

Abstract
Phosphorylation is ubiquitous in control of protein activity, yet its effects on protein structure are poorly understood. Here we investigate the effect of serine phosphorylation in the interior of an α-helix when a salt bridge is present between the phosphate group and a positively charged side chain (in this case lysine) at i,i + 4 spacing. The stabilization of the helix is considerable and can overcome the intrinsically low preference of phosphoserine for the interior of the helix. The effect is pH dependent, as both the lysine and phosphate groups are titratable, and so calculations are given for several charge combinations. These results, with our previous work, highlight the different, context-dependent effects of phosphorylation in the α-helix. The interaction between the phosphate2− group and the lysine side chain is the strongest yet recorded in helix–coil studies. The results are of interest both in de novo design of peptides and in understanding the structural modes of control by phosphorylation.
Phosphorylation and dephosphorylation have long been known as control mechanisms for various processes in biochemistry, and roughly one-third of all proteins in eukaryotes are estimated to undergo reversible phosphorylation (1). Since the middle of the last century it has been known that protein activity can be controlled by the addition or removal of a phosphate group (2–5). The control of gene expression, macromolecule production, and cellular proliferation have since been shown to be orchestrated through multiple intracellular signal transduction pathways. These pathways, or protein kinase cascades, propagate signals received at the plasma membrane to the interior of the cell through a series of phosphorylation-controlled events.
The mechanisms of action of phosphorylation are, however, still relatively poorly understood at the structural level, though many studies of individual systems now show some conformational differences between proteins and synthetic peptides before and after phosphorylation (see, e.g., refs 6–13). Previous work from our own group (14) and others (15–17) has shown that phosphorylation at the N-terminus stabilizes α-helices but also that the effect is position dependent. When interior serine residues are phosphorylated, the effect is greatly destabilizing, but at the three N-terminal positions it is greatly stabilizing (phosphoserine at the N2 position in a helix is the most stabilizing interaction yet found at this position). More recent work has suggested that protein phosphorylation sites, especially those for serine and threonine, are predominantly disordered prior to phosphorylation and that phosphorylation sites resemble natively unstructured proteins in terms of their charge, hydrophobicity, and amino acid composition. Some of the exceptions to these sites may be crystallization artifacts, and in others it may be that the substrate undergoes an order–disorder transition prior to binding (18). However, 9 of 13 annotated phosphoserine residues, e.g., Ser57 in SpoIIAA from Bacillus subtilis (PDB files 1H4X and 1H4Z), have the phosphorylation site in the N-terminal region of an α-helix, which we have shown previously to be a very stabilizing location in synthetic peptides (14). One way in which phosphorylation can modify protein structure is thus via inducing or breaking helices. A second method, which we investigate here, is for the phosphoserine to form salt bridges to positively charged groups.
Salt bridges have previously been shown to stabilize structure in several proteins and helical peptides (19–29). Marqusee and Baldwin (21) showed that, in synthetic helices, glutamate–lysine salt bridges at i,i + 3 and i,i + 4 stabilized the helices with those at i,i + 4 giving the greater stabilization. In light of these findings we thought it possible that peptides having a phosphate group in the interior of a helix could be stabilized by including a possible salt bridge interaction with an appropriately placed positively charged side chain (lysine).
Peptide Design
Four peptides were designed (see Table 1), based around a control peptide having the sequence AAKAAAAKAAAASAAAAKAGY, with the N-terminus as an acetyl group and the C-terminus as an amide. This peptide has no i,i + 3 or i,i + 4 side-chain interactions except for a possible Lys–Tyr i,i + 3 interaction at the C-terminus at high pH. This will be present in all peptides and is distant from the area under investigation. The tyrosine residue is to allow concentration determination using absorbance spectroscopy. Glycine, commonly known as a “helix breaker”, is used to remove the tyrosine residue from the helical part of the peptide to minimize any errors in concentration determination. Lysine residues are present to aid solubility. This sequence could then be made with the serine replaced by phosphoserine, to allow a check on the effect of the phosphorylation in the helix interior.
Table 1.
Peptides Used in This Study
| peptide | sequencea | test for |
|---|---|---|
| KS5 | XAKAAAAKAAAASAAAAKAGYZ | control |
| KpS5 | XAKAAAAKAAAApSAAAAKAGYZ | effect of phosphoserine |
| KS4 | XAKAAAAKAAASAAAAAKAGYZ | lysine–serine i,i + 4 |
| KpS4 | XAKAAAAKAAApSAAAAAKAGYZ | lysine–phosphoserine i,i + 4 interaction |
X denotes an acetyl group, Z denotes an amide group, and pS denotes a phosphoserine.
Theoretical helicities were calculated using modified Lifson–Roig helix–coil theory (30), implemented to include capping and side-chain interaction parameters. The helix propagation values used are given in Table 2. The statistical mechanics program SCINT2 was used for these calculations (available from http://www.bi.umist.ac.uk/users/mjfajdg/scint.htm) (31–33). Initial helix contents for the four peptides were calculated with a default side-chain interaction (p) value of 1. This could then be adjusted in the light of circular dichroism measurements to give the stabilization energies of the interactions. To simulate different pH conditions, the helicities were calculated with combinations of both charged and uncharged lysine side chains and with phosphoserine with −1 and −2 charges. Calculated helicities are presented in Table 3 alongside the experimental values.
Table 2.
Helix Propagation Parameters Used in Calculations with SCINT2
Table 3.
Experimental and Calculated Percentage Helix Content for the Studied Peptides under Different Charge Conditions
| peptide | pH range | mean % helix (exptl) | charge on K | charge on pSer | calcd % helix |
|---|---|---|---|---|---|
| KS5 | <9.5 | 64.9 (±0.6) | +1 | n/a | 68.6 |
| KS5 | >10.7 | 70.9 (±0.7) | 0 | n/a | 76.9 |
| KS4 | <8.5 | 73.4 (±0.5) | +1 | n/a | 68.8 |
| KS4 | >11 | 76.8 (±0) | 0 | n/a | 77.4 |
| KpS5 | <5 | 58.9 (±1.5) | +1 | −1 | 56.5 |
| KpS5 | 7–9 | 45.7 (±1.8) | +1 | −2 | 43.6 |
| KpS5 | >11 | 59.2 (±1.4) | 0 | −2 | 54.8 |
| KpS4 (p = 1) | <6 | 83.2 (±0.9) | +1 | −1 | 56.0 |
| KpS4 (p = 1) | >11.5 | 67.8 (±0.7) | +1 | −2 | 41.6 |
MATERIALS AND METHODS
Peptide Synthesis
Peptides were synthesized on an Applied Biosystems 431A peptide synthesizer using Fmoc (9-fluorenylmethoxycarbonyl) solid-phase chemistry. Suitably protected amino acids were coupled to rink amide resin using HCTU [2-(6-chloro-1H-benzotriazol-1-yl)-1,1,3,3-tetra-methyluronium hexafluorophosphate] with an N-methyl-pyrrolidone (NMP) solvent. Acetylation of N-termini was carried out with pyridine and acetic anhydride. Cleavage from the resin and removal of Ser, Ser(P), Lys, and Tyr side-chain protecting groups were accomplished with 95% trifluoroacetic acid (TFA) and 5% anisole. Solvents were purchased from Applied Biosystems and other chemicals from Novabiochem.
The crude peptides were then washed and precipitated in cold diethyl ether and then lyophilized before purification using reverse-phase HPLC with a C18 column (Phenomenex, Luna 250 × 10 mm, 5 μm particle size). The peptide identity was confirmed by MALDI mass spectrometry. Peptide purity was confirmed to be >95% by analytical C18 reverse-phase HPLC using an Agilent 1100 HPLC system. The eluents were (A) water/TFA (99.9:0.1 v/v) and (B) acetoinitrile/TFA (99.9:0.1 v/v). These were run as a gradient of increasing (B) from 5% to 50%.
Circular Dichroism Measurements
A Jasco J-810 spectropolarimeter fitted with a Peltier temperature control system was used for circular dichroism measurements. Measurements were taken at a temperature of 273 K in a 1 mm path length cell. Samples were dissolved in a mixed buffer system containing 10 mM NaCl, 1 mM sodium phosphate, 1 mM sodium borate, and 1 mM sodium citrate. A blank consisting of buffer only was run during each session. To check for any self-association of the peptides, helicity was measured as a function of concentration in the range 10–150 μM. If there were no self-association, one would expect helicity to be essentially invariant over this range, whereas self-association would give a significant change with increasing concentration.
The concentrations of peptide solutions were determined using the UV absorbance of tyrosine, using the molar extinction coefficient є280 = 1209 M−1·cm−1 (35). The common units for CD measurements are mean residue ellipticity, [θ], in deg·cm·dmol−1 which, at 222 nm wavelength, can be converted to helix content (fH) using the equations (36, 37)
where T is experimental temperature (°C) and Nr is the number of residues in the peptide chain. θC and θH are the baseline ellipticities of random coil and total helix, respectively.
Titrations were performed between pH 2 and pH 12 while monitoring the ellipticity at 222 nm. These data were fitted to the Henderson–Hasselbach equation in order to determine pKa values for the various titratable groups. Two forms of the equation were used for fitting, for either one or two pKa’s in the titration range. For a fit to two pKa values, the equation
was used, whereas for titrations showing only one pKa value, we employed the equation
All fitting was performed using ProFit (v5.6.2, Quantum Software, Zurich, Switzerland) with custom-written fitting functions.
RESULTS AND DISCUSSION
Self-Association
For all four peptides studied the helicity was essentially constant over the range investigated (data not shown), indicating that no self-association or aggregation is occurring in this range. Titrations were therefore performed at concentrations of approximately 20 μM.
Peptide Titrations
The changes in [θ]222 of the peptides during titration are shown in Figures 1–4.
Figure 1.
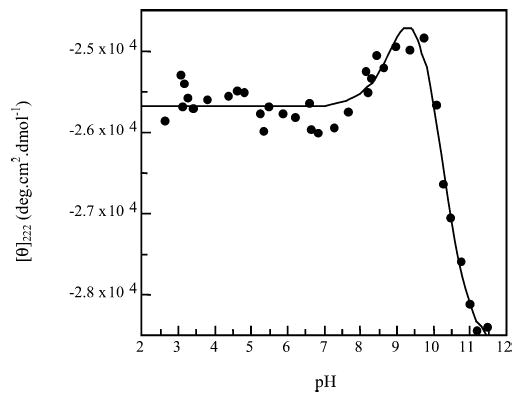
Titration of peptide KS5.
Figure 4.
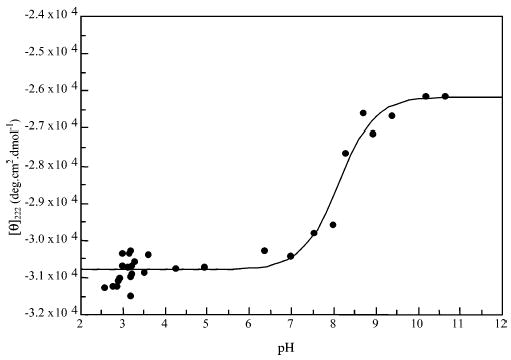
Titration of peptide KpS4.
Peptide KS5
The titration of this peptide shows two transitions. The first has a pKa of 9.1 (± 0.3) where [θ]222 increases slightly. The second has a pKa of 10.1 (±0.2) and [θ]222 decreases heavily. A positive change in [θ]222 indicates a decrease in helix content. As the tyrosine side chain has a lower pKa than that of the є-amino group on the lysine side chain, it is likely that the first transition is deprotonation of the tyrosine side chain.
The introduction of a negative charge at the C-terminus would normally destabilize the helix due to helix–macrodi-pole interactions. There is destabilization here, but at the C-terminus of the peptide there is a lysine at i − 3 from the tyrosine, which carries a positive charge on this residue. This would enable an i,i + 3 side-chain interaction to stabilize the helical C-terminus, the increased stability offsetting the effect of the negative charge somewhat.
The second transition is of the lysine side chains in the peptide. Uncharged lysine has a higher helix propagation factor (w) than that carrying a positive charge, and this could account for the increased stability at higher pH. When the side chains of the lysine residues are neutral, there is also an increased likelihood of peptide aggregation.
Peptide KS4
With the KS4 peptide we see very similar titration, with pKa values of 9.2 (±0.4) and 9.9 (±0.4). This is unsurprising as the only difference between this peptide and peptide KS5 is that the serine residue has been moved one position toward the N-terminus of the peptide to place it at i + 4 from a lysine residue in peptide KS4. This should give a very similar helix content.
Peptide KpS5
Here, the serine has been phosphorylated, and there are two transitions in the titration. The first, with pKa = 5.90 (±0.14), markedly destabilizes the helix. This is the transition from singly charged to doubly charged phosphate on the phosphoserine residue. This transition decreases the stability of the helix as, from our previous work, the helix propagation (w) value for phosphoserine with a double negative charge is very low (w = 0.045) when compared to that for singly charged phosphoserine (w = 0.125) (14). Both of these values are much smaller than that for serine alone (w = 0.36) (34), so at the lower pH range the KpS5 peptide is less helical than peptides KS4 and KS5.
The second transition stabilizes the helix and is at a pKa of 10.49 (±0.14). This is likely to be the tyrosine–lysine i,i + 3 interaction described above.
Peptide KpS4
This shows only one transition at a pKa of 8.11 (±0.10). The transition is most likely that from singly charged to doubly charged phosphate. At low pH values this peptide has a markedly lower [θ]222 than the others, indicating a much higher helix content (83%). The predicted helix content with a side-chain interaction parameter (p = 1) is 56% (see Table 3). This peptide has the possibility for an i,i + 4 salt bridge between the positively charged lysine side chain and the singly negatively charged phosphate group on phosphoserine. The very low calculated percent helix, compared to the experimental value in these instances, indicates that a value of p = 1 is too low to account for the side-chain interaction, which should markedly stabilize the helix, negating the deleterious effect of the phosphoserine, and so a value of p > 1 is required. As pH is increased and the phosphoserine becomes doubly charged, the ellipticity increases. It might be thought that the double charge on the phosphate group would increase the strength of the salt bridge and thus increase the helicity of the peptide. However, the w value for doubly charged phosphoserine is considerably lower than that for the singly charged residue, and so the helix content of the peptide is actually reduced (68%). A side-chain interaction parameter can also be estimated from this value.
Side-Chain Interaction Parameters
Using SCINT2 and altering the side-chain interaction parameter (p) for the lysine–phosphoserine1− interaction (low pH) gives a very high p value. The predicted helicity enters the experimental range at approximately p = 5 and begins to plateau at approximately p = 25 (Figure 5) before the mean experimental helicity value is reached. Increasing the value of p to infinity would not actually achieve the 83% helix content measured by experiment.
Figure 5.
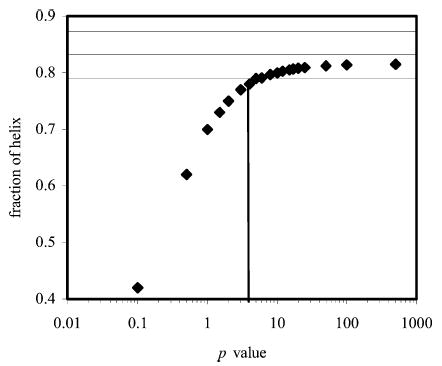
Plot showing the variation in the computed helix fraction of peptide KpS4 with the p value, as calculated using SCINT2 and singly charged phosphoserine. Horizontal lines indicate the error range of the experimental values.
Free Energy Calculations
Using ΔG = –RT ln p, a value of p = 5 gives ΔG = −0.9 kcal·mol−1 so the interaction has this free energy change as a minimum value. No maximum value could be estimated. With phosphoserine2−the variation in helix fraction with p is shown in Figure 6. A range of p values from 8 to 50 pass within the experimental range of the helix fraction. These correspond to free energy changes between −1.2 and −2.2 kcal·mol−1, respectively, so the interaction between phosphoserine2− and lysine appears stronger than that involving phosphoserine1−.
Figure 6.
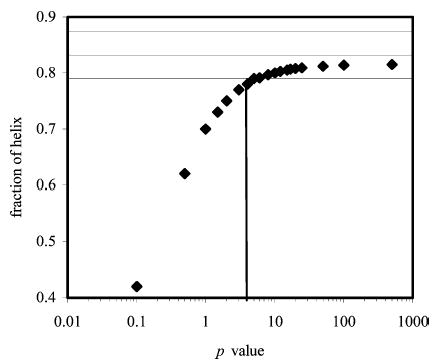
Plot showing the variation in the computed helix fraction of peptide KpS4 with the p value, as calculated using SCINT2 and doubly charged phosphoserine. Horizontal lines indicate the error range of the experimental values.
While the experimental data can still be fitted within error, there can be no upper limit of p identified in the case of pSer−1. There is no way, in the current implementation, that propagation of the effect of i,i + 4 side-chain interactions outside the five-residue region in which they are present can be accounted for. Once the intervening residues have reached maximal helicity, there is no way to increase the overall helicity of the peptide. The difference between experiment and theory in this case is therefore most likely due to the increased helicity induced outside the region bounded by the lysine–phosphoserine salt bridge.
It is also possible to calculate the change in free energy using the change in pKa for the transition from singly to doubly charged phosphate as in Andrew et al. (14):
Using this equation and the difference in pKa between peptides KpS4 and KpS5 (having phosphoserine in the singly charged state) a value of ΔG = −2.8 (±0.3) kcal·mol−1 is obtained. This value is much higher than that calculated above but is not describing precisely the same situation. The p value above is describing the change from peptide KS4 to KpS4, i.e., the phosphorylation of the serine residue already placed at i,i + 4 from the lysine. The value derived from ΔpKa is describing the energy difference upon moving the already phosphorylated serine from i,i + 5 to i,i + 4. The former position is already known to be destabilizing, with a ΔG of 1.2 kcal·mol−1 in the helix interior (14). Accounting for this gives us a free energy change for the side-chain interaction of −1.6 kcal·mol−1, which is still significantly higher than the value calculated from p. ΔG = −1.6 kcal·mol−1 is equivalent to a p value of 18, which is within in the range indicated from the calculations above and is inside the range estimated for the interaction with doubly charged phosphoserine.
This interaction energy is much higher than the only value calculated previously in synthetic peptides for a similar arginine to phosphoserine salt bridge (38) of 0.45 kcal·mol−1. However, this value was calculated using very different w values for the phosphoserine than we have found in our studies. In that study a w value of 0.46 was used for phosphoserine, whereas our previous work (14) gave values of 0.125 and 0.045 for phosphoserine with −1 and −2 charges, respectively. Our values give helicity predictions for the peptides without side-chain interactions in this study that are close to experimental values (see Table 3), confirming their accuracy. Given that a much higher helix propagation parameter was used in the earlier study, a lower p value and thus side-chain interaction energy would naturally be expected. This discrepancy can be attributed to i,i + 3 phosphoserine–arginine interactions in their peptides that are assumed to be insignificant, and this may lead them to overestimate w values and hence underestimate p for the i,i + 4 interaction. The w value that they derive for phosphoserine in this way is 0.46, which is higher than the value of 0.36 already reported for nonphosphorylated serine (34) and is an order of magnitude greater than our reported values (see above). Again, the stabilization of helical structure by phosphorylation of an internal serine is contradicted by experiment. The authors also do not use a control (nonphosphorylated) peptide as a check on their own values, which may have alerted them to this problem.
The interaction energies derived in this study are highly stabilizing and the strongest salt bridges in synthetic helix studies. In our previous work (14) a free energy change of −2.3 kcal·mol−1 was found for the inclusion of phosphoserine2− at the helix N3 position, while the value at N2 could not be calculated, though is clearly also very high. Other values measured for salt bridges range from 0.5 to 5 kcal·mol−1 (39, 40), with the strongest salt bridge being that between Asp (70) and His (31) of the bacteriophage T4 lysozyme (39). This is not within one α-helix but between an α-helix and a region of β-sheet.
In a de novo designed peptide based upon the Lac repressor tetramerization domain, Signarvic and DeGrado (13) showed how phosphorylation at the N-terminus, with appropriate design of electrostatic interactions, could stabilize a four-helix bundle by up to 4.6 kcal·mol−1. Vinson and coworkers (16) showed 1.4 kcal·mol−1 (of dimer) stabilization of a leucine zipper by phosphorylation of an interior serine residue with electrostatic interactions. These results agree well with our own findings here and show that, with the appropriate neighboring residues, phosphorylation in α-helices can be highly stabilizing. In contrast, Brooks and coworkers (41) showed how phosphorylation of a serine residue in bovine prolactin could destabilize a modeled α-helix and result in reduced activity. Their supposition was that phosphorylation of Ser90 would place a phosphate group in a steric clash with an already stabilizing, highly conserved, salt bridge between the neighboring Arg89 and Asp93 residues. Further analysis using a helical wheel representation (available at http://www.site.uottawa.ca/~turcotte/resources/HelixWheel/) indicates that the phosphate group would not be in a position to interfere with the salt bridge as it is roughly 70° around the helix axis from this salt bridge. Thus the destabilization is most probably due to the highly unfavorable pSer in the helix interior overcoming the stabilization of the Arg–Asp salt bridge.
In terms of the effects of phosphorylation on protein structure and function, the results of our studies, and those of others mentioned, show that a range of possibilities can occur. The phosphorylation can induce extra stability, either through interactions at helix termini or through salt bridge networks. It can also reduce stability, especially in α-helices, resulting in conformational changes. It can form salt bridge networks to enable protein–protein interactions, affecting the ability to form complexes or their stability. These effects depend largely upon the position and context of the phosphorylated residue and any neighboring residues with which interactions can be formed and are all potentially very large.
This helps to understand the widespread nature of phosphorylation as a control mechanism in nature. The effects of phosphorylating an amino acid side chain are diverse and depend on local context. As seen above, the presence of a nearby side chain that allows salt bridge formation can change the effect of phosphorylation from very destabilizing (>1 kcal·mol−1) to very stabilizing (<−2 kcal·mol−1).
Figure 2.
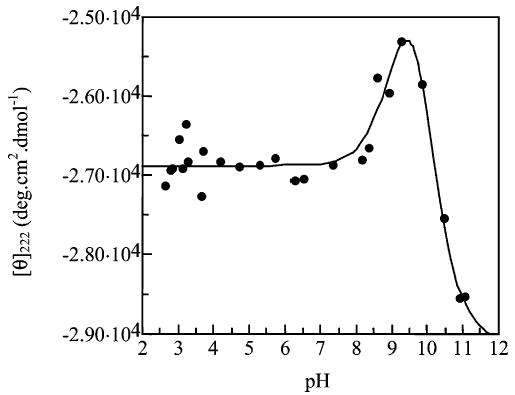
Titration of peptide KS4.
Figure 3.
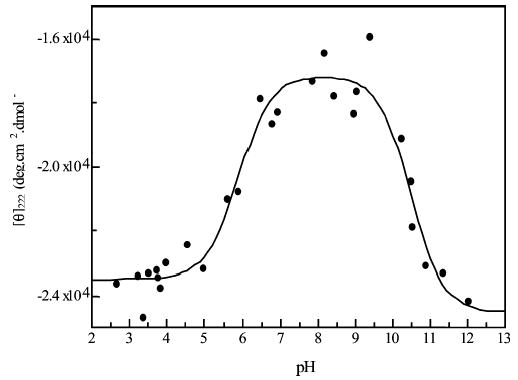
Titration of peptide KpS5.
Acknowledgments
The authors acknowledge the assistance of the Michael Barber Center for Mass Spectrometry, UMIST (now The University of Manchester), for analysis of synthetic peptides.
Footnotes
This work was funded by The Wellcome Trust (award reference 065106).
References
- 1.Marks, F. (1996) Protein Phosphorylation, VCH, Weinheim, New York, Basel, Cambridge, and Tokyo.
- 2.Burnett G, Kennedy EP. The enzymatic phosphorylation of proteins. J Biol Chem. 1954;211:969–980. [PubMed] [Google Scholar]
- 3.Fischer EH, Krebs EG. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J Biol Chem. 1955;216:121–132. [PubMed] [Google Scholar]
- 4.Krebs EG, Fischer EH. Phosphorylase activity of skeletal muscle extracts. J Biol Chem. 1955;216:113–120. [PubMed] [Google Scholar]
- 5.Green AA, Cori GT. Crystalline muscle phosphorylase. I Preparation, properties and molecular weight. J Biol Chem. 1943;151:21–29. [Google Scholar]
- 6.Fujitani N, et al. Structure determination and conformational change induced by tyrosine phosphorylation of the N-terminal domain of the α-chain of pig gastric H+,K+-ATPase. Biochem Biophys Res Commun. 2003;300:223–229. doi: 10.1016/s0006-291x(02)02794-8. [DOI] [PubMed] [Google Scholar]
- 7.Fernando P, Megeny LA, Heikkila JJ. Phosphorylation-dependent structural alterations in the small hsp30 chaperone are associated with cellular recovery. Exp Cell Res. 2003;286:175–785. doi: 10.1016/s0014-4827(03)00067-3. [DOI] [PubMed] [Google Scholar]
- 8.Krupa A, Preethi G, Srinivasan N. Structural modes of stabilization of permissive phosphorylation sites in protein kinases: Distinct strategies in Ser/Thr and Tyr Kinases. J Mol Biol. 2004;339:1025–1039. doi: 10.1016/j.jmb.2004.04.043. [DOI] [PubMed] [Google Scholar]
- 9.Zor T, et al. Roles of phosphorylation and helix propensity in the binding of the KIX domain of CREB-binding protein by constitutive (c-Myb) and inducible (CREB) activators. J Biol Chem. 2002;277:42241–42248. doi: 10.1074/jbc.M207361200. [DOI] [PubMed] [Google Scholar]
- 10.Gaudet R, et al. A molecular mechanism for the phosphorylation-dependent regulation of heterotrimeric G proteins by phosducin. Mol Cell. 1999;3:649–660. doi: 10.1016/s1097-2765(00)80358-5. [DOI] [PubMed] [Google Scholar]
- 11.Tokmakov AA, Sato K-I, Fukami Y. Phosphorylation sensitive secondary structure in a synthetic peptide corresponding to the activation loop of MAP kinase. Biochem Biophys Res Commun. 1997;236:243–247. doi: 10.1006/bbrc.1997.6941. [DOI] [PubMed] [Google Scholar]
- 12.Hamuro Y, et al. Phosphorylation driven motions in the COOH-terminal Src kinase, Csk, revelaed through enhanced hydrogen–deuterium exchange and mass spectrometry (DXMS) J Mol Biol. 2002;323:871–881. doi: 10.1016/s0022-2836(02)01003-3. [DOI] [PubMed] [Google Scholar]
- 13.Signarvic RS, DeGrado WF. De novo design of a molecular switch: phosphorylation dependent association of designed peptides. J Mol Biol. 2003;334:1–12. doi: 10.1016/j.jmb.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 14.Andrew CD, et al. Effect of phosphorylation on α-helix stability as a function of position. Biochemistry. 2002;41:1897–1905. doi: 10.1021/bi0113216. [DOI] [PubMed] [Google Scholar]
- 15.Smart JL, McCammon JA. Phosphorylation stabilizes the N-termini of α-helices. Biopolymers. 1999;49:225–233. doi: 10.1002/(SICI)1097-0282(199903)49:3<225::AID-BIP4>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 16.Szilak L, et al. Phosphorylation destabilizes alpha-helices. Nat Struct Biol. 1997;4:112–114. doi: 10.1038/nsb0297-112. [DOI] [PubMed] [Google Scholar]
- 17.Pullen K, et al. Phosphorylation of serine-46 in HPr, a key regulatory protein in bacteria, results in stabilization of its solution structure. Protein Sci. 1995;4:2478–2486. doi: 10.1002/pro.5560041204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iakoucheva LM, et al. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32:1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee WS, Park CH, Byun SM. Streptomyces griseus trypsin is stabilized against autolysis by the cooperation of a salt bridge and cation-pi interaction. J Biochem. 2004;135:93–99. doi: 10.1093/jb/mvh011. [DOI] [PubMed] [Google Scholar]
- 20.Luo R, et al. Strength of solvent-exposed salt bridges. J Phys Chem B. 1999;103:727–736. [Google Scholar]
- 21.Marqusee S, Baldwin RL. Helix stabilization by Glu−···Lys+ salt bridges in short peptides of de novo design. Proc Natl Acad Sci USA. 1987;84:8898–8902. doi: 10.1073/pnas.84.24.8898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marqusee S, Sauer RT. Contributions of a hydrogen bond/salt bridge network to the stability of secondary and tertiary structure in λrepressor. Protein Sci. 1994;3:2217–2225. doi: 10.1002/pro.5560031207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marti D, Bosshard N, Bosshard R. Electrostatic interactions in leucine zippers: Thermodynamic analysis of the contributions of Glu and His residues and the effect of mutating salt bridges. J Mol Biol. 2003;330:621–637. doi: 10.1016/s0022-2836(03)00623-5. [DOI] [PubMed] [Google Scholar]
- 24.McClain D, Gurnon G, Oakley GM. Importance of potential interhelical salt-bridges involving interior residues for coiled-coil stability and quaternary structure. J Mol Biol. 2002;324:257–270. doi: 10.1016/s0022-2836(02)01072-0. [DOI] [PubMed] [Google Scholar]
- 25.Meier M, et al. Removing an interhelical salt bridge abolishes coiled-coil formation in a de novo designed peptide. J Struct Biol. 2002;137:65–72. doi: 10.1006/jsbi.2002.4467. [DOI] [PubMed] [Google Scholar]
- 26.Olsen CA, et al. Cooperative helix stabilization by complex Arg-Glu salt bridges. Proteins: Struct Funct, Genet. 2001;44:123–132. doi: 10.1002/prot.1079. [DOI] [PubMed] [Google Scholar]
- 27.Shi Z, et al. Stabilisation of α-helix structure by side-chain interactions: Complex salt-bridges, cation–πinteractions and C–H···O H-bonds. Biopolymers. 2001;60:366–380. doi: 10.1002/1097-0282(2001)60:5<366::AID-BIP10177>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Strop PS, Mayo L. Contribution of surface salt-bridges to protein stability. Biochemistry. 2000;39:1251–1255. doi: 10.1021/bi992257j. [DOI] [PubMed] [Google Scholar]
- 29.Takano K, et al. Contribution of salt-bridges near the surface of a protein to the conformational stability. Biochemistry. 2000;39:12375–12381. doi: 10.1021/bi000849s. [DOI] [PubMed] [Google Scholar]
- 30.Lifson S, Roig AJ. On the helix-coil transition in polypeptides. J Chem Phys. 1961;34:1963–1974. [Google Scholar]
- 31.Doig AJ, et al. Determination of free energies of N-capping in α-helices by modification of the Lifson-Roig helix-coil theory to include N- and C-capping. Biochemistry. 1994;33:3396–3403. doi: 10.1021/bi00177a033. [DOI] [PubMed] [Google Scholar]
- 32.Chakrabartty A, Kortemme T, Baldwin RL. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 1994;3:843–852. doi: 10.1002/pro.5560030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stapley BJ, Rohl CA, Doig AJ. Addition of side chain interactions to modified Lifson-Roig helix-coil theory: Application to energetics of phenylalanine-methionine interactions. Protein Sci. 1995;4:2383–2391. doi: 10.1002/pro.5560041117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rohl C, Chakrabartty AA, Baldwin RL. Helix propagation and N-cap propensities of the amino acids measured in alanine-based peptides in 40 volume percent trifluoroethanol. Protein Sci. 1996;5:2623–2637. doi: 10.1002/pro.5560051225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du H, et al. PhotochemCAD: A computer-aided design and research tool in photochemistry. Photochem Photobiol. 1998;68:141–142. [Google Scholar]
- 36.Rohl CA, Baldwin RL. Comparison of NH exchange and circular dichroism as techniques for measuring the parameters of the helix-coil transition in peptides. Biochemistry. 1997;36:8345–8442. doi: 10.1021/bi9706677. [DOI] [PubMed] [Google Scholar]
- 37.Luo P, Baldwin RL. Mechanism of helix induction by trifluoroethanol: A framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water. Biochemistry. 1997;36:8413–8421. doi: 10.1021/bi9707133. [DOI] [PubMed] [Google Scholar]
- 38.Liehr S, Chenault HK. A comparison of the α-helix forming propensities and hydrogen bonding properties of serine phosphate and α-amino-γ-phoshphonobutyric acid. Bioorg Med Chem Lett. 1999;9:2759–2762. doi: 10.1016/s0960-894x(99)00469-2. [DOI] [PubMed] [Google Scholar]
- 39.Anderson DE, Becktel WJ, Dahlquist FW. pH-induced denaturation of proteins: A single salt bridge contributes 3–5 kcal/mol to the free energy of folding of T4 lysozyme. Biochemistry. 1990;29:2403–2408. doi: 10.1021/bi00461a025. [DOI] [PubMed] [Google Scholar]
- 40.Sun D-p, et al. Contributions of engineered surface salt bridges to the stability of T4 lysozyme determined by direct mutagenesis. Biochemistry. 1991;30:7142–7153. doi: 10.1021/bi00243a015. [DOI] [PubMed] [Google Scholar]
- 41.Schenck EJH, Canfield JM, Brooks CL. Functional relationship of serine 90 phosphorylation and the surrounding putative salt bridge in bovine prolactin. Mol Cell Endocrinol. 2003;204:117–125. doi: 10.1016/s0303-7207(03)00123-0. [DOI] [PubMed] [Google Scholar]