

Abstract
Models for the diversity and evolution of pathogens have branched into two main directions: the adaptive dynamics of quantitative life-history traits (notably virulence) and the maintenance and invasion of multiple, antigenically diverse strains that interact with the host's immune memory. In a first attempt to reconcile these two approaches, we developed a simple modelling framework where two strains of pathogens, defined by a pair of life-history traits (infectious period and infectivity), interfere through a given level of cross-immunity. We used whooping cough as a potential example, but the framework proposed here could be applied to other acute infectious diseases. Specifically, we analysed the effects of these parameters on the invasion dynamics of one strain into a population, where the second strain is endemic. Whereas the deterministic version of the model converges towards stable coexistence of the two strains in most cases, stochastic simulations showed that transient epidemic dynamics can cause the extinction of either strain. Thus ecological dynamics, modulated by the immune parameters, eventually determine the adaptive value of different pathogen genotypes. We advocate an integrative view of pathogen dynamics at the crossroads of immunology, epidemiology and evolution, as a way towards efficient control of infectious diseases.
Keywords: epidemiological model, strain dynamics, pertussis, infectious period, waning immunity
1. Introduction
Mathematical modelling provides an efficient way to synthesize current epidemiological and evolutionary knowledge on infectious diseases. Epidemiological models have proven helpful in deciphering epidemics, notably childhood diseases (Bartlett 1953; Schenzle 1984; Anderson & May 1991; Grenfell et al. 2001; Bjørnstad et al. 2002), AIDS (Anderson & May 1986), SARS (Anderson et al. 2004), influenza (Mills et al. 2004) and foot and mouth disease (Keeling et al. 2003). A major current preoccupation is to integrate pathogen diversity into epidemiological models and simulate strain dynamics and evolution (May & Anderson 1983; Gupta et al. 1994; Ferguson et al. 1999; Day & Proulx 2004; Stollenwerk et al. 2004).
A first approach assumes that quantitative traits of the pathogen evolve as a response to intrinsic trade-offs and extrinsic pressures from its host (ranging from demography to vaccination). The most common assumption states that virulence (i.e. the deleterious effects of the pathogen on its host) is positively correlated with transmission ability (May & Anderson 1983; van Baalen & Sabelis 1995). Accordingly, pathogens are predicted to evolve an optimal, non-zero level of virulence. Although the underlying assumptions are still debated (Ebert & Bull 2003a,b; Elliot 2003; Gandon & Day 2003), this ecological framework recently provided new insights into the potential effects of imperfect vaccines (Gandon et al. 2001) and the evolution of malaria agents (Mackinnon & Read 2004).
A second, parallel field addresses the dynamics and evolution of cross-immunity. This explores the mechanisms and implications underlying the observation that infection by a first strain of the pathogen often modulates susceptibility to subsequent infections by related strains of pathogens, due to the host's acquired immunity. Typical models consider two or more strains with identical phenotypes but different immune profiles, represented by distances in the antigenic space (Gog & Swinton 2002; Gomes et al. 2002). Models and observations have been used to explore strain dynamics in a number of systems, including influenza (Andreasen et al. 1997; Gog & Grenfell 2002), malaria (Gupta et al. 1994), dengue (Ferguson et al. 1999) and meningitis (Kamo & Sasaki 2002).
Thus, life-history variation and cross-immunity appear as two key factors driving pathogen evolution. However, they have generally not been combined in an integrated study of a single pathogen—the former approach usually predicts the advent of a particular phenotype, the latter often aims at tracking the distribution of strains circulating in a population. Here, we draw these threads together in a study motivated by the dynamics of pertussis.
Research triggered by the recent re-emergence of whooping cough in developed countries points to Bordetella pertussis as a relevant example for this challenge. Indeed, genetic typing shows that the strains from which most existing vaccines have been designed since the 1950s have been replaced by novel variants (Mooi et al. 1999; Cassiday et al. 2000; Guiso et al. 2001; Weber et al. 2001). Experiments in mice showed that existing vaccines may indeed be less protective against some of these new strains (King et al. 2001; Gzyl et al. 2004). In addition, acquired immunity against pertussis (whether elicited by vaccination or primary infection) may last for only a few years (Olin et al. 2003; Broutin et al. 2004), which could have contributed to recent epidemic outbreaks (van Boven et al. 2000). Given the lack of information on the antigenic and phenotypic variability among existing strains of B. pertussis, mathematical models are needed to investigate different possible scenarios for the invasion of novel strains and changes in epidemiology.
In this paper, we investigate how life-history variability and cross-immunity can modulate the ability of a novel pathogenic strain to invade and persist in a population, where an endemic strain is already present. Whereas adaptive dynamics traditionally rely on deterministic invasion rates to decide the outcome of competition, we demonstrate the need for stochastic simulations of the whole epidemic process to determine whether and when either strain (the resident or the invader) may go extinct. Thus we focus on one single step of the evolutionary process, rather than the long-term iterative dynamics. We use pertussis as a hypothetical (but plausible) case study of an acute infection, but our main rationale is to present a conceptual framework that can then be adapted to various acute diseases.
2. Material and methods
(a) Description of the model
The model derives from a simple susceptible–infectious–recovered (Anderson & May 1979) framework, in which the host population is divided into compartments following the steps of the infectious cycle. Infection occurs through random contacts between susceptible (S) and infected (I) individuals, hence the rate of infection is modelled by mass-action law: βSI/N, where N is the size of the population and β is the transmission rate. We assume equality of birth and death rates μ, and all newborns are initially susceptible to infection. We then consider two strains of the pathogen, labelled 1 and 2. Following primary infection by one strain and recovery, individuals are susceptible to secondary infection by the other strain only (leading to compartments and ), albeit with a probability reduced by a factor (1−φ), where φ∈[0,1] is the intensity of cross-immunity. For simplicity, both primary and secondary infections are equally infectious and recover at the same rate (γ1 and γ2 for either strain). Recovery from secondary infection leads to compartment R′. Last, we allow for loss of acquired immunity (from R1, R2 and R′), leading back to the susceptible state S. This loss occurs at a constant rate σ, the same for all infections (we made this simplistic assumption in view of the scarcity of quantitative data on the loss of immunity). A diagram of the model is shown in figure 1.
Figure 1.

Diagram of the compartmental model used in this paper. Expressions next to arrows show the per capita flow rates between compartments. Births and deaths are not shown. See main text for symbols.
We used two mathematical implementations of the model. The deterministic version relies on a classical system of differential equations
| 2.1 |
where N is the constant total population size and is the force of infection of strain i={1,2}. In subsequent analyses, we take N=1 without loss of generality, so that dynamic variables (S, I1, etc.) represent the expected proportions of each compartment in an infinite population.
For the stochastic version, we used a Monte Carlo algorithm (Bartlett 1953; Gillespie 1977) which tracks the succession of discrete events (birth, death, infection, etc.) that affect the numbers of individuals in specific compartments. Each possible event is given a weight, equal to the corresponding rate in the deterministic model (e.g. μN for a birth, γ1I1 for a recovery from primary infection with strain 1, etc.; refer to the electronic supplementary material, Section II, for the full list). The size of each compartment is now an integer which describes the actual number of individuals therein. At each time step the next event is chosen at random, according to the relative weights of the possible events, and the sizes of the compartments are updated accordingly. Extinction of either strain occurs whenever the number of its carriers reaches zero. This simple model captures the main features of pertussis biology and dynamics (van Boven et al. 2000; Keeling et al. 2001; Rohani et al. 2002).
(b) Analyses
Thanks to the simplicity of the model framework, we have only four parameters of interest. The first two (transmission rate β and recovery rate γ) are strain-specific, while the latter two (cross-immunity φ and loss of immunity σ) are assumed to be identical for the two strains.
We chose the baseline values of the strain-specific parameters in accordance with classical estimates for whooping cough (Anderson & May 1991): the infectious period (1/γ in our model) is around three weeks on average, and the basic reproductive ratio (roughly the ratio β/γ) is around 17. The latter parameter, known as R0, is a theoretical estimate of the number of secondary cases a single carrier can cause in a fully susceptible population. Interestingly, measles has a similar R0 but a much shorter infectious period (typically less than a week), hence a much higher transmission rate than pertussis (Anderson & May 1991). In evolutionary terms, this feature can be seen as a trade-off between transmission rate and infectious period (Grenfell 2001), i.e. between an ‘aggressive’ (for measles) and a ‘prudential’ (for pertussis) strategy. Recently, van Ballegooijen & Boerlijst (2004) showed that a similar trade-off can emerge in a spatially explicit model of pathogen evolution. Following these arguments, we considered two strains with equal reproductive ratios but different infectious periods. Although the variability of these traits has not yet been investigated among pertussis strains, it is important to assess potential selective pressures with the help of mathematical models (van Boven et al. 2005).
The values of the immunological parameters were unconstrained a priori due to the scarcity of data. Acquired immunity against pertussis can wane within 10 years of primary infection or vaccination, although there may be wide variability. Besides, we are not aware of any data providing estimates of cross-immunity between existing pertussis strains. We therefore considered wide ranges of variation (0≤φ≤1, immune period from 5 to 50 years).
Thus, for every pair of values of the immunological parameters, we investigated the dynamics of invasion of a novel strain with a given phenotype (infectious period and transmission rate). Practically, with the deterministic model, we assumed that the resident strain had reached its endemic equilibrium (electronic supplementary material, Section I) and introduced a fraction of one carrier of the second strain per million individuals. We used the same initial conditions for stochastic simulations, with a population size of one million to ensure maintenance of the endemic strain in the absence of a competitor (electronic supplementary material, Section III). For each set of parameter values, we ran 1000 stochastic simulations and recorded the frequencies and dates of strain extinctions. We did not reintroduce strains at any point, so each one could go extinct only once per simulation.
3. Results
(a) Deterministic model
The deterministic two-strain model has two possible outcomes. If either strain has a much lower basic reproductive ratio than its competitor, its prevalence in the population asymptotically approaches zero. This case of ‘deterministic extinction’ actually requires extreme discrepancies between the two strains (electronic supplementary material, Section I). Otherwise, the system converges towards steady coexistence of the two strains. Previous models found similar possible outcomes at equilibrium (Dietz 1979; Lipsitch 1997). Nevertheless, certain features of the transient dynamics in the latter case suggest possible strain extinctions in a finite population (figure 2). Before actually analysing stochastic simulations in the next section, we use the deterministic dynamics as a semi-quantitative predictor of extinction hazards.
Figure 2.
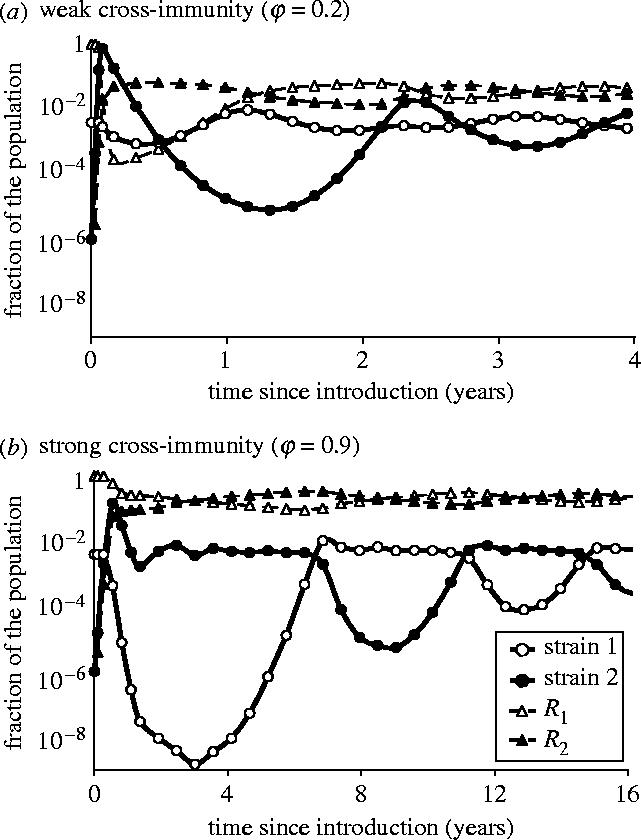
Two examples of deterministic dynamics following introduction of strain 2. Immune period: 1/σ=20 years. For both strains: R0=17, infectious period 1/γ=21 days. Life-expectancy: 1/μ=50 years. (a) Weak cross-immunity (φ=0.2). (b) Strong cross-immunity (φ=0.9).
First, we analysed the initial growth rate of strain 2. Its analytical expression can be easily derived from the differential equations (electronic supplementary material, Section I). Assuming that strain 2 has the same basic reproductive ratio (R0) as strain 1 (with possibly different infectious periods and transmission rates), the invasion of strain 2 is made possible by cross-infections only , with an initial rate equal to
| 3.1 |
So the spread of strain 2 will be all the faster because (i) the infectious period of strain 2 (1/γ2) is shorter, traded off for a higher rate of transmission, (ii) cross-immunity (φ) is weaker, and (iii) the immune period (1/σ) is longer. The first two points obey intuition and the third can be easily understood as follows: strain 1 being endemic, a longer immune period results in a higher fraction of the population in compartment R1, which can then be only infected by strain 2 (see figure 1 and electronic supplementary material, Section I). At this point, one can expect these factors to reduce the probability of initial extinction of strain 2 in the stochastic model.
Second, following invasion of strain 2, both strains run out of susceptible individuals and their prevalences drop within a few months, before bouncing back and converging towards equilibrium (figure 2). With strong cross-immunity (figure 2b), strain 1 experiences a very deep trough shortly after the epidemic peak, while strain 2 remains around endemic level until strain 1 re-invades, a few years later. Depending on the actual size of the population, deep troughs might lead to stochastic extinction of either strain.
We recorded the depths of these post-epidemic troughs of both strains for a range of parameter values (figure 3). A longer immune period causes deeper troughs for both strains. Indeed, it promotes rapid invasion of strain 2 and then slows down the replenishment of the susceptible pool for both strains. In contrast, strong cross-immunity (high φ) causes deep troughs of strain 1 only and has no clear effect on strain 2. This discrepancy is due to the dynamics of invasion (figure 2). As stated above, strain 2 initially spreads by cross-infecting individuals immune to strain 1 only (R1). Meanwhile it also depletes the pool of fully susceptible individuals (S) and, after recovery, populates the compartment of those susceptible to strain 1 only (R2). Then strong cross-immunity will prevent strain 1 from fully exploiting the latter pool, which results in a deeper trough. Strain 2 initially goes through a shallow trough if cross-immunity is strong (figure 2b), but later on it will experience a deeper trough as strain 1 bounces back. The depth of the latter trough with strong cross-immunity roughly matches that of the initial trough with weak cross-immunity (figure 2a), hence the nearly vertical isoclines for strain 2 in figure 3d–f.
Figure 3.

Minimum prevalence (troughs) of strain 1 (upper row, a–c) or strain 2 (lower row, d–f) reached after introduction of strain 2, using the deterministic model. Horizontally: immune period (1/σ), where ‘Inf’ stands for life-long immunity. Vertically: cross-immunity (φ). Left-hand column (a and d): infectious period of strain 2 (1/γ2) 14 days; middle column (b and e): 21 days; right-hand column (c and f): 28 days. Infectious period of strain 1 : 21 days. For both strains: R0=17.
The depth of troughs also depends on the genotype of strain 2. Still assuming that the two strains have equal basic reproductive ratios R0, figure 3 shows that it may be advantageous for strain 2 to have a longer infectious period than the resident strain. Indeed, this results in shallower troughs for strain 2 and deeper ones for strain 1. However, as stated above, the cost of a longer infectious period (associated with lower transmission rate) is a reduced initial growth rate. Actually, the shallower trough is due to the slower exploitation of the susceptible pool by strain 2.
Thus, in evolutionary terms, a mutant strain is expected to face a trade-off between initial extinction, favoured by low transmission rate, and post-epidemic extinction, favoured by high transmission rate. We now confront predictions from the deterministic models with the results of the stochastic model.
(b) Stochastic model
We recorded the extinctions occurring during stochastic simulations for 48 combinations of the same three parameters as before: average duration of immunity (1/σ), cross-immunity (φ) and infectious period of strain 2 (1/γ2), assuming that both strains have the same basic reproductive rate (R0=17) and that strain 1 has an infectious period of 21 days. The results are displayed in figure 4 for an initial population size of one million. We distinguished ‘initial extinctions’ of strain 2, which occurred within 100 days of introduction and did not trigger an epidemic, and ‘trough extinctions’ which occurred at least 200 days after introduction and followed an epidemic peak (within each series of simulations, there was a clear gap of around 200 days between initial and trough extinctions). The other three possible outcomes were replacement of strain 1 by strain 2, eradication of both strains in post-epidemic troughs, and coexistence. We assessed the frequencies of these five possible outcomes by running 1000 simulations for each set of parameter values.
Figure 4.

Distribution of outcomes for 48 series of 1000 stochastic simulations with different values of cross-immunity (vertical axis), infectious period of strain 2 and immune period (horizontal axis). Infectious period of strain 1 : 21 days. For both strains: R0=17. Population size: 106.
As expected, initial extinctions were more frequent with stronger cross-immunity. However, immune and infectious periods had no visible effect on initial extinctions. Indeed, equation (3.1) shows that varying the immune period from 5 to 50 years, when infection only lasts a few weeks, causes changes in the initial growth rate of around 1% only. In contrast, doubling the infectious period (28 versus 14 days) was expected to increase initial extinctions by halving the initial growth rate (equation (3.1)), but the stochastic simulations proved this prediction wrong. In fact, the time series of initial extinctions (not shown) indicate that, during the first 5–10 days that follow introduction, a novel strain with a longer infectious period (28 days) has a lower frequency of extinctions than one with a shorter infectious period (14 days). Within a month, the difference has vanished. So there appears to be an initial advantage to longer infectious period, specific to the stochastic model, which is eventually balanced by the cost of lower transmission rate.
Let us now consider those simulations in which strain 2 escaped initial extinction and spread. The distribution of post-epidemic extinctions of strain 1 or 2 shows complex interactions between the three parameters of interest. Short immune periods of up to 10 years mainly resulted in steady coexistence of the two strains, whereas long immune periods of 50 years always led to extinction of at least one strain. In the latter case, strong cross-immunity (φ≥0.6) often caused eradication of the pathogen, due to trough extinctions of both strains. Competitive exclusion of strain 1 (‘replacement’) only occurred with strong cross-immunity (φ≥0.6), while that of strain 2 (‘trough extinctions’) was more likely with weaker cross-immunity. So far, these patterns of extinctions correlate very well with the depth gradient of post-epidemic troughs in the deterministic model (figure 3), but we have only considered cross-immunity and the immune period.
The phenotype of strain 2 (expressed equivalently as infectious period or transmission rate, since their product R0 is fixed) had a more complex effect on extinctions. First, with an immune period of 20 years, an increase in the infectious period dramatically reduced the probability of trough extinctions of strain 2. Second, with strong cross-immunity (φ=0.8), an increase in the infectious period strongly promoted competitive exclusion of strain 1. Elsewhere, the genotype of strain 2 had only marginal effects, if any. This pattern contrasts with the smooth drift of trough depths in the deterministic model (figure 3) caused by similar variations in the genotype of strain 2. However, it makes sense when one considers the numerical values of these troughs, bearing in mind the population size of one million that we used in the stochastic simulations. Indeed, an incidence of less than 10−6 of either strain in the deterministic model represents an expected value of less than one infected individual. Note that this correspondence was conserved when we used smaller population sizes (see results for N=10 000 in the electronic supplementary material, Section III). Figure 3d–f shows that, with an immune period of 20 years, the expected minimum incidence of strain 2 in a population of one million increases from around one to around 100 as infectious period increases from 14 to 28 days. Corresponding stochastic simulations show a drop in the probability of trough extinctions of strain 2 from around 0.95 to nearly 0. A similar numerical argument, combining the troughs of both strains, explains why elimination of strain 1 (‘replacement’) is particularly sensitive to the genotype of strain 2 when cross-immunity φ is around 0.8. Naturally, this means that the exact frequencies of extinctions as shown in figure 4 is sensitive to the population size (electronic supplementary material, Section III). However, the qualitative effects of the three parameters of interest and their interactions are expected to be quite general.
4. Discussion
We analysed the dynamics of invasion of a novel pathogenic strain in a population, where a previous strain was endemic. Our model includes three main features which had not, to our knowledge, been combined before: cross-immunity between the two strains, waning immunity and life-history variability among strains. This begins to reconcile two previously separated approaches to strain dynamics—one inspired by immunology, the other by evolutionary ecology—including them in an epidemiological framework. Whereas a simple invasion analysis always predicts stable coexistence of the two strains, stochastic simulations show that extinctions of either strain (or even both of them) can occur after a successful invasion of the novel strain. However, certain dynamical features of the deterministic model help predict the most likely outcomes of stochastic simulations. This study brings forward novel predictions for the evolution of acute infectious diseases such as pertussis.
(a) The dynamics of invasion and extinction
Most existing models for the evolution of pathogens' life history rely on game theory to determine an evolutionarily stable strategy (Dietz 1979; Anderson & May 1982; Bremermann & Pickering 1983; Lenski & May 1994; van Baalen & Sabelis 1995; van Baalen 1998; Gandon et al. 2001). They consider the long-term evolution of pathogens, assuming that invasion automatically leads to fixation of a fitter genotype (Day & Proulx 2004). Here, we showed that invasion by a mutant pathogen—in an acute disease such as whooping cough—creates epidemics followed by troughs which can lead to extinction, thus preventing the ultimate fixation predicted by the game-theoretical approach. These considerations raise two questions.
(i) Is there a relation between the initial epidemic growth rate and the depth of the post-epidemic troughs across strains?
Equation (3.1) shows that the initial growth rate increases with longer immune period and weaker cross-immunity. Under the assumption that both strains have the same R0, a shorter infectious period (associated with a higher transmission rate) also increases the initial growth rate; otherwise, the higher R0, the higher the growth rate. The corresponding effects on the depth of troughs are contrasted. Whereas longer immune period and shorter infectious period deepen the invader's trough, cross-immunity has more complex effects. Indeed, although strong cross-immunity tends to reduce the depth of the immediate post-epidemic trough, deeper troughs can occur later when strain 1 bounces back (figure 2b). With the latter restriction, a higher growth rate of the novel strain is likely to cause a deeper post-epidemic trough, in line with analysis of the simple SIR model (Bailey 1975). In contrast, there is no simple relation between the depth of the endemic strain's trough and the invading strain's growth rate.
(ii) Do deterministic dynamics accurately predict the risks of extinction?
We expected that slow invasion rates and deep troughs would favour extinctions in the stochastic model. Again, our study demonstrated an imperfect correspondence. On the one hand, lower cross-immunity increased both the initial growth rate and the probability of initial survival of strain 2. On the other hand, a twofold increase in the infectious period, which halves the deterministic invasion rate, did not alter the frequency of initial extinctions. Besides, the depth of the troughs experienced by both strains after invasion proved a good predictor of the patterns of post-epidemic extinctions. For example, in regions of the parameter space associated with a deep trough of the endemic strain only, the latter was replaced by the invading strain in a high proportion of the stochastic simulations. The range of parameter values we explored covered all possible outcomes, from eradication of both strains to stable coexistence. Interestingly, for certain values of the immunological parameters, the invading strain's genotype had a critical effect on its probability of trough extinction. In these cases, a longer infectious period (traded for a lower transmission rate) favoured the survival of the invading strain or the elimination of the resident strain.
(b) Evolution of pathogen life history
The latter results suggest that, in certain circumstances, there can be selective pressure on the pathogen to reduce transmission rate and increase infectious period. This pressure appeared as a consequence of the immunological and epidemiological context of the model, relevant to acute infectious diseases. To our knowledge, there is no conclusive evidence of genetic variability of the traits considered here in any pathogen species (Gog et al. 2003), but it may be for lack of investigation. However, the trade-off between transmission rate and infectious period has already been considered (cf. §1). In particular, van Ballegooijen & Boerlijst (2004) showed that, when the two traits are free to evolve independently in a lattice-based model, mutants with higher rate of infection and shorter infectious period tend to take over. Indeed, the latter benefit from shorter inter-epidemic periods, allowing them to dominate the spatial contest. Since our study shows that such genotypes suffer from higher extinction rates when imperfect cross-immunity is taken into account, it would be interesting to confront the two approaches in a unified framework. Although the trade-off as we modelled it may remain speculative, the hypothesis has a conceptual value: it shows that models for pathogen evolution need to go beyond the basic reproductive ratio. The ecological dynamics of infection can create selective pressure that distinguishes strains with the same R0 but different combinations of life-history traits.
(c) General perspectives
We presented results for a single evolutionary step, following the introduction of a new genotype. The full, long-term evolutionary dynamics are basically an endless iteration of this process. Of course, there may be other factors arising, such as the number of strains simultaneously present or constraints on the variability of pathogenic traits. A thorough study of these long-term dynamics is needed to achieve unification between epidemiology, immunology and evolution.
Although some of the numerical values we used were inspired by whooping cough, the framework and the qualitative results can apply to other acute infectious diseases. Strain diversity has been established in many pathogen species, but mainly on genetic grounds. In many cases, whether these strains have different antigenic or phenotypic characteristics remains an open question. We hope that future advances in experimental and clinical research will provide directions for specific applications exploring these issues. Conversely, the predictions we formulated here could drive experimental research towards integrative approaches, beyond simple genomic or serologic typing of strains. Strain diversity must be apprehended in a dynamical context, including epidemiology, immunology and evolution.
An important extension of our modelling framework will be to include vaccination (van Boven et al. 2005). We plan to investigate the selective pressures created by a vaccine on pathogen strains, depending, again, on their antigenic and phenotypic characteristics. Thus, we hope to gain better understanding of the recent dynamics of pertussis in vaccinated countries. It is also important to cast these results in a spatial (metapopulation) framework, to account for local extinction and migrations among populations with different vaccination policies and different locally circulating strains. Ultimately, we will also need to allow explicitly for pathogen genetics to complete a thorough description of pathogen dynamics (Grenfell et al. 2004).
Acknowledgments
This research was funded by a Wellcome Trust grant to B.G. and a Lavoisier Fellowship (Ministère des Affaires Etrangères, France) to O.R., with additional support from BBSRC and the Cambridge Infectious Diseases Consortium. We are grateful to P. Rohani and to two anonymous referees for their helpful comments.
Supplementary Material
References
- Anderson R.M, May R.M. Population biology of infectious diseases: part I. Nature. 1979;280:361–367. doi: 10.1038/280361a0. doi:10.1038/280361a0 [DOI] [PubMed] [Google Scholar]
- Anderson R.M, May R.M. Coevolution of hosts and parasites. Parasitology. 1982;85:411–426. doi: 10.1017/s0031182000055360. [DOI] [PubMed] [Google Scholar]
- Anderson R.M, May R.M. The invasion, persistence and spread of infectious diseases within animal and plant communities. Phil. Trans. R. Soc. B. 1986;314:533–570. doi: 10.1098/rstb.1986.0072. [DOI] [PubMed] [Google Scholar]
- Anderson R.M, May R.M. Oxford University Press; Oxford, UK: 1991. Infectious diseases of humans: dynamics and control. [Google Scholar]
- Anderson R.M, Fraser C, Ghani A.C, Donnelly C.A, Riley S, Ferguson N.M, Leung G.M, Lam T.H, Hedley A.J. Epidemiology, transmission dynamics and control of SARS: the 2002–2003 epidemic. Phil. Trans. R. Soc. B. 2004;359:1091–1105. doi: 10.1098/rstb.2004.1490. doi:10.1098/rstb.2004.1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen V, Lin J, Levin S.A. The dynamics of cocirculating influenza strains conferring partial cross-immunity. J. Math. Biol. 1997;35:825–842. doi: 10.1007/s002850050079. doi:10.1007/s002850050079 [DOI] [PubMed] [Google Scholar]
- Bailey N.T.J. 2nd edn. Griffin; London: 1975. The mathematical theory of infectious diseases. [Google Scholar]
- Bartlett M.S. Stochastic processes or the statistics of change. Appl. Stat. 1953;2:44–64. [Google Scholar]
- Bjørnstad O.N, Finkenstädt B, Grenfell B.T. Dynamics of measles epidemics: estimating scaling of transmission rates using a time series SIR model. Ecol. Monogr. 2002;72:169–184. [Google Scholar]
- Bremermann H.J, Pickering J. A game-theoretical model of parasite virulence. J. Theor. Biol. 1983;100:411–426. doi: 10.1016/0022-5193(83)90438-1. doi:10.1016/0022-5193(83)90438-1 [DOI] [PubMed] [Google Scholar]
- Broutin H, Rohani P, Guégan J.-F, Grenfell B.T, Simondon F. Loss of immunity to pertussis in a rural community in Senegal. Vaccine. 2004;22:594–596. doi: 10.1016/j.vaccine.2003.07.018. [DOI] [PubMed] [Google Scholar]
- Cassiday P.K, Sanden G.N, Heuvelman K, Mooi F.R, Bisgard K.M, Popovic T. Polymorphism in Bordetella pertussis pertactin and pertussis toxin virulence factors in the United States, 1935–1999. J. Infect. Dis. 2000;182:1402–1408. doi: 10.1086/315881. doi:10.1086/315881 [DOI] [PubMed] [Google Scholar]
- Day T, Proulx S.S. A general theory for the evolutionary dynamics of virulence. Am. Nat. 2004;163:E40–E63. doi: 10.1086/382548. doi:10.1086/382548 [DOI] [PubMed] [Google Scholar]
- Dietz K. Epidemiologic interference of virus populations. J. Math. Biol. 1979;8:291–300. doi: 10.1007/BF00276314. [DOI] [PubMed] [Google Scholar]
- Ebert D, Bull J.J. Challenging the trade-off model for the evolution of virulence. Trends Microbiol. 2003a;11:15–20. doi: 10.1016/s0966-842x(02)00003-3. doi:10.1016/S0966-842X(02)00003-3 [DOI] [PubMed] [Google Scholar]
- Ebert D, Bull J.J. Response to: Elliot and Gandon and Day: revisiting virulence management. Trends Microbiol. 2003b;2003:208–209. [Google Scholar]
- Elliot S.L. The virulence–transmission trade-off and virulence management. Trends Microbiol. 2003;11:205–206. doi: 10.1016/s0966-842x(03)00071-4. [DOI] [PubMed] [Google Scholar]
- Ferguson N, Anderson R.M, Gupta S. The effect of antibody-dependent enhancement on the transmission dynamics and persistence of multiple-strains pathogens. Proc. Natl Acad. Sci. USA. 1999;96:790–794. doi: 10.1073/pnas.96.2.790. doi:10.1073/pnas.96.2.790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandon S, Day T. Understanding and managing pathogen evolution: a way forward. Trends Microbiol. 2003;11:206–207. doi: 10.1016/s0966-842x(03)00074-x. [DOI] [PubMed] [Google Scholar]
- Gandon S, Mackinnon M.J, Nee S, Read A.F. Imperfect vaccines and the evolution of pathogen virulence. Nature. 2001;414:751–756. doi: 10.1038/414751a. doi:10.1038/414751a [DOI] [PubMed] [Google Scholar]
- Gillespie D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977;81:2340–2361. doi:10.1021/j100540a008 [Google Scholar]
- Gog J, Grenfell B.T. Dynamics and selection of many-strain pathogens. Proc. Natl Acad. Sci. USA. 2002;99:17 209–17 214. doi: 10.1073/pnas.252512799. doi:10.1073/pnas.252512799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gog J, Swinton J. A status-based approach to multiple strain dynamics. J. Math. Biol. 2002;44:169–184. doi: 10.1007/s002850100120. doi:10.1007/s002850100120 [DOI] [PubMed] [Google Scholar]
- Gog J.R, Rimmelzwaan G.F, Osterhaus A.D.M.E, Grenfell B.T. Population dynamics of rapid fixation in cytotoxic T lymphocyte escape mutants of influenza A. Proc. Natl Acad. Sci. USA. 2003;100:11 143–11 147. doi: 10.1073/pnas.1830296100. doi:10.1073/pnas.1830296100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes M.G.M, Medley G.F, Nokes D.J. On the determinants of population structure in antigenically diverse pathogens. Proc. R. Soc. B. 2002;269:227–233. doi: 10.1098/rspb.2001.1869. doi:10.1098/rspb.2001.1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenfell B.T. Dynamics and epidemiological impact of microparasites. In: Smith G.L, Irving W.L, McCauley J.W, Rowlands D.J, editors. New challenges to health. Cambridge University Press; Cambridge, UK: 2001. pp. 33–52. [Google Scholar]
- Grenfell B.T, Bjørnstad O.N, Kappey J. Travelling waves and spatial hierarchies in measles epidemics. Nature. 2001;414:716–723. doi: 10.1038/414716a. doi:10.1038/414716a [DOI] [PubMed] [Google Scholar]
- Grenfell B.T, Pybus O.G, Gog J.R, Wood J.L.N, Daly J.M, Mumford J.A, Holmes E.C. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. doi:10.1126/science.1090727 [DOI] [PubMed] [Google Scholar]
- Guiso N, Boursaux-Eude C, Weber C, Hausman S.Z, Sato H, Iwaki M, Kamachi K, Konda T, Burns D. Analysis of Bordetella pertussis isolates collected in Japan before and after introduction of acellular pertussis vaccines. Vaccine. 2001;19:3248–3252. doi: 10.1016/s0264-410x(01)00013-5. doi:10.1016/S0264-410X(01)00013-5 [DOI] [PubMed] [Google Scholar]
- Gupta S, Swinton J, Anderson R.M. Theoretical studies of the effects of heterogeneity in the parasite population on the transmission dynamics of malaria. Proc. R. Soc. B. 1994;256:231–238. doi: 10.1098/rspb.1994.0075. [DOI] [PubMed] [Google Scholar]
- Gzyl A, Augustynowicz E, Gniadek G, Rabczenko D, Dulny G, Slusarczyk J. Sequence variation in pertussis S1 subunit toxin and pertussis genes in Bordetella pertussis strains used for the whole-cell pertussis vaccine produced in Poland since 1960. Efficiency of the DTwP vaccine-induced immunity against currently circulating B. pertussis isolates. Vaccine. 2004;22:2122–2128. doi: 10.1016/j.vaccine.2003.12.006. doi:10.1016/j.vaccine.2003.12.006 [DOI] [PubMed] [Google Scholar]
- Kamo M, Sasaki A. The effects of cross-immunity and seasonal forcing in a multi-strain epidemic model. Physica D. 2002;165:228–241. doi:10.1016/S0167-2789(02)00389-5 [Google Scholar]
- Keeling M.J, Rohani P, Grenfell B.T. Seasonally forced disease dynamics explored as switching between attractors. Physica D. 2001;148:317–335. doi:10.1016/S0167-2789(00)00187-1 [Google Scholar]
- Keeling M.J, Woolhouse M.E.J, May R.M, Davies G, Grenfell B.T. Modelling vaccination strategies against foot-and-mouth disease. Nature. 2003;421:136–1452. doi: 10.1038/nature01343. doi:10.1038/nature01343 [DOI] [PubMed] [Google Scholar]
- King A.J, Berbers G, van Oirschot H.F.L.M, Hoogerhout P, Knipping K, Mooi F.R. Role of the polymorphic region 1 of the Bordetella pertussis protein pertactin in immunity. Microbiology. 2001;147:2885–2895. doi: 10.1099/00221287-147-11-2885. [DOI] [PubMed] [Google Scholar]
- Lenski R.E, May R.M. The evolution of virulence in parasites and pathogens: reconciliation between two competing hypotheses. J. Theor. Biol. 1994;169:253–265. doi: 10.1006/jtbi.1994.1146. doi:10.1006/jtbi.1994.1146 [DOI] [PubMed] [Google Scholar]
- Lipsitch M. Vaccination against colonizing bacteria with multiple serotypes. Proc. Natl Acad. Sci. USA. 1997;94:6571–6576. doi: 10.1073/pnas.94.12.6571. doi:10.1073/pnas.94.12.6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon M.J, Read A.F. Virulence in malaria: an evolutionary viewpoint. Phil. Trans. R. Soc. B. 2004;359:965–986. doi: 10.1098/rstb.2003.1414. doi:10.1098/rstb.2003.1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May R.M, Anderson R.M. Epidemiology and genetics in the coevolution of parasites and hosts. Proc. R. Soc. B. 1983;219:281–313. doi: 10.1098/rspb.1983.0075. [DOI] [PubMed] [Google Scholar]
- Mills C.E, Robins J.M, Lipsitch M. Transmissibility of 1918 pandemic influenza. Nature. 2004;432:904–906. doi: 10.1038/nature03063. doi:10.1038/nature03063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooi F.R, He Q, van Oirschot H, Mertsola J. Variation in the Bordetella pertussis virulence factors pertussis toxins and pertactin in vaccine strains and clinical isolates in Finland. Infect. Immun. 1999;67:3133–3134. doi: 10.1128/iai.67.6.3133-3134.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olin P, Gustafsson L, Barreto L, Hessel L, Mast T.C, van Rie A, Bogaerts H, Storsaeter J. Declining pertussis incidence in Sweden following the introduction of acellular pertussis vaccine. Vaccine. 2003;21:2015–2021. doi: 10.1016/s0264-410x(02)00777-6. doi:10.1016/S0264-410X(02)00777-6 [DOI] [PubMed] [Google Scholar]
- Rohani P, Keeling M.J, Grenfell B.T. The interplay between determinism and stochasticity in childhood diseases. Am. Nat. 2002;159:469–481. doi: 10.1086/339467. doi:10.1086/339467 [DOI] [PubMed] [Google Scholar]
- Schenzle D. An age-structured model of pre- and post-vaccination measles transmission. IMA J. Math. Appl. Med. Biol. 1984;1:169–191. doi: 10.1093/imammb/1.2.169. [DOI] [PubMed] [Google Scholar]
- Stollenwerk N, Maiden M.C.J, Jansen V.A.A. Diversity in pathogenicity can cause outbreaks of meningococcal disease. Proc. Natl Acad. Sci. USA. 2004;101:10 229–10 234. doi: 10.1073/pnas.0400695101. doi:10.1073/pnas.0400695101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baalen M. Coevolution of recovery ability and virulence. Proc. R. Soc. B. 1998;265:317–325. doi: 10.1098/rspb.1998.0298. doi:10.1098/rspb.1998.0298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baalen M, Sabelis M.W. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 1995;146:881–910. doi:10.1086/285830 [Google Scholar]
- van Ballegooijen W.M, Boerlijst M.C. Emergent trade-offs and selection for outbreak frequency in spatial epidemics. Proc. Natl Acad. Sci. USA. 2004;101:18 246–18 250. doi: 10.1073/pnas.0405682101. doi:10.1073/pnas.0405682101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Boven M, De Melker H.E, Schellekens J.F.P, Kretzschmar M. Waning immunity and sub-clinical infection in an epidemic model: implications for pertussis in The Netherlands. Math. Biosci. 2000;164:161–182. doi: 10.1016/s0025-5564(00)00009-2. doi:10.1016/S0025-5564(00)00009-2 [DOI] [PubMed] [Google Scholar]
- van Boven M, Mooi F.R, Schellekens J.F.P, De Melker H.E, Kretzschmar M. Pathogen adaptation under imperfect vaccination: implications for pertussis. Proc. R. Soc. B. 2005;272:1617–1624. doi: 10.1098/rspb.2005.3108. doi:10.1098/rspb.2005.3108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber C, Boursaux-Eude C, Coralie G, Caro V, Guiso N. Polymorphism of Bordetella pertussis isolates circulating for the last 10 years in France, where a single effective whole-cell vaccine has been used for more than 30 years. J. Clin. Microbiol. 2001;39:4396–4403. doi: 10.1128/JCM.39.12.4396-4403.2001. doi:10.1128/JCM.39.12.4396-4403.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.