mechanism must be compromised and that strict coordination of the motor domains must be disrupted. A series of mantADP release experiments were used to dissect the pathway of motor interaction with the microtubule for wild type dimeric kinesin (12, 13, 32, 36). These experiments showed that head 1 binds to the microtubule with rapid release of mantADP, and it is ATP binding at this empty site that signals the second motor domain to bind the microtubule and quickly release its mantADP. ADP binding at head 1 does not elicit rapid release of mantADP from head 2. We pursued these same experiments with K401-4 to examine the degree of cooperative interactions between the motor domains of the mutant. Our results show that there was rapid release of mantADP from head 2; however, both ADP and ATP binding at head 1 can induce rapid dissociation of mantADP from this high affinity site (Fig. 6). These results indicate that the mechanism of communication between the motor domains of K401-4 is defective. However, the results also show that there is some degree of communication between the motor domains, because mantADP release from head 2 does require nucleotide binding (ADP or ATP) at head 1. Buffer will not stimulate rapid release of mantADP from head 2. These results imply that K401-4 cannot sense the absence of the γ-phosphate and that the breakdown in signal transmission between the motor domains results in an ATPase mechanism in which the motor domains are no longer strictly coordinated.
Another key difference in the ATPase cycle of K401-4 that probably contributes to disruption of the wild type alternating site ATP hydrolysis mechanism (Schemes 1 and 2) is the more rapid rate of ATP hydrolysis observed in the mutant. Our mantADP release experiments have documented mantADP release from the high affinity site (head 2) at 70–100 s−1, and both ATP and ADP can activate rapid mantADP release from head 2. With ATP hydrolysis by head 1 occurring at 257 s−1, is possible that subsequent release of head 1 from the microtubule occurs before head 2 can bind the microtubule, resulting in detachment of the dimer from the microtubule (Scheme 2, path 1). An alternative possibility is that because signaling between the motor domains is aberrant, the first head does not detach after ATP hydrolysis and the second head binds the microtubule followed by concomitant ADP release (Scheme 2, path 2). Path 2 results in a kinesin intermediate in which both heads are bound to the microtubule and both active sites are empty. Thus, the motor domains of the dimer are in phase with each other after ATP turnover rather than out of phase where the two motor domains are always at different stages of the ATPase cycle as required by the alternating site ATPase mechanism of wild type kinesin.
To discriminate between these two paths, we performed ATP-promoted detachment assays at low salt conditions (50 mm; Fig. 8). In low salt, K401-wt remains bound for several ATP turnover cycles before detaching, and the observed kinetics of motor detachment appear quite slow. However, if path 1 were the mechanism, the motor should detach immediately after the first turnover with the rate constant at 60 s−1. In low salt, K401-wt dissociates with an observed rate constant of 1.7 s−1, indicating that the dimeric motor is in association with the microtubule for 0.6 s. This transit time suggests multiple steps along the microtubule, each coupled to a single ATP turnover. For K401-4, the kinetics are more complex and biphasic, with the initial exponential phase at 1.4 s−1 and the second slower phase at 0.1 s−1. Our interpretation is that K401-4 remains associated with the microtubule for multiple ATP turnovers; therefore, these kinetics exclude path 1 and favor path 2 as the mechanism of the K401-4 ATPase. Although the results show prolonged association with the microtubule at these conditions, the mechanistic basis of K401-4 microtubule association is more difficult to interpret and is probably not reflecting processive ATP movement with one ATP turnover tightly coupled to one productive step.
Fig. 8.
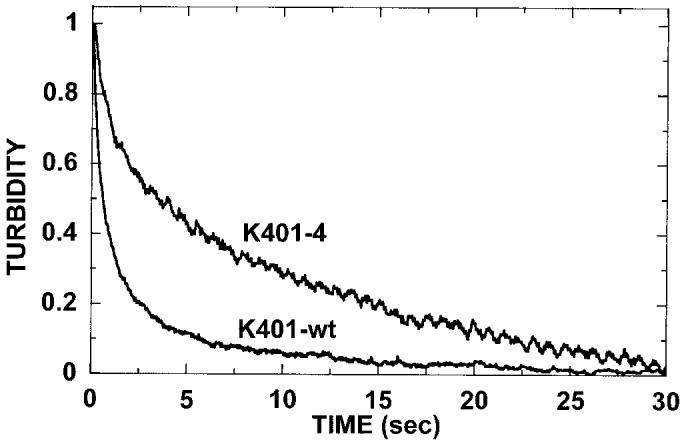
ATP-promoted dissociation at low salt. The preformed Mt·K401-wt and Mt·K401-4 complexes (3 μm K401-4, 2.9 μm tubulin) were rapidly mixed with 1 mm MgATP, and turbidity was monitored in the stopped-flow apparatus. For this experiment, additional salt was not added to the ATP syringe to evaluate whether the mutant motor detaches directly after ATP hydrolysis (Scheme 2, path 1) or the mutant motor follows path 2 and results in an intermediate with both heads bound to the microtubule and at the same stage in the ATPase cycle (Scheme 2, path 2). The dissociation kinetics of K401-wt at 1.7 s−1 indicate that the wild type motor is in association with the microtubule for 0.6 s (transit time = 1/kobs). For K401-4, the dissociation kinetics are biphasic with the exponential phase at 1.4 s−1 (transit time = 0.7 s) followed by a second slower phase at 0.1 s−1.
In Drosophila, the T291M mutation is recessive lethal (23). The timing of lethality and the prelethal, paralytic behavioral phenotype indicates a complete loss of function (1). The earliest cellular defects seen in mutants is the stochastic accumulation of mitochondria, vesicles, and other organelles in large “traffic jams” that force axonal swellings (24). Disruption of fast axonal transport by kinesin mutations causes motor neuron disease phenotypes in Drosophila. Therefore, the in vivo effects, the slow motility promoted by K401-4 in microtubule-gliding assays in vitro (1), and the kinetics presented here, lead us to believe that processive movement is severely impaired by the T291M mutation.
Thr291, which is conserved throughout the kinesin superfamily, is located in a hydrophobic pocket, internal to the microtubule-binding site at Loop12 (Fig. 9). Scrutiny of KHC crystal structures suggests that Thr291 forms a hydrogen bond with the carbonyl group of Tyr285 (HIPYR), which is also highly conserved and located near the end of Loop12. Upon substitution of Thr291 with methionine, interactions with Tyr285 as well as with a number of hydrophobic residues (Leu294, Leu268, Leu298, and Phe89) must be altered (Fig. 9).
Fig. 9.
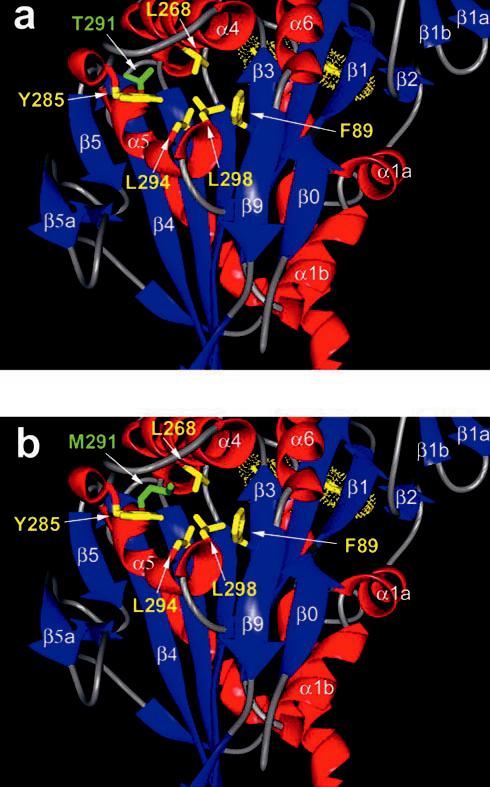
Location of the K401-4 mutation. A, crystal structure of the kinesin motor domain (rat 2kin; Ref. 4). This view has the microtubule-binding surface forward, and the active site in the rear (ADP is yellow). The side chain of Thr291 on α5 is identified in green (amino acid designation based on Drosophila KHC sequence). Thr291 appears to be part of a hydrophobic pocket that includes (in yellow) Tyr285, Leu298, Leu268, Leu294, and Phe89. All of these residues are highly conserved throughout the kinesin superfamily, suggesting that the fine structure of this pocket is critical for normal function. Note that Loop12, important in microtubule binding, is located between α4 and α5 to the top left in this view. B, view identical to that in A with the side chain of Thr291 substituted with methionine and identified in green. Note that the methionine is longer and must disrupt the critical contacts that the threonine normally makes. We propose a disruption of the hydrogen bond between Thr291 and the carbonyl of Tyr285 as well as disruption of the hydrophobic core involving Leu298, Leu268, Leu294, and Phe89.
Disturbance of this hydrophobic pocket by the T291M mutation may explain the kinetics of K401-4. It has been proposed that α4/Loop11 is analogous to the G-protein switch II region that senses the γ-phosphate of ATP (9). In vitro mutagenesis studies of KHC in the switch II region have documented the importance of the conserved glycine and glutamic acid (VDLAGSE) for ATP hydrolysis and the conformational change necessary to generate force and movement (22). The authors proposed that the glycine in kinesin forms a hydrogen bond with the γ-phosphate and triggers a conformation change between the ATP and ADP states. Further experimental work suggests that this region is important in the coupling of the nucleotide pocket and microtubule-binding surface (1, 21). Thus, the observed weak binding of ATP and microtubules by K401-4 may reflect a shift in the switch II element at the beginning of Loop11. Disruption of the hydrogen bond between Thr291 and Tyr285 could cause a structural “ripple effect” altering the positions of the adjacent Loop12, α4, and Loop11. Shifting the switch II element at the beginning of Loop11 could affect its hydrogen bond interaction with the γ-phosphate, explaining the insensitivity of K401-4 to the absence of the γ-phosphate during microtubule-stimulated ADP release.
Another consequence of the T291M substitution is the lowered affinity for ATP. One of the residues in the hydrophobic pocket, Phe89 (Fig. 9), is located on β3, two amino acids away from the P-loop consensus sequence in Loop4 which is critical for contact with the α, β phosphates of ATP (9). Disruption of the hydrophobic pocket could disrupt the position of Phe89 and alter the conformation or position of the P-loop, changing contacts with the α- and β-phosphates of the nucleotide and altering both ATP binding and switch I-switch II interactions for ATP hydrolysis.
In summary, we have identified a single amino acid change that has a dramatic effect on the ATPase cycle of kinesin by altering communication between the motor domains. The results presented for T291M illustrate the importance of understanding the functionally relevant transition states and the structural requirements for motor coordination and processive movement. Through the use of this multifaceted approach, we hope to advance the understanding of the structural requirements for motor coordination and processive movement.
Acknowledgments
We thank John M. Rosenberg for assistance with molecular modeling and structural interpretation and Eckhard Mandelkow for stimulating discussions about KHC structure. We particularly thank Jack Correia and the University of Mississippi Medical Center Analytical Ultracentrifuge Facility for assistance with the sedimentation studies.
Footnotes
This work was supported by National Institutes of Health Grants GM-54141 (to S. P. G.) and GM-46295 (to W. M. S.), American Cancer Society (ACS) Grant IRG-58-35 (to S. P. G.), and March of Dimes Birth Defects Foundation Grant 5-FY95-1136 (to S. P. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: KHC, kinesin heavy chain; Khc, Drosophila kinesin heavy chain gene; K401, KHC fragment containing the N-terminal 401 amino acids; K401-wt, wild type K401; K401-4, K401 with T291M amino acid substitution; AMP-PNP, 5′-adenylyl imidodiphosphate; Mt, microtubule; Mt·K401, microtubule-K401 complex; mantATP, 2′(3′)-O-(N-methylanthraniloyl)-adenosine 5′-triphosphate; mantADP, 2′(3′)-O-(N-methylanthraniloyl)-adenosine 5′-diphosphate.
Greene, E. A., and Henikoff, S., The kinesin home page. 2000.http://www.blocks.fhcrc.org/∼kinesin/.
REFERENCES
- 1.Brendza KM, Rose DJ, Gilbert SP, Saxton WM. J. Biol. Chem. 1999;274:31506–31514. doi: 10.1074/jbc.274.44.31506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kull FJ, Vale RD, Fletterick RJ. J. Muscle Res. Cell Motil. 1998;19:877–886. doi: 10.1023/a:1005489907021. [DOI] [PubMed] [Google Scholar]
- 3.Kozielski F, Sack S, Marx A, Thormählen M, Schönbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 4.Sack S, Müller A, Marx M, Thormählen M, Mandelkow E-M, Brady ST, Mandelkow E. Biochemistry. 1997;36:16155–16165. doi: 10.1021/bi9722498. [DOI] [PubMed] [Google Scholar]
- 5.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sablin EP, Kull FJ, Cooke R, Vale RD, Fletterick RJ. Nature. 1996;380:555–559. doi: 10.1038/380555a0. [DOI] [PubMed] [Google Scholar]
- 7.Sablin EP, Case RB, Dai SC, Hart CL, Ruby A, Vale RD, Fletterick RJ. Nature. 1998;395:813–816. doi: 10.1038/27463. [DOI] [PubMed] [Google Scholar]
- 8.Gulick AM, Song H, Endow SA, Rayment I. Biochemistry. 1998;37:1769–1776. doi: 10.1021/bi972504o. [DOI] [PubMed] [Google Scholar]
- 9.Vale RD. J. Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furch M, Fujita-Becker S, Geeves MA, Holmes KC, Manstein DJ. J. Mol. Biol. 1999;290:797–809. doi: 10.1006/jmbi.1999.2921. [DOI] [PubMed] [Google Scholar]
- 11.Hackney DD. Proc. Natl. Acad. Sci. 1994;91:6865–6869. doi: 10.1073/pnas.91.15.6865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma YZ, Taylor EW. J. Biol. Chem. 1997;272:724–730. doi: 10.1074/jbc.272.2.724. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert SP, Moyer ML, Johnson KA. Biochemistry. 1998;37:792–799. doi: 10.1021/bi971117b. [DOI] [PubMed] [Google Scholar]
- 14.Block SM. J. Cell Biol. 1998;140:1281–1284. doi: 10.1083/jcb.140.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandelkow E, Johnson KA. Trends Biochem. Sci. 1998;23:429–433. doi: 10.1016/s0968-0004(98)01278-x. [DOI] [PubMed] [Google Scholar]
- 16.Müller J, Marx A, Sack S, Song Y-H, Mandelkow E. Biol. Chem. 1999;380:981–982. doi: 10.1515/BC.1999.122. [DOI] [PubMed] [Google Scholar]
- 17.Woehlke G, Ruby AK, Hart CL, Ly B, Hom-Booher N, Vale RD. Cell. 1997;90:207–216. doi: 10.1016/s0092-8674(00)80329-3. [DOI] [PubMed] [Google Scholar]
- 18.Romberg L, Pierce DW, Vale RD. J. Cell Biol. 1998;140:1407–1416. doi: 10.1083/jcb.140.6.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoenger A, Sack S, Thormählen M, Marx A, Müller J, Gross H, Mandelkow E. J. Cell Biol. 1998;141:419–430. doi: 10.1083/jcb.141.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore JD, Song H, Endow SA. EMBO J. 1996;15:3306–3314. [PMC free article] [PubMed] [Google Scholar]
- 21.Song HB, Endow SA. Nature. 1998;396:587–590. doi: 10.1038/25153. [DOI] [PubMed] [Google Scholar]
- 22.Rice S, Lin AW, Safer D, Hart CL, Naber N, Carragher BO, Cain SM, Pechatnikova E, Wilson-Kubalek EM, Whittaker M, Pate E, Cooke R, Taylor EW, Milligan RA, Vale RD. Nature. 1999;402:778–784. doi: 10.1038/45483. [DOI] [PubMed] [Google Scholar]
- 23.Saxton WM, Hicks J, Goldstein LSB, Raff EC. Cell. 1991;64:1093–1102. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- 24.Hurd DD, Saxton WM. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Philo JS. Anal. Biochem. 2000;279:151–163. doi: 10.1006/abio.2000.4480. [DOI] [PubMed] [Google Scholar]
- 26.Correia JJ, Gilbert SP, Moyer ML, Johnson KA. Biochemistry. 1995;34:4898–4907. doi: 10.1021/bi00014a047. [DOI] [PubMed] [Google Scholar]
- 27.Stafford WF., III Anal. Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 28.Philo JS. Biophys. J. 1997;72:435–444. doi: 10.1016/S0006-3495(97)78684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert SP, Johnson KA. Biochemistry. 1993;32:4677–4684. doi: 10.1021/bi00068a028. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert SP, Johnson KA. Biochemistry. 1994;33:1951–1960. doi: 10.1021/bi00173a044. [DOI] [PubMed] [Google Scholar]
- 31.Sadhu A, Taylor EW. J. Biol. Chem. 1992;267:11352–11359. [PubMed] [Google Scholar]
- 32.Gilbert SP, Webb MR, Brune M, Johnson KA. Nature. 1995;373:671–676. doi: 10.1038/373671a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma Y-Z, Taylor EW. J. Biol. Chem. 1997;272:717–723. doi: 10.1074/jbc.272.2.717. [DOI] [PubMed] [Google Scholar]
- 34.Moyer ML, Gilbert SP, Johnson KA. Biochemistry. 1998;37:800–813. doi: 10.1021/bi9711184. [DOI] [PubMed] [Google Scholar]
- 35.Moyer ML, Gilbert SP, Johnson KA. Biochemistry. 1996;35:6321–6329. doi: 10.1021/bi960017n. [DOI] [PubMed] [Google Scholar]
- 36.Ma YZ, Taylor EW. Biochemistry. 1995;34:13242–13251. doi: 10.1021/bi00040a040. [DOI] [PubMed] [Google Scholar]