

Abstract
Dendritic cells (DC) play a central role in immune responses by presenting antigenic peptides to CD4+ T cells through MHCII molecules. Here, we demonstrate a TRIF–GEFH1–RhoB pathway is involved in MHCII surface expression on DC. We show the TRIF (TIR domain-containing adapter inducing IFNβ)- but not the myeloid differentiation factor 88 (MyD88)-dependent pathway of lipopolysaccharide (LPS)-signaling in DC is crucial for the MHCII surface expression, followed by CD4+ T-cell activation. LPS increased the activity of RhoB, but not of RhoA, Cdc42, or Rac1/2 in a manor dependent on LPS-TRIF- but not LPS-Myd88-signaling. RhoB colocalized with MHCII+ lysosomes in DC. A dominant-negative (DN) form of RhoB (DN-RhoB) or RhoB's RNAi in DC inhibited the LPS-induced MHCII surface expression. Moreover, we found GEFH1 associated with RhoB, and DN-GEFH1 or GEFH1's RNAi suppressed the LPS-mediated RhoB activation and MHCII surface expression. DN-RhoB attenuated the DC's CD4+ T-cell stimulatory activity. Thus, our results provide a molecular mechanism relating how the MHCII surface expression is regulated during the maturation stage of DC. The activation of GEFH1-RhoB through the TRIF-dependent pathway of LPS in DC might be a critical target for controlling the activation of CD4+ T cells.
Keywords: CD4+ T cells, dendritic cells, GEF, MHC class II, Rho, TLR
Introduction
The initial CD4+ T-cell activation is induced by the presentation of antigen peptides on the MHCII molecules of dentritic cell (DC) (Steinman and Inaba, 1985). In immature DC, MHCII molecules accumulate in late endosomal and lysosomal compartments near the nucleus. Vesicles carrying MHCII molecules are considered to be a primary site for MHCII-peptide complex formation (Peters et al, 1991). The activation stages of CD4+ T cells are regulated by a T-cell receptor (TCR)-induced signaling pathway that is initiated by an interaction with MHCII-peptide complexes on DC; the expression level of the surface MHCII-peptide complexes on DC is therefore important for determining the properties of CD4+ T cells (Robinson and Delvig, 2002). After the DC receive maturation signals through a Toll-like receptor (TLR), huge amounts of MHCII-peptide complexes first appear in lysosome-like compartments and subsequently on the DC plasma membrane, where CD4+ T cells are able to recognize them (Steinman et al, 1997). Several studies have focused on how the MHCII-peptide complexes in MHCII+ lysosome vesicles reach the cell surface. It has been suggested that cytoskeletal components play a critical role in the vesicle trafficking in many types of cells, including DC (Rodriguez-Boulan et al, 2005). It is not clear, however, how the TLR signals molecularly control the surface expression of MHCII molecules in DC.
Members of the TLR family recognize conserved microbial structures and activate both innate and adaptive immune responses through the activation of DC (Medzhitov and Janeway, 1997; Schnare et al, 2001). After TLR4 binds lipopolysaccharide (LPS), at least two signaling pathways are activated. One is dependent on myeloid differentiation factor 88 (MyD88) and the other on TRIF (TIR domain-containing adapter inducing IFNβ) (Beutler, 2004). Myd88, a TLR-binding protein, contributes to the LPS-induced activation of NF-κB and mitogen-activated protein kinases. Previous studies indicated the LPS-mediated activation of Myd88-dependent signaling induces inflammatory cytokines such as IL-6, TNFα, and IL-1. Myd88-deficient DC normally show an enhanced surface expression of MHCII and costimulatory molecules after LPS treatment (Horng et al, 2002). On the other hand, TRIF-dependent signaling, which is identified as Myd88-independent signaling from the TLR4 receptor, leads to IFNβ expression via activation of IRF3 (Hoebe et al, 2003a). The TRIF–IFNβ pathway induces the upregulation of costimulatory molecules such as CD86, CD80, and CD40; thus, IFNαβR-deficient macrophages fail to enhance costimulatory molecule expression after LPS stimulation (Hoebe et al, 2003b). Recently, the modification of actin organization by LPS-TLR4-Myd88-independent signaling was shown to play an important role in antigen uptake by DC (West et al, 2004). It is also demonstrated that TRIF-mediated signaling is regulated by TRAF6, TRAF3, RIP, TBK1, IKK-ɛ, and IRF3 (Barton and Medzhitov, 2004; Hacker et al, 2005; Oganesyan et al, 2005). However, involvement of TRIF pathway in MHCII vesicle movement has not been described.
The Rho family of GTPases, Rho, Rac, and Cdc42, play key roles in intracellular vesicle trafficking, including endocytosis and exocytosis, through dynamic regulation of the actin and tubulin cytoskeleton (Ridley, 2001). RhoA, RhoB, and RhoC are all similar in primary structure, but they appear to have some functional differences (Ridley, 2001; Burridge and Wennerberg, 2004). Activated RhoB molecules inhibit the transport of vesicles carrying EGF receptors to lysosomes (Gampel et al, 1999). Some Rho family members have been shown to function in DC (Garrett et al, 2000; Burns et al, 2001). Garrett et al (2000) showed Cdc42 is critical for endocytosis in immature DC, and Benvenuti et al (2004) showed Rac1/2 control mature DC migration towards and contact with T cells.
The activation of Rho family proteins is regulated by their binding with GDP or GTP, which is mediated by various cell-surface receptors (Kjoller and Hall, 1999). Cycling between the GDP- and GTP-bound state of Rho family members is controlled primarily by two classes of regulatory molecules: GTPase-activating proteins (GAPs), which enhance the relatively slow intrinsic GTPase activity of Rho proteins, and guanine nucleotide-exchange factors (GEFs), which catalyze the exchange of GDP for GTP. GAPs suppress Rho activity, whereas GEFs promote it. GEFH1, which associates with tubulin in a manner dependent on a Zn-finger motif, is a unique regulator of the Rho family (Ren et al, 1998). GEFH1 is known to activate Rho A, B, C, and Rac1 (Ren et al, 1998). Although roles of this molecule in DC have not been investigated, GEFH1 is known to play a role in morphological changes and cell–cell interaction in other cell types (Benais-Pont et al, 2003; Aijaz et al, 2005; Birukova et al, 2006).
Here, we showed TRIF-dependent but Myd88-independent activation of a GEFH1–RhoB pathway by LPS is involved in the surface expression of MHCII molecules, which induces the optimal CD4+ T cell activation in DC.
Results
LPS–TRIF pathway is critical for the upregulation of MHCII molecules on DC, followed by CD4+ T-cell activation
LPS-mediated TLR4 signaling stimulates two pathways: (i) MyD88-mediated and (ii) TRIF-mediated (Hoebe et al, 2003a; Barton and Medzhitov, 2004). We investigated which pathway was important for the LPS-mediated upregulation of MHCII molecules on DC. We prepared DC from TRIFKO, Myd88KO, and control mice. As TRIF-deficiency might reduce total amount of MHCII molecules in DC, we investigated total MHCII level in DC by Western blotting as described previously (Kitamura et al, 2005) and showed MHCII levels were comparable between DC derived from TRIFKO, Myd88KO, and control mice (Supplementary Figure S1). Therefore, we stimulated them with LPS to investigate MHCII expression. LPS induced the upregulation of MHCII molecules on DC from control and Myd88KO mice but not on DC from TRIFKO mice (Figure 1A and B). Moreover, we showed LPS-mediated upregulation of MHCII suppressed in TRIFKO DCs compared to other ones from 6 h after stimulation (Figure 1C).
Figure 1.
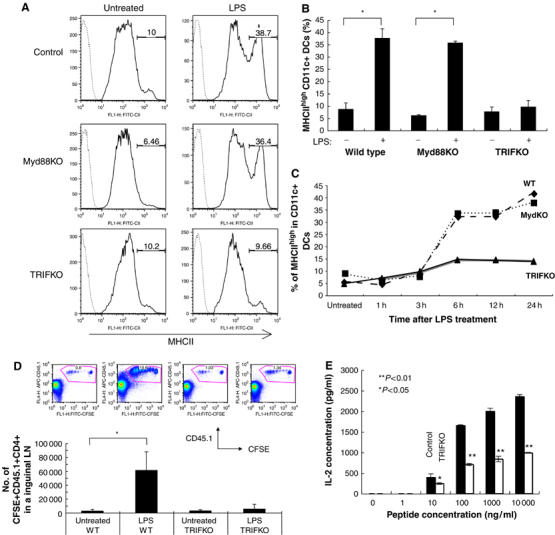
A TRIF-mediated pathway is critical for the LPS-mediated upregulation of MHCII molecules on DC and division of CD4+ T cells in vivo. (A) BMDC were prepared from TRIFKO, Myd88KO, and control mice, and stimulated with LPS. The surface expression of MHCII on CD11c+7AAD− cells was measured by a flow cytometer. The values on the profiles are the percentage of cells represented by the MHCIIhigh population. (B) The average of the percent MHCIIhigh CD11c+7AAD− cells with or without LPS treatment in five independent experiments is shown with error bars indicating 1 s.d. *P<0.05) was calculated by Student's t-test. (C) The percent MHCIIhigh CD11c+7AAD− cells with or without LPS treatment is shown. These experiments were performed four times independently, and representative data are shown. (D) BMDC were prepared from TRIFKO and control mice, stimulated with LPS and loaded with peptide-P25. The cells were injected at footpads of C57BL/6 mice that were transferred with CFSE-P25 TCR-Tg CD4+ T cells (CD45.1+). T-cell division in the inguinal lymphonodes was analyzed by CFSE profiles of CD45.1+CD4+ cells. The values in the FACS profiles are the percentage of cells represented by the CD45.1+CFSE+ population. The average of the number of CFSE+CD45.1+CD4+T cells in the each condition of three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (E) CD4+ T cells of p25 TCR-Tg mice were co-cultured with BMDC that were prepared from TRIFKO and control mice, and incubated with or without LPS in the presence of the antigenic peptide (1–10 000 ng/ml) for 2 days. IL-2 in the culture supernatants was measured by ELISA. These experiments were performed three times, independently, and representative data are shown. The average IL-2 production is shown with error bars indicating 1 s.d. The P-value (<0.05 or <0.01) was calculated by Student's t-test and is indicated by * or **.
We next investigated whether TRIFKO-DC have normal level of CD4+ T-cell stimulatory activity. LPS-stimulated wild-type or TRIFKO DC were pulsed with peptide-P25 and injected in footpads of C57BL6 mice, which were transferred with CFSE (5- and 6-carboxyfluorescein diacetate succinimidyl ester)-labeled p25-CD4+ T cells (CD45.1+). While the wild type DC induced strong division of p25-CD4+ T cells in vivo, the TRIFKO ones hardly enhanced T-cell division (Figure 1D). We also showed both TRIFKO DCs and control DCs reached almost similarly to inguinal lymphnodes in this experiment (Supplementary Figure S2). However, the defect of in vivo T-cell activation could be due to too many other factors besides MHCII expression. To rule out these possibilities better, we performed an in vitro assay of peptide presentation and showed TRIFKO DCs had reduced T-cell stimulatory activity compared to the control ones at any concentration of the antigen peptide (Figure 1E). From these results, it is reasonable that defect of LPS-mediated MHCII upregulation is at least in part involved in reduced T-cell stimulatory activity of TRIFKO DCs, although we also observed TRIFKO DCs defect LPS-mediated upregulation of costimulatory molecules (Supplementary Figure S3). All these results suggested that a TRIF-dependent pathway of TLR4 signaling in DC is critical for the surface expression of MHCII, which at least in part plays a role for activation of CD4+ T cells.
Activity of RhoB but not of RhoA, Cdc42, or Rac1/2 is enhanced in DC by LPS
The Rho family of small GTPases plays a pivotal role in the dynamic regulation of vesicle transport through the remodeling of cytoskeletal components (Camera et al, 2003). We confirmed that cytoskeletal components were rearranged after LPS treatment in DC (West et al, 2004) (data not shown). We hypothesized that the TRIF-mediated pathway activates Rho family member(s), which are necessary for the alteration of cytoskeletal components to traffic intracellular MHCII+ lysosome vesicles toward the cell surface of DC after LPS stimulation. RhoB protein level was increased until 3 h after LPS stimulation (Figure 2A). Importantly, LPS-mediated RhoB expression and RhoB activity per cells dramatically increased 3 h after LPS stimulation (Figure 2A). As we wish to analyze the activity of each Rho family member per unit of RhoB protein, the samples were adjusted so that each lane contained the same amount of total RhoB, based on the results of Western blotting before the real part. We found that the RhoB-activity per unit was enhanced starting at 1.5 h and peaked 3 h at least until 6 h after LPS treatment in DC (Figure 2B and C). In contrast, we did not observe any increase in RhoA, Cdc42, or Rac1/2 activity per unit in DC (Figure 2B and C). The Cdc42-activity per unit gradually decreased after LPS treatment, as described by Mellman's group (Garrett et al, 2000; Figure 2B and C). We also analyzed the activity of RhoGTPases short time points, 5 and 15 min, after LPS stimulation and showed they did not significantly alter their activity within the short time periods (Supplementary Figure S4). Consistent with this result, Watts and co-workers demonstrated that LPS-mediated signal slightly increase Cdc42 but did not increase Rac1/2 activity short time periods (West et al, 2004). Moreover, as described above, we observed that RhoB mRNA and its product increased after LPS treatment (Figure 2A and data not shown); the results indicated the total RhoB activity in one dendritic cell dramatically increased after LPS treatment (see also Figure 2A, right panel). Together with the results shown in Figure 1, these data suggested that RhoB activation by LPS might play an important role in the upregulation of MHCII molecules in DC.
Figure 2.
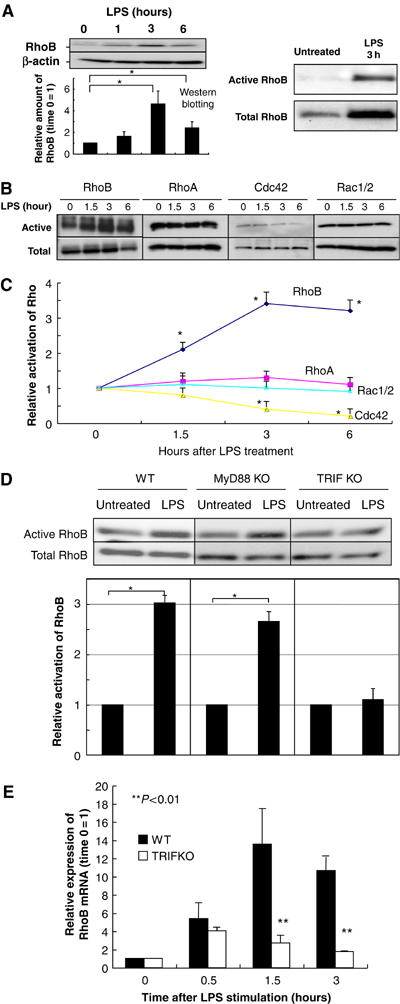
RhoB activity is enhanced by the LPS–TRIF pathway in DC. (A) (Left panel) BMDC were prepared from wild-type mice and stimulated with LPS. The protein levels of RhoB was investigated. We put the same number of CD11c+ cells that were magnetically sorted in each lane. Western blotting for RhoB is performed and relative expression of RhoB is shown in the figure. We calculated ‘time 0' as 1. Representative data from three independent experiments are shown. The relative amount of RhoB is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (Right panel) BMDC were prepared from wild-type mice and stimulated with LPS for 3 h. The protein levels of RhoB (activated or total) was investigated. The same number of CD11c+ cells that were magnetically sorted is applied in each lane. These experiments were performed at least three times independently, and representative data are shown. (B, C) BMDC were prepared from wild-type mice and stimulated with LPS for the indicated time period. CD11c cells were sorted and the activities of Rho family were investigated. The amount of total RhoB was adjusted to be the same in each lane in this experiment. The activities were compared with each other by defining the LPS-untreated control as 1. These experiments were performed three times independently, and representative data are shown. The relative activation level is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (D) BMDC were prepared from TRIFKO, Myd88KO, and control mice and stimulated with LPS for 6 h. CD11c cells were sorted and the activities of RhoB were investigated. The activities were compared with each other by defining the LPS-untreated control as 1. These experiments were performed three times independently, and representative data are shown. The relative activation level is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (E) BMDC were prepared from wild type and TRIFKO mice and stimulated with LPS. Real-time PCR of RhoB was performed. We showed relative expression of RhoB in the figure. We calculated ‘time 0' as 1. Representative data from three independent experiments are shown. The relative amount of RhoB is shown with error bars indicating 1 s.d. The P-value (<0.01) was calculated by Student's t-test and is indicated by **.
RhoB activation by LPS is dependent on the TRIF-mediated pathway in DC
We next investigated whether the TRIF-dependent but Myd88-independent pathway enhanced the RhoB activity in DC after LPS stimulation. We prepared DC from TRIFKO, Myd88KO, and wild-type mice and stimulated them with LPS. We again analyzed the activity of RhoB per unit. RhoB activation per unit increased in the DC derived from MyD88KO but not TRIFKO mice after LPS stimulation (Figure 2D). We also confirmed that RhoA, Cdc42, or Rac1/2 activity did not increase in DC from TRIFKO mice after LPS treatment (data not shown). Moreover, we observed that LPS-mediated increase of RhoB mRNA and its product was dependent on TRIF-mediated signaling (Figure 2E and data not shown). All these results clearly showed the TRIF-mediated pathway of LPS signaling is important for RhoB activation in DC.
RhoB colocalized with MHCII in DC
The data described above suggested that RhoB might play a key role in the LPS-TRIF-mediated surface expression of MHCII molecules in DC. This hypothesis was supported by the observation that RhoB distribution was highly correlated with the localization of MHCII+ lysosomes either with or without LPS treatment (Figure 3A, see arrow heads). Moreover, a detailed colocalization analysis of MHCII and RhoB was performed with the profiles using TCS-SP2 and we confirmed a strong colocalization between MHCII and RhoB in LPS-treated DC (Figure 3B). We also counted BMDC after staining with the anti-RhoB antibody and showed a significant colocalization between RhoB and MHCII+ lysosome tubules after LPS treatment in the outside of the perinuclear area in DC (Figure 3B). In contrast, RhoA was mainly found in the cytoplasm, but partially colocalized with MHCII+ lysosomes regardless of LPS treatment (data not shown). All these data support the idea that RhoB is involved in a regulatory mechanism of the surface expression of MHCII on DC after LPS treatment.
Figure 3.
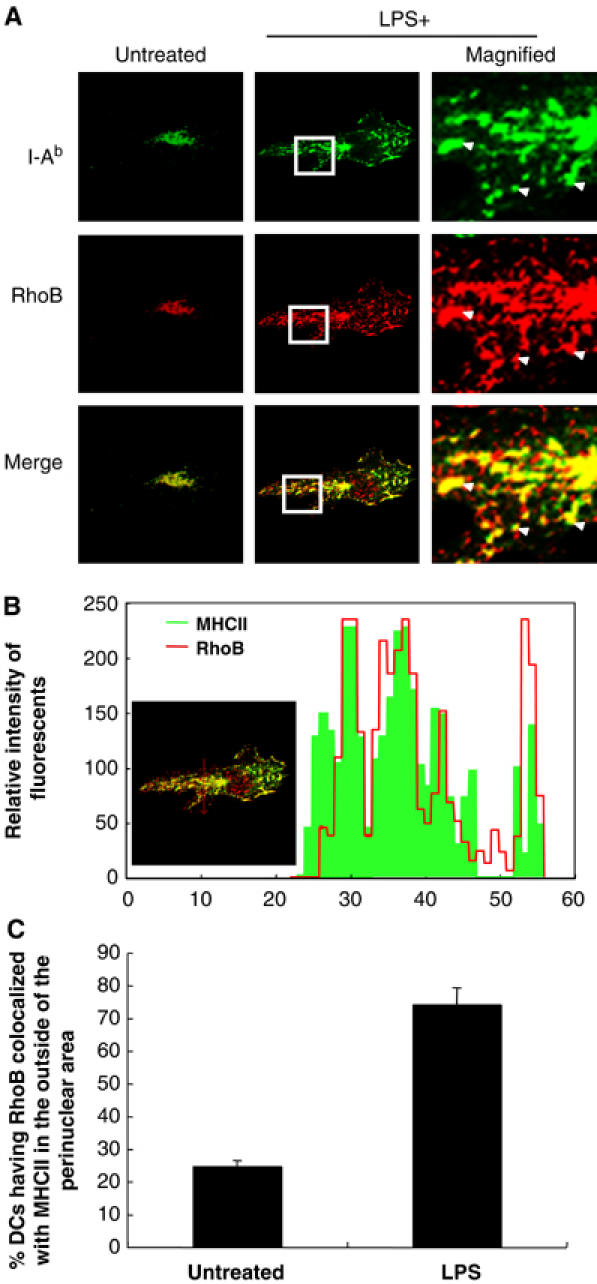
RhoB colocalized with MHCII+ lysosomes in DC. (A) BMDC were prepared from wild-type mice, incubated with or without LPS for 3 h, and fixed. The I-Ab and RhoB molecules were analyzed by confocal microscopy. Arrowheads indicate MHCII+ lysosomes colocalized with RhoB. Representative data from more than five independent experiments are shown. (B) A detailed colocalization analysis of MHCII and RhoB was performed with the profiles using TCS-SP2 (Leica) (upper graph). (C) The amount of DC that had RhoB molecules colocalized with MHCII+ lysosomes in the outside of the perinuclear area after LPS treatment was calculated by the confocal microscopy results (bottom graph). We analyzed total 50–100 MHCII+ cells in three independent experiments of day 6-BMDC cultures and shown with error bars indicating 1 s.d.
A dominant negative form of RhoB (DN-RhoB) and RNAi of RhoB suppressed the LPS-mediated surface expression of MHCII molecules on DC
To confirm the importance of RhoB activity for the surface expression of MHCII, we used a retroviral gene delivery system for DN-RhoB. The infection efficiency, as evaluated by Thy1.1 expression on the cell surface, was routinely 3–6% in CD11c+ cells (data not shown) and we used only Thy1.1+ cells, which were sorted by Moflo, in the analysis described below. DN-Rho molecules might be cytotoxic as reported previously (Gomez et al, 1997), but DN-RhoB expression in DC did not increase apoptotic cells (7AAD+ cells) (data not shown). Then, we analyzed the activity of RhoB per unit. DN-RhoB, but not a control vector (Mock), significantly inhibited the RhoB activity in DC after LPS treatment (more than 50% reduction of RhoB activity) (Figure 4A). As shown in Figure 4B, DN-RhoB but not DN-RhoA, or Mock infection significantly suppressed the LPS-mediated surface expression of MHCII molecules, indicating that RhoB, but not RhoA, is critical for the LPS-mediated surface expression of MHCII. This was consistent with the fact that LPS stimulation activated RhoB but not RhoA as shown in Figure 2B. Moreover, we observed that siRNA of RhoB significantly decreased both RhoB mRNA level and LPS-induced surface expression of MHCII on DC (Figure 4C and Supplementary Figure S5) like DN-RhoB transduction (Figure 4B). Thus, we concluded that effects of the RhoB's RNAi and the DN-RhoB are the same at least on MHCII movement in DCs.
Figure 4.
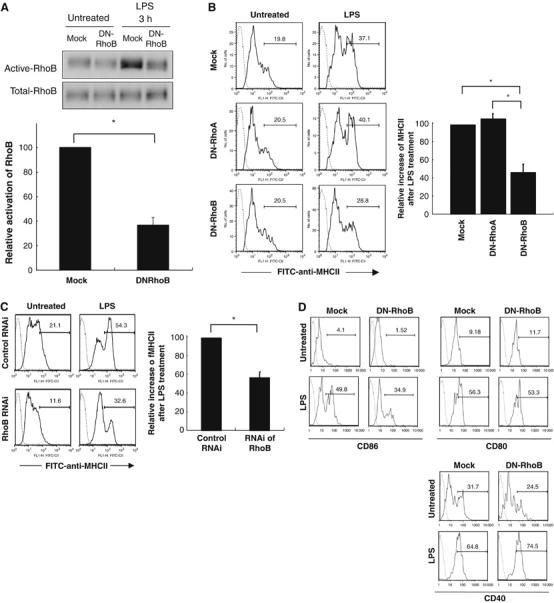
DN-RhoB expression and RNAi of RhoB treatment inhibited the LPS-mediated surface expression of MHCII in DC. BMDC were prepared from wild-type mice, infected with Thy1.1+ retrovirus carrying or lacking DN-RhoB or carrying DN-RhoA, and then incubated with or without LPS stimulation for 12 h. (A) The RhoB activity was evaluated. We calculated the increase amount of activated RhoB by intensity of the bands. We calculated ‘the intensity increased after LPS stimulation in DC Mock-infected' as 100. The average relative activation level from three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (B) The resulting DC were processed by flow cytometer. The values on the profiles are the percentage of cells represented by the MHCIIhigh population in the gate for Thy1.1+CD11c+ cells. These experiments were performed five times independently, and representative data are shown. The average relative increase of MHCIIhigh population after LPS treatment in five independent experiments is shown in the figure with error bars indicating 1 s.d. The increases were compared with each other by defining the Mock-infected control as 1. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (C) BMDC were prepared from wild-type mice, treated them with RNAi for RhoB from 36 h before LPS treatement, and then incubated with or without LPS stimulation for 12 h. Surface expression of MHCII was analyzed in the presence or absence of LPS. We calculated the increase of MHCIIhigh population in each profile (see the bar). We calculated ‘the percent increased after LPS stimulation in DC control RNAi treated ‘ as 100. The average relative activation level from three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (D) BMDCs were prepared from wild-type mice, infected with Thy1.1+ retrovirus carrying or lacking DN-RhoB, and then incubated with or without LPS stimulation for 12 h. The resulting DCs were processed by flow cytometer. These experiments were performed four times independently, and representative data are shown.
Although we showed a transduction of constitutive active form (CA) RhoB in DC altered actin organization and MHCII+ vesicle positioning (Supplementary Figure S6), surface expression of MHCII molecules just slightly increased (Supplementary Figure S6), suggesting that another molecular mechanism in addition to activation of RhoB is necessary to induce full expression of MHCII on DC.
The inhibitory effect of DN-RhoB was also observed in LPS-induced expression of CD86 but not CD80 and CD40, while transduction of DN-RhoA had no effect on these expressions (Figure 4D and data not shown). This result suggested that not only MHCII but also CD86 surface expression is regulated by RhoB molecules and, in other words, RhoB-mediated regulation is specific at least for MHCII and CD86 but not for CD80 and CD40 after LPS stimulation.
GEFH1 associated and colocalized with RhoB in DC
We next wished to identify molecules that might regulate RhoB in DC. We therefore investigated molecules that were associated with RhoB in DC. We focused on GEF, which usually have around 100 kDa, because Rho activity is mainly enhanced by GEFs (Rossman et al, 2005). We prepared a GST-fusion protein of the activated form of RhoB (GST-RhoB) and incubated it with the lysates of LPS-stimulated DC. A 100-kDa molecule was associated with GST-RhoB in the lysates from LPS-stimulated DC (see arrowhead, Supplementary Figure S7). MALDI-TOF mass analysis revealed that the 100-kDa molecule was GEFH1 (Ren et al, 1998). These results suggested that GEFH1 is associated with RhoB in DC. Additionally, a GST-fusion protein of the wild type of RhoB was also associated with GEFH1 (data not shown), suggesting GEFH1 associates with both active and inactive forms of Rho as described previously (Ren et al, 1998). We further investigated whether GEFH1 was colocalized with RhoB molecules in DC. We prepared a polyclonal GEFH1 antibody and confirmed its specificity by ELISA and immunohistochemistry (Koga et al, 2004) (data not shown). Confocal microscopy analysis revealed that GEFH1 colocalized with RhoB in DCs regardless of LPS stimulation (see Figure 5A, arrow heads). These results showed GEFH1 is associated and colocalized with RhoB molecules in DC.
Figure 5.
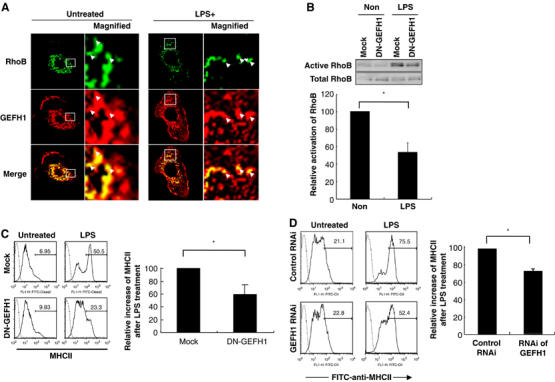
RhoB colocalized with GEFH1 and DN-GEFH1 expression or RNAi of GEFH1 suppressed the LPS-mediated RhoB activation and surface expression of MHCII in DC. (A) BMDC were prepared from wild-type mice, incubated with or without LPS for 3 h, and fixed. Expression of RhoB and GEFH1 were analyzed by confocal microscopy. Arrowheads indicate MHCII+ lysosomes colocalized with GEFH1. We performed at least five independent experiments, analyzed more than 30 MHCII+ cells in each experiment, and showed a representative result. (B) BMDC were prepared from wild-type mice, infected with control or DN-GEFH1-carrying retroviral vectors, and incubated with or without LPS. The RhoB activity was evaluated. The average relative activation level from three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (C) The control or DN-GEFH1-carrying DC were processed by flow cytometer. The values on the profiles are the percentage of cells represented by the MHCIIhigh population. These experiments were performed three times, independently, and representative data are shown. The average frequency of MHCIIhigh cells in the Thy1.1+CD11c+ cells from three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *. (D) BMDC were prepared from wild-type mice, treated them with RNAi for GEFH1 from 36 h before LPS treatement, and then incubated with or without LPS stimulation for 12 h. Surface expression of MHCII was analyzed in the presence or absence of LPS. We calculated the increase of MHCIIhigh population in each profile (see the bar). We calculated ‘the percent increased after LPS stimulation in DC control RNAi treated ‘as 100. The average relative activation level from three independent experiments is shown with error bars indicating 1 s.d. The P-value (<0.05) was calculated by Student's t-test and is indicated by *.
DN-GEFH1 and RNAi of GEFH1 inhibited the LPS-mediated RhoB activation and surface expression of MHCII in DC
We next investigated whether GEFH1 activity was involved in the LPS-induced RhoB activation in DC. We used a DN-GEFH1 that had a point mutation in the Dbl-homology domain, which causes it to lose its GEF activity (Krendel et al, 2002). We analyzed the activity of RhoB per unit after infection of DN-GEFH1 in DCs. The samples were adjusted so that each lane contained the same amount of total RhoB, based on the results of Western blotting before the real part. We transduced DN-GEFH1 into DC by retroviral vector, and found that DN-GEFH1 inhibited the LPS-mediated RhoB activation per unit in DC (Figure 5B). Importantly, DN-GEFH1 significantly suppressed the surface level of MHCII expression after LPS treatment in DC (Figure 5C) as did DN-RhoB (Figure 4B). RNAi of GEFH1 significantly but partially inhibited LPS-mediated surface expression of MHCII molecules in DCs (Figure 5D). Possible reasons of this partial inhibitory effect is that (i) the GEFH1 RNAi partially inhibits expression of GEFH1 mRNA in DCs (Supplementary Figure S5) and/or (ii) GEFH1 protein is relatively stable and has a long half-life. All these results demonstrated that GEFH1 is involved in both LPS-mediated RhoB activation and the surface expression of MHCII in DC.
Expression of DN-RhoB but not DN-RhoA in DC inhibited their CD4+ T-cell stimulatory activity
We finally investigated whether DN-RhoB expression in DC reduced their T cells stimulatory activity. CD4+ T cells were isolated from CD4+ TCR transgenic mice, p25 (Tamura et al, 2004), and cocultured with DC that had been transfected with DN-RhoB, DN-RhoA, or empty vector (Mock), in the presence or absence of various concentration of the antigenic peptide. The activation of p25 CD4+ T cells by DC in the presence of TCR-specific peptide was monitored by the production of IL-2. The IL-2 secretion from p25 T cells was significantly attenuated in DC expressing DN-RhoB compared with Mock controls or DN-RhoA transduction after LPS stimulation (Figure 6). Together with the results that CD86 expression is dependent on LPS-mediated RhoB activation (Figure 2D), these results indicated the LPS-mediated increase in RhoB activity in DC is critical for CD4 T-cell activation at least via regulation of the DC surface expression level of MHCII and CD86 costimulatory molecules.
Figure 6.
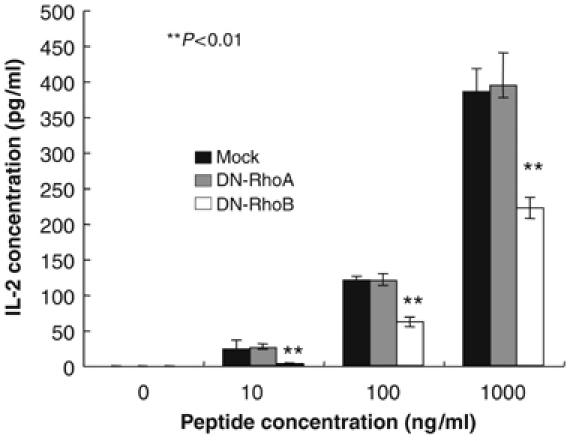
DC containing DN-RhoB failed to stimulate CD4+T cells. CD4+T cells of p25 TCR-Tg mice were co-cultured with BMDC that were infected with retroviral vectors carrying DN-RhoB, DN-RhoA, or lacking it (Mock), and incubated with or without LPS in the presence of an antigenic peptide for 2 days. IL-2 in the culture supernatants was measured by ELISA. These experiments were performed three times, independently, and representative data are shown. The average IL-2 production is shown with error bars indicating 1 s.d. The P-value (<0.01) was calculated by Student's t-test and is indicated by **.
Discussion
Here we demonstrated that a TRIF–GEFH1–RhoB pathway is involved in the LPS-mediated surface expression of MHCII molecules. This molecular mechanism at least in part explains how the surface expression of MHCII is regulated in the maturation stage of DC. Furthermore, we showed RhoB activation by LPS stimulation plays a role for the activation of CD4 T–cells, suggesting that the TRIF–GEFH1–RhoB pathway is involved in a signaling pathway for inducing the adaptive immune response via DC after LPS stimulation.
What is the role of RhoB activation in the surface expression of MHCII molecules? It is well accepted that Rho family members regulate intracellular vesicle trafficking by controlling cytoskeletal components (Ridley, 2001). MHCII molecules that are stored in lamp2+ lysosomes localize to the perinuclear area of immature DC. Activation signals, such as those mediated by TLR, induce the movement of MHCII+ lysosomes to the plasma membrane. The reorganization of tubulin filaments in DC is reported to be important for the tubulation of MHCII+ lysosomes after the engagement of the DC with T cells (Boes et al, 2002), the dynamic changes in actin organization modified by TLR4-LPS signaling play an important role in antigen uptake into DC (West et al, 2004) and we confirmed that the reorganizations of cytoskeletal components were induced by LPS treatment (data not shown). All these data suggested that cytoskeletal components are involved in the regulation of antigen presentation via MHCII+ lysosome trafficking.
RhoB has been shown to localize to intracellular vesicles, while other family members, RhoA and RhoC are largely cytoplasmic (Adamson et al, 1992). Here, we show RhoB molecules highly colocalized with MHC Class II+ vesicles in DC (Figure 3A). In addition, we observed that cytoskeletal components at least in part colocalized with MHCII+ lysosomes after LPS stimulation (data not shown). Therefore, it is possible that RhoB controls MHCII+ lysosome trafficking from the perinuclear area to the plasma membrane in DC by regulating cytoskeleton components. Moreover, it should be pointed out that RhoB-mediated regulation is specific at least for MHCII and CD86 but not for CD80 and CD40 molecules after LPS treatment (Figure 4D). Thus, this result strongly suggested that not all recycling endocytic molecules could be controlled by RhoB-mediated regulation after LPS stimulation.
As Turley et al (2000) showed the importance of actin organization for recruitment of MHCII to the cell surface, it is reasonable that RhoB mainly regulates actin organization. However, Neefjes and co-workers demonstrated that microtubule-mediated movement of MHCII-containing lysosomes was regulated by Rab7 and dynein-dynactin motors (Wubbolts et al, 1999; Jordens et al, 2001). It is also possible that RhoB regulates tubulin filaments to activate Rab7 and dynein-dynactin motor pathway. Since Neefjes and co-workers also reported that Dia1 is required for the formation of the actin coat around endosomes downstream of RhoB (Fernandez-Borja et al, 2005), we transfected DN-Dia1 (Arakawa et al, 2003) in DC and investigated LPS-mediated surface expression of MHCII molecules. However, DN-Dia1 did not affect the MHCII expression in the DC (data not shown). Further analysis is needed to identify effecter molecule(s) in downstream of RhoB for MHCII+ vesicle movement in DCs. Anyway, activation of RhoB alone is not enough for surface expression of MHCII on DC; expression of active form of RhoB failed to induce full expression of surface MHCII molecules (Supplementary Figure S6), although actin organization was likely to be altered and MHCII vesicles was spread out from the perinuclear area (Supplementary Figure S6). These results suggested the involvement of additional LPS-activated signaling in the full expression of surface MHCII molecules. To investigate critical step(s) for the RhoB-mediated MHCII vesicle movement in DCs, we analyzed the localization of MHCII molecules at various time points after LPS stimulation in the presence or absence of RhoB RNAi. We showed movement of MHCII+ vesicles at least partially suppressed (i) between MHCII+Lamp2+ and MHCII+Lamp2− vesicles (see 6 h data in Supplementary Figure S8) and (ii) between MHCII+Lamp2− vesicles and surface MHCII+ expression (see 12 h data in Supplementary Figure S8). These results suggested that RhoB plays a role from early to late steps of MHCII vesicle trafficking in DCs.
As we demonstrated that (i) GEFH1 was associated and colocalized with RhoB in DC (Supplementary Figures S5A and 7), (ii) the LPS-mediated activation of RhoB was suppressed by DN-GEFH1 and RNAi of GEFH1 (Figure 5B and C), (iii) DN-RhoB, RNAi of RhoB, DN-GEFH1, and RNAi of GEFH1 showed the inhibitory effect on LPS-induced surface expression of MHCII molecules (Figures 4B, C , 5C and D) and (iv) LPS activated RhoB but not RhoA, Rac1/2 nor Cdc42 (Figure 2B and C), we concluded that GEFH1 that is associated with RhoB plays a role for the activation of RhoB molecules in DC. The data here indicated GEFH1-mediated RhoB activation is at least in part involved in the regulation of MHCII surface expression in DC, although we are not completely able to neglect a possibility that RhoB plus other Rho GTPases that might be controlled by GEFH1 are involved in regulation of MHCII+ compartment movement.
A unique feature of GEFH1 is that it binds to tubulin filaments through a Zn-finger motif in its N-terminal end (Krendel et al, 2002). GEFH1 can be activated when it departs from tubulin filaments (Krendel et al, 2002). Therefore, it is interesting that LPS stimulation induced the alteration of tubulin molecules near the MHCII+ lysosomes at the perinuclear region during the early period after LPS stimulation (data not shown). These data suggest that the LPS-mediated alteration of tubulin filaments might induce GEFH1 activation, which could trigger MHCII+ lysosome trafficking in DC. Another unique feature of this molecule is that GEFH1 binds both active RhoA (RhoA-GTP) and inactive one (RhoA-GDP) (Ren et al, 1998), while GEFs in general bind most tightly to nucleotide-free or DN Rho proteins, and bind very poorly to active Rho proteins (Jaffe and Hall, 2005). Consistent with this data, we observed active form of RhoB also bound to GEFH1 (Supplementary Figure S7 and data not shown).
TRIF-mediated signaling is known to induce the surface expression of costimulatory molecules via IFNβ-mediated signaling (Hoebe et al, 2003b). We investigated whether LPS-mediated signaling directly or via some factor(s), including IFNβ, induced RhoB activation. Treatment of DC with cycloheximide (CHX), an inhibitor of translational events, inhibited both the expression of IL-12 mRNA and the LPS-induced RhoB activity almost completely (data not shown), suggesting that the RhoB activation by LPS is dependent on translational events. As type I-IFNs are reported to be critical targets for TRIF-mediated signaling (Hoebe et al, 2003a), and type I-IFNs are known activation factors for DC (Montoya et al, 2002), we first hypothesized that the TRIF-mediated upregulation of MHCII molecules is dependent on type I-IFNs. However, this was not the case: LPS stimulation enhanced the surface level of MHCII molecules as well as the RhoB activation almost normally in both IFNα/βRKO and control DC (Supplementary Figure S9 and data not shown), suggesting that the LPS-mediated surface expression of MHCII molecules was mainly independent on an LPS–IFNβ-mediated pathway. Consistent with this result, deficiency of IRF3 molecules did not show any effect on LPS-mediated surface expression of MHCII molecules (data not shown). It was reported that increase of the RhoB protein level might be important for RhoB activation (Wheeler and Ridley, 2004). As expected, both the RhoB mRNA and protein levels increased after LPS treatment in DC (Figure 2A and E). Moreover, increase of RhoB mRNA and protein levels was dependent on TRIF-mediated pathway of LPS (Figure 2E and data not shown). Although we showed the involvement of GEFH1 for the activation of RhoB molecules after LPS stimulation, we also hypothesize that an increase in RhoB molecules themselves by LPS plays a role in triggering RhoB activation in DC. Further study is needed to test this hypothesis.
The transduction of DN-RhoB into DC significantly suppressed both the RhoB activity and the LPS-mediated enhancement of the MHCII surface expression on DC compared to mock- and DN-RhoA-infected ones (Figure 4A, B, and data not shown). For this experiment, the specificity of the DN-RhoB is critical, since Rho family members have a high level of homology (Wheeler and Ridley, 2004). Several groups have used the same DN-RhoB, we used here and showed that it strongly abrogated the RhoB activity (Vardouli et al, 2005; Zhang et al, 2005). We also observed that the transduction of DN-RhoB only minimally affected the basal activity levels of RhoA and Cdc42 in DC compared to Mock transduction (data not shown). Moreover, we always used a retrovirus vector carrying DN-RhoA or an empty Mock vector as controls for DN-RhoB experiments and showed these controls had little effects compared to DN-RhoB transduction in DC. Importantly, we also observed that siRNA of RhoB decreased both RhoB mRNA level and LPS-induced surface expression of MHCII on DC (Figures 4C and 5). We further showed the transduction of DN-GEFH1 and RNAi of GEFH1 significantly suppressed both the RhoB activity and the LPS-mediated enhancement of the MHCII surface expression on DC (Figure 5B–D and Supplementary Figure S5). Taking these findings together, we concluded that LPS-mediated RhoB activation, which is at least in part controlled by GEFH1, is involved in the regulation of surface expression of MHCII in DC.
Materials and methods
Mice
C57BL/6 mice were purchased from SLC (Shizuoka, Japan). C57BL/6/MyD88 KO (Kaisho et al, 2001) and C57BL/6/TRIFKO (Yamamoto et al, 2003) (backcrossed more than five times to C57BL/6) were used. IFNα/βRKO were purchased from B & K Universal (Hull, UK). C57BL/6/P25 TCR-Tg were a gift from Dr Takatsu (University of Tokyo) (Tamura et al, 2004). C57BL6/SJL were purchased from the Jackson Laboratory (Bar Harbar, ME). The mice were maintained at the Institute of Experimental Animal Sciences at Osaka University Medical School.
BMDC
BMDC were generated as described (Park et al, 2004). We have shown a typical flowcytometer phenotype of DC before and after LPS stimulation (Kitamura et al, 2005).
Antibodies and reagents
Antibodies against RhoA (119), RhoB (119), and RhoB (C-5) (for confocal analysis) were purchased from Santa Cruz. We checked the specificity of the RhoB antibody using RhoA or RhoB fusion protein and showed anti-RhoB antibody (119) from Santa Cruz reacted against only GST-RhoB (Supplementary Figure S10), suggesting the RhoB antibody made by Santa Cruz is a RhoB specific. Antibodies against Rac1/2 (23A8) and Cdc42 (C-20) were purchased from Upstate Biotechnology and Santa Cruz, respectively. Rhotekin-Rho-Binding-Domain-agarose, PAK-Rac/cdc42-binding-domain-sepharose, and Mg2+ Lysis/Wash Buffer were purchased from Upstate Biotechnology. The anti-Thy1.1 (OX-7) was from BD. Anti-I-Ab (M5/114.15.2) was from eBioscience, and anti I-Ab (KH74) was purchased from Biolegend. CFSE, Phalloidin, goat anti-rabbit IgG, and anti-mouse IgG were from Molecular Probes. LPS from Escherichia coli, serotype:055:B5 were from Sigma. RNAi of RhoB and GEFH1 were purchased from Dharmacon.
Flow cytometer analysis
BMDC were incubated with LPS (100 ng/ml) for 12 h. The cells were stained with antibodies for MHC class II and CD11c. The resulted cells were analyzed by flow cytometry (BD, Tokyo).
Measurement of GTPase activity
The activities of Rho family members were analyzed using small GTPase activation assay kits (Upstate Biotechnology).
Confocal microscopy
BMDC on glass slides were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature (RT), and then washed twice with PBS. BMDC were sometimes sorted by Moflo and the resulted cells that were adhered to poly-L-lysine-coated glass slides (BD) for 10 min were fixed with the 4% paraformaldehyde and then washed twice with PBS. For immunostaining, BMDC were permeabilized with BD Perm/Wash Buffer (BD) then blocked with 1% BSA in BD Perm/Wash Buffer. Primary antibodies were diluted in BD Perm/Wash Buffer containing 1% BSA, then added to the BMDC and left for 1 h at RT. The cells were washed three times with PBS, then labeled with secondary antibodies using the same procedure as for the primary antibodies. We used the following concentrations of antibodies: anti-GEFH1, 1:20 dilution, anti-I-Ab, 1:50 dilution; anti-RhoB, 1:20; Phalloidin, 0.8 U/sample; goat anti-mouse IgG, goat anti-rabbit IgG; 1/100. The glass plates were mounted in Dako-Cytomation-Fluorescent Mounting Medium (DAKO) and viewed using the LSM 5 Pas confocal microscopy system (Carl Zeiss). Imaging was performed using a Plan-Apochromat 63 × /1.4 Oil DIC I lens. Laser lines at 488 and 543 nm were used for excitation of FITC, Alexa Fluor-488, PE, Alexa Fluor-594, and emissions wavelengths were separated by band pass (505–530 nm) or long pass (560 nm) filters, respectively. The pinhole size was set to 1.2–1.6 Airy Units, and the frame scan rate was 7.86 s. Images were optimized using LSM 5 Pas software release 3.2 (Carl-Zeiss), and transferred to Photoshop 7.0 (Adobe Systems) to produce the final figures. The confocal data shown in this study are representative of at least four independent experiments.
DNA constructs
pDrive-GEFH1 was made by inserting full-length GEFH1 cDNA into pDrive vector (QIAGEN) (Supplementary Table 1), and sequenced. DN-GEFH was prepared by site-directed mutagenesis to generate a Tyr-to-Ala mutation at residue 395. After the construct vector was digested with NotI and ClaI, the full-length DN GEFH1 cDNA was inserted into MSCV-Thy1.1 retroviral-vector. DN-RhoB, CA-RhoB, and DN-RhoA were constructed by PCR, creating ClaI and NotI sites. The PCR products were cloned into the pDrive-vector, and sequenced. The resulting-constructs were digested with ClaI and NotI and cloned into MSCV-Thy1.1 vector. GST-tagged CA-RhoB expression vector was constructed by PCR, creating HindIII and SalI sites. The PCR products were cloned into the pDrive-vector, and sequenced. After the construct vector was digested with SalI and HindIII, the CA-RhoB cDNA was inserted into the pGEX-6p-3 vector (Amersham-Biosciences).
Transduction of DN-GEFH1, DN-RhoA, and DN-RhoB
The retroviral vector containing the DN-GEFH1, DN-RhoA, or DN-RhoB gene was transduced into DC as described (Park et al, 2004). CD11c+Thy1.1+ cells were identified as infected BMDC. For calculation of the relative increase of MHCII after LPS treatment in Figures 4B and 5C, we calculated the increased percentage of MHCIIhigh cells in Mock-infected DCs before and after LPS treatment as 100 ng/ml.
RNAi transfection in DCs
RNAi for RhoB and GEFH1 were obtained from Dharmacon and transfected in DCs for 24 or 48 h by Geneporter (Gene therapy systems).
DC migration assay
DCs were labeled with CFSE and injected into the footpad (3 × 106 DCs each). Draining lymph nodes were isolated and the total number of CFSE+ cells was measured by FACS.
RT–PCR and real-time PCR
BMDC were stimulated with LPS. The CD11c-positive cells were purified by a Dendritic Cell Enrichment System (BD). The purity of the CD11c+ BMDC was routinely >95%. RT–PCR of RhoB and GEFH1 was performed using specific primers (Supplementary Table 1). The DNAs were amplified at 25 cycles (94 → 58 → 72 degree) with a thermal cycler system (Perkin Elmer).
Real-time PCR was performed using a SYBR Green PCR Master Mix (Applied Biosystems) using RhoB specific primers (Supplementary Table 1). PCRs were performed using the GeneAmp 7000 Sequence Detection System (Applied-Biosystems). The amounts of the transcripts were normalized to the HPRT transcript.
Western blotting of RhoB
The CD11c+ BMDC were lysed with NP40 buffer and subjected to SDS–PAGE and Western blotting with anti-RhoB antibody.
Preparation of GST fusion protein
GST-fusion CA-RhoB (GST-RhoB in Supplementary Figure S6), GST-fusion normal RhoA, GST-fusion normal RhoB, or control GST molecules was prepared using a general method.
Identification of proteins by peptide mass mapping
BMDC were stimulated with LPS for 3 h, and the CD11c-positive cells were magnetically sorted. The resulting CD11c cells were lysed with Triton X-100 buffer and immunoprecipitated with GST-RhoB. The precipitated samples were subjected to SDS–PAGE and transferred onto a Mini Problot membrane (Applied Biosystems), which was then stained with Colloidal Gold Total Protein Stain (Bio-Rad). The PVDF-immobilized proteins were reduced, S-carboxymethylated, and digested in situ with Achromobacter protease I (Iwamatsu and Yoshida-Kubomura, 1996). Molecular mass analyses was performed by matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry using an ABI PerSeptive Biosystem Voyager DE/RP (Applied Biosystems). Identification of the proteins was conducted by comparing the molecular mass determined by MALDI-TOF/MS with theoretical peptide masses from the proteins registered in NCBInr.
Anti-GEFH1 antibody
An anti-GEFH1 antibody was prepared as described previously (Koga et al, 2004).
CD4+ T-cell stimulation in vitro
Purified P25 CD4 T cells were stimulated with 1–10 000 or 10–1000 ng/ml peptide-25 in the presence of 2000-rad-irradiated DC (5 × 103 or 1 × 105/culture). After 48 h, the culture supernatants were subjected to ELISA.
CD4+ T-cell stimulation in vivo
Purified P25 CD4 T cells from C57BL/6/p25 TCR Tg/SJL were labeled with CFSE, and 5 × 106 cells were transferred into C57BL/6 by intravenous injection. For immunization, LPS-stimulated BMDC from wild type or TRIFKO mice were sorted and loaded with 20 μg/ml of pepetide-P25. The cells (5 × 105) were injected at a footpad of adoptively transferred mice. At 4 days after injection, we analyzed T-cell division in the inguinal-lymphonodes by CFSE profiles of CD45.1+CD4+ cells.
ELISA
Concentrations of IL-2 in the culture supernatant were measured by ELISA (Biosource).
Statistical testing
Student's t-test was used for the statistical testing between two groups.
Supplementary Material
Supplementary Figures S1 to S10
Supplementary Table 1
Acknowledgments
We thank Dr Kiyoshi Takatsu and Dr Thomas Mitchell for P25 TCR Transgenic mice and pMSCV-IRES-Thy1.1 vector, respectively. We thank Ms Ryoko Masuda, Ayako Kubota, Emi Iketani, and Takayo Hayashi for their excellent technical assistance. This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan, and by the Uehara Foundation and the Osaka Foundation for the Promotion of Clinical Immunology.
References
- Adamson P, Paterson HF, Hall A (1992) Intracellular localization of the P21rho proteins. J Cell Biol 119: 617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aijaz S, D'Atri F, Citi S, Balda MS, Matter K (2005) Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev Cell 8: 777–786 [DOI] [PubMed] [Google Scholar]
- Arakawa Y, Bito H, Furuyashiki T, Tsuji T, Takemoto-Kimura S, Kimura K, Nozaki K, Hashimoto N, Narumiya S (2003) Control of axon elongation via an SDF-1alpha/Rho/mDia pathway in cultured cerebellar granule neurons. J Cell Biol 161: 381–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R (2004) Toll signaling: RIPping off the TNF pathway. Nat Immunol 5: 472–474 [DOI] [PubMed] [Google Scholar]
- Benais-Pont G, Punn A, Flores-Maldonado C, Eckert J, Raposo G, Fleming TP, Cereijido M, Balda MS, Matter K (2003) Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J Cell Biol 160: 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, Tybulewicz VL, Amigorena S (2004) Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science 305: 1150–1153 [DOI] [PubMed] [Google Scholar]
- Beutler B (2004) Inferences, questions and possibilities in Toll-like receptor signalling. Nature 430: 257–263 [DOI] [PubMed] [Google Scholar]
- Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AA (2006) GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 290: L1139–L1145 [DOI] [PubMed] [Google Scholar]
- Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL (2002) T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature 418: 983–988 [DOI] [PubMed] [Google Scholar]
- Burns S, Thrasher A, Blundell MP, Machesky L, Jones GE (2001) Configuration of human dendritic cell cytoskeleton by Rho GTPases, the WAS protein, and differentiation. Blood 98: 1142–1149 [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K (2004) Rho and Rac take center stage. Cell 116: 167–179 [DOI] [PubMed] [Google Scholar]
- Camera P, da Silva JS, Griffiths G, Giuffrida MG, Ferrara L, Schubert V, Imarisio S, Silengo L, Dotti CG, Di Cunto F (2003) Citron-N is a neuronal Rho-associated protein involved in Golgi organization through actin cytoskeleton regulation. Nat Cell Biol 5: 1071–1078 [DOI] [PubMed] [Google Scholar]
- Fernandez-Borja M, Janssen L, Verwoerd D, Hordijk P, Neefjes J (2005) RhoB regulates endosome transport by promoting actin assembly on endosomal membranes through Dia1. J Cell Sci 15: 2661–2670 [DOI] [PubMed] [Google Scholar]
- Gampel A, Parker PJ, Mellor H (1999) Regulation of epidermal growth factor receptor traffic by the small GTPase rhoB. Curr Biol 9: 955–958 [DOI] [PubMed] [Google Scholar]
- Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, Galan JE, Mellman I (2000) Developmental control of endocytosis in dendritic cells by Cdc42. Cell 102: 325–334 [DOI] [PubMed] [Google Scholar]
- Gomez J, Martinez C, Giry M, Garcia A, Rebollo A (1997) Rho prevents apoptosis through Bcl-2 expression: implications for interleukin-2 receptor signal transduction. Eur J Immunol 27: 2793–2799 [DOI] [PubMed] [Google Scholar]
- Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M (2005) Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439: 204–207 [DOI] [PubMed] [Google Scholar]
- Hoebe K, Du X, Georgel P, Janssen E, Tabeta K, Kim SO, Goode J, Lin P, Mann N, Mudd S, Crozat K, Sovath S, Han J, Beutler B (2003a) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424: 743–748 [DOI] [PubMed] [Google Scholar]
- Hoebe K, Janssen E, Kim SO, Alexopoulou L, Flavell RA, Han J, Beutler B (2003b) Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat Immunol 4: 1223–1229 [DOI] [PubMed] [Google Scholar]
- Horng T, Barton GM, Flavell RA, Medzhitov R (2002) The adaptor molecule TIRAP provides signalling specificity for Toll-like receptors. Nature 420: 329–333 [DOI] [PubMed] [Google Scholar]
- Iwamatsu A, Yoshida-Kubomura N (1996) Systematic peptide fragmentation of polyvinylidene difluoride(PVDF)-immobilized proteins prior to microsequencing. J Biochem (Tokyo) 120: 29–34 [DOI] [PubMed] [Google Scholar]
- Jaffe AB, Hall A (2005) Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol 21: 247–269 [DOI] [PubMed] [Google Scholar]
- Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, Janssen H, Wubbolts R, Neefjes J (2001) The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol 11: 1680–1685 [DOI] [PubMed] [Google Scholar]
- Kaisho T, Takeuchi O, Kawai T, Hoshino K, Akira S (2001) Endotoxin-induced maturation of MyD88-deficient dendritic cells. J Immunol 166: 5688–5694 [DOI] [PubMed] [Google Scholar]
- Kitamura H, Kamon H, Sawa S-I, Park S-J, Katunuma N, Ishihara K, Murakami M, Hirano T (2005) IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity 23: 491–502 [DOI] [PubMed] [Google Scholar]
- Kjoller L, Hall A (1999) Signaling to Rho GTPases. Exp Cell Res 253: 166–179 [DOI] [PubMed] [Google Scholar]
- Koga H, Shimada K, Hara Y, Nagano M, Kohga H, Yokoyama R, Kimura Y, Yuasa S, Magae J, Inamoto S, Okazaki N, Ohara O (2004) A comprehensive approach for establishment of the platform to analyze functions of KIAA proteins: generation and evaluation of anti-mKIAA antibodies. Proteomics 4: 1412–1416 [DOI] [PubMed] [Google Scholar]
- Krendel M, Zenke FT, Bokoch GM (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 4: 294–301 [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Janeway CA (1997) Innate immunity: impact on the adaptive immune response. Curr Opin Immunol 9: 4–9 [DOI] [PubMed] [Google Scholar]
- Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF (2002) Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 99: 3263–3271 [DOI] [PubMed] [Google Scholar]
- Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G (2005) Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439: 208–211 [DOI] [PubMed] [Google Scholar]
- Park SJ, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S, Kamimura D, Ueda N, Iwakura Y, Ishihara K, Murakami M, Hirano T (2004) IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J Immunol 173: 3844–3854 [DOI] [PubMed] [Google Scholar]
- Peters PJ, Neefjes JJ, Oorschot V, Ploegh HL, Geuze HJ (1991) Segregation of MHC class II molecules from MHC class I molecules in the Golgi complex for transport to lysosomal compartments. Nature 349: 669–676 [DOI] [PubMed] [Google Scholar]
- Ren Y, Li R, Zheng Y, Busch H (1998) Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem 273: 34954–34960 [DOI] [PubMed] [Google Scholar]
- Ridley AJ (2001) Rho proteins: linking signaling with membrane trafficking. Traffic 2: 303–310 [DOI] [PubMed] [Google Scholar]
- Robinson JH, Delvig AA (2002) Diversity in MHC class II antigen presentation. Immunology 105: 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Boulan E, Kreitze G, Musch A (2005) Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol 6: 233–347 [DOI] [PubMed] [Google Scholar]
- Rossman KL, Der CJ, Sondek J (2005) GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 6: 167–180 [DOI] [PubMed] [Google Scholar]
- Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2: 947–950 [DOI] [PubMed] [Google Scholar]
- Steinman RM, Inaba K (1985) Stimulation of the primary mixed leukocyte reaction. Crit Rev Immunol 5: 331–348 [PubMed] [Google Scholar]
- Steinman RM, Pack M, Inaba K (1997) Dendritic cells in the T-cell areas of lymphoid organs. Immunol Rev 156: 25–37 [DOI] [PubMed] [Google Scholar]
- Tamura T, Ariga H, Kinashi T, Uehara S, Kikuchi T, Nakada M, Tokunaga T, Xu W, Kariyone A, Saito T, Kitamura T, Maxwell G, Takaki S, Takatsu K (2004) The role of antigenic peptide in CD4+ T helper phenotype development in a T cell receptor transgenic model. Int Immunol 16: 1691–1699 [DOI] [PubMed] [Google Scholar]
- Turley SJ, Inaba K, Garrett WS, Ebersold M, Unternaehrer J, Steinman RM, Mellman I (2000) Transport of peptide-MHC class II complexes in developing dendritic cells. Science 288: 522–527 [DOI] [PubMed] [Google Scholar]
- Vardouli L, Moustakas A, Stournaras C (2005) LIM-kinase 2 and cofilin phosphorylation mediate actin cytoskeleton reorganization induced by transforming growth factor-beta. J Biol Chem 280: 11448–11457 [DOI] [PubMed] [Google Scholar]
- West MA, Wsllin R, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C (2004) Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science 305: 1153–1157 [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ (2004) Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res 301: 43–49 [DOI] [PubMed] [Google Scholar]
- Wubbolts R, Fernandez-Borja M, Jordens I, Reits E, Dusseljee S, Echeverri C, Vallee RB, Neefjes J (1999) Opposing motor activities of dynein and kinesin determine retention and transport of MHC class II-containing compartments. J Cell Sci 112: 785–795 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301: 640–643 [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhu J, Bu X, Cushion M, Kinane TB, Avraham H, Koziel H (2005) Cdc42 and RhoB activation are required for mannose receptor-mediated phagocytosis by human alveolar macrophages. Mol Biol Cell 16: 824–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures S1 to S10
Supplementary Table 1