

Abstract
This study identified CD63, a member of the tetraspanin family, as a TIMP-1 interacting protein by yeast two-hybrid screening. Immunoprecipitation and confocal microscopic analysis confirmed CD63 interactions with TIMP-1, integrin β1, and their co-localizations on the cell surface of human breast epithelial MCF10A cells. TIMP-1 expression correlated with the level of active integrin β1 on the cell surface independent of cell adhesion. While MCF10A cells within a three-dimensional (3D) matrigel matrix form polarized acinar-like structures, TIMP-1 overexpression disrupted breast epithelial cell polarization and inhibited caspase-mediated apoptosis in centrally located cells, necessary for the formation and maintenance of the hollow acinar-like structures. Small hairpin RNA (shRNA)-mediated CD63 downregulation effectively reduced TIMP-1 binding to the cell surface, TIMP-1 co-localization with integrin β1, and consequently reversed TIMP-1-mediated integrin β1 activation, cell survival signaling and apoptosis inhibition. CD63 downregulation also restored polarization and apoptosis of TIMP-1 overexpressing MCF10A cells within a 3D-matrigel matrix. Taken together, the present study identified CD63 as a cell surface binding partner for TIMP-1, regulating cell survival and polarization via TIMP-1 modulation of tetraspanin/integrin signaling complex.
Keywords: apoptosis, cell survival signaling, epithelial cell polarization, integrin, tetraspanin
Introduction
The family of tissue inhibitors of metalloproteinases (TIMPs), consisting of four members (TIMP-1 to -4), regulates dynamic processes of extracellular matrix (ECM) turnover and remodeling as well as activities of growth factors and their cell surface receptors, in part through inhibition of matrix metalloproteinases (MMPs). However, mounting evidence suggests that TIMPs can regulate angiogenesis, cell survival, proliferation and apoptosis independent of their MMP inhibitory activity via interactions with cell adhesion molecules or growth factor receptors (Guedez et al, 1998; Airola et al, 1999; Slee et al, 1999; Liu et al, 2003, 2005; Qi et al, 2003). TIMP-2 was shown to interact with α3β1 integrin, dissociating protein tyrosine phosphatase (PTP) and the SH2-containing PTP (SHP-1) from the β1 integrin complex, which in turn negatively regulates tyrosine kinase receptor signal transduction pathways critical for cell proliferation and angiogenesis (Slee et al, 1999). MMP-independent TIMP-3 inhibition of angiogenesis was also suggested as shown by its interaction with vascular endothelial growth factor (VEGF) receptor-2, blocking the binding of VEGF to its receptor (Qi et al, 2003).
TIMP-1 was discovered almost two decades ago as an MMP inhibitor with its identity to the erythroid-potentiating activity protein, a humoral factor that enhances the proliferation of human erythroid progenitors and certain cancer cells (Avalos et al, 1988; Nguyen et al, 1994). Since then, it has been suggested that TIMP-1 has pleiotropic activities in the regulation of proliferation, cell survival, and differentiation independent of its MMP inhibitory activity. Consistently, recent studies show that administration of TIMP-1 can suppress both intrinsic and extrinsic cell death pathways via an MMP-independent mechanism in lymphocytes and breast epithelial cells (Guedez et al, 1998; Airola et al, 1999; Liu et al, 2003, 2005). However, a cell surface binding protein responsible for mediating TIMP-1 survival signaling has not been identified.
In this study, we identified CD63 as a TIMP-1 interacting protein by yeast two-hybrid screening. CD63 is a member of the tetraspanins, a group of hydrophobic proteins containing four transmembrane α-helices, two extracellular loops, and a short cytoplasmic tail. CD63 is present in late endosomes, lysosomes, secretory vesicles, and in the plasma membrane. At the plasma membrane, the members of the tetraspanin family including CD63 are known to interact with cell adhesion molecules such as integrins, regulating intracellular signal transduction pathways including cell adhesion, motility, and survival (Berditchevski, 2001; Hemler, 2001; Yunta and Lazo, 2003). Here, we demonstrated a CD63-dependent TIMP-1 association with integrin β1 on the cell surface of human breast epithelial MCF10A cells. TIMP-1 maintained the activated conformation of integrin β1 (one of the main tetraspanin-interacting integrins) in a CD63-dependent manner regardless of cell anchorage, resulting in activation of cell survival signaling and inhibition of apoptosis. Within a three-dimensional (3D) matrigel matrix, MCF10A cells formed polarized acinar-like structures that resembled normal breast glandular epithelial structure, while TIMP-1 overexpression disrupted breast epithelial cell polarization and inhibited caspase-mediated apoptosis in centrally located cells in a CD63-dependent manner. Taken together, the present study identified CD63 as a cell surface binding partner for TIMP-1, addressing a new paradigm of cell signaling critical for breast epithelial cell polarization and survival via TIMP-1 modulation of a tetraspanin/integrin complex, in addition to extracellular TIMP-1's regulation of ECM turnover and remodeling through MMP inhibition.
Results
To identify TIMP-1 binding proteins, we performed yeast two-hybrid (Y2H) screening of a human placenta cDNA library using the bait plasmid encoding the full-length mature form of the human TIMP-1 protein lacking the signal peptide sequences. DNA sequencing analysis revealed that, out of 1.2 × 107 diploid colony-forming units screened, six positive colonies contained prey plasmids encoding for the CD63 protein, a member of the tetraspanin family of proteins. The purified CD63 prey plasmid was introduced back into yeast cells and tested for interactions with full-length TIMP-1, the N-terminal MMP-inhibitory domain of TIMP-1, the C-terminal domain of TIMP-1, or TIMP-2 bait by Y2H interaction mating assays. As shown in Figure 1A, CD63 interacted with TIMP-1 but not with TIMP-2, although TIMP-1 and TIMP-2 are highly homologous in their protein structures and their MMP inhibitory activities are mostly interchangeable. Importantly, CD63 interacted with the C-terminal, but not with the N-terminal domain of TIMP-1, suggesting TIMP-1 interaction with CD63 is independent of its MMP-inhibitory activity. In addition, incubating MCF10A lysates with biotinylated rTIMPs demonstrated that anti-CD63 antibody co-immunoprecipitated endogenous CD63 with recombinant TIMP-1 (rTIMP-1) but not with rTIMP-2 (Figure 1B). An immuno-complex of CD63 with endogenous TIMP-1 was also detected from lysates of T29 cells, a previously established MCF10A cell clone engineered to overexpress TIMP-1 (Airola et al, 1999; Figure 1C). The CD63 protein with 30–70% of its size (30–60 kDa) being heavily glycosylated (Stipp et al, 2003) exhibited a diffuse distribution on SDS–PAGE under a nonreducing condition (Figure 1C, left panel). CD63 interaction with TIMP-1 on the cell surface was assessed by confocal microscopic analysis. Immunofluorescence staining of nonpermeabilized parental MCF10A cells with antibodies to CD63 and TIMP-1 showed a punctate co-staining pattern on the cell periphery consistent with cell surface co-localization (Figure 1D).
Figure 1.
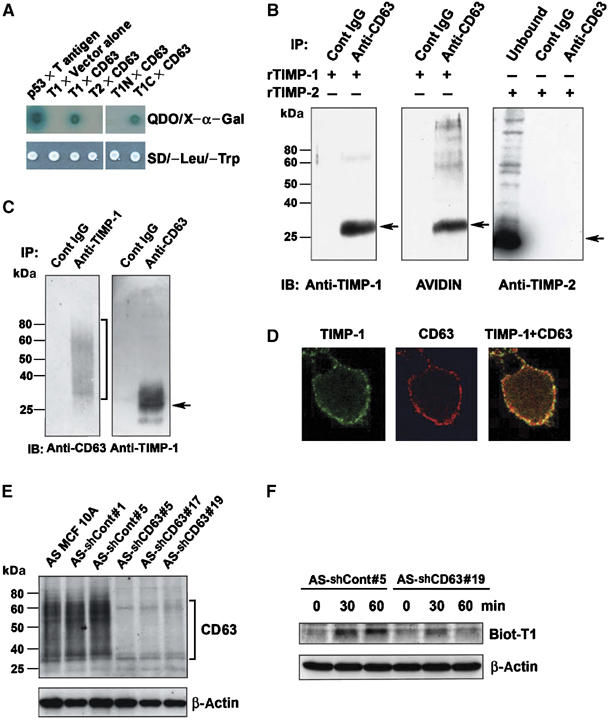
TIMP-1-specific interaction with CD63. (A) Yeast AH109 expressing p53 bait (as a positive control), full-length TIMP-1 (amino acids 1–184) lacking signal peptide (T1), full-length TIMP-2 (T2), N-terminal domain (amino acids 1–125) of TIMP-1 (T1N), or C-terminal domain (amino acids 126–184) of TIMP-1 (T1C) was mated with Y187 expressing large T antigen prey (as a positive control), vector alone (as a negative control), or full-length CD63. Diploids grow on SD/−Leu/−Trp selective media. The specific interaction between bait and prey proteins was detected on quadruple dropout (QDO) media SD/−Leu/−Trp/−His/−Ade containing X-α-Gal. (B) Anti-CD63 immunoprecipitates of MCF10A cell lysates in the presence of 500 ng/ml biotinylated rTIMP-1 (left and middle panel) or rTIMP-2 (right panel) were analyzed by Western blot analysis with anti-TIMP-1 monoclonal antibody, avidin-HRP, and anti-TIMP-2 polyclonal antibody. (C) TIMP-1 overexpressing MCF10A (T29) cell lysates were immunoprecipitated with anti-TIMP-1 polyclonal antibody or anti-CD63 monoclonal antibody, followed by immunoblot analysis using anti-CD63 under nonreducing condition or anti-TIMP-1 antibody under reducing condition, respectively. (D) MCF10A cells were grown on the coverslips overnight, blocked with PBS containing 10% horse serum 1% BSA, and co-stained with anti-CD63 Ab/Texas red conjugated secondary Ab (red staining), or with anti-TIMP-1 Ab/FITC conjugated secondary Ab (green staining). Co-localization of TIMP-1 and CD63 is shown as yellow staining (live cell staining). (E) Cell lysates of AS MCF10A clones stably transfected with control vector (AS-shCont#1 and AS-shCont#5) or with shRNA vector targeting CD63 (AS-shCD63#5, AS-shCD63#17 and AS-shCD63#19) were subjected to immunoblot analysis with an anti-CD63 antibody in a nonreducing condition. The bottom panel shows the β-actin levels of the same blot reprobed with an anti-human β-actin antibody. (F) AS-shCont#5 and AS-shCD63#19 cells were incubated with biotinylated TIMP-1 (500 ng/ml) for 0, 30, and 60 min at 37°C. For the 0 min time point, cells were washed right after the addition of biotinylated TIMP-1. Cells lysates (20 μg/lane) were subjected to immunoblot analysis with avidin-HRP.
To evaluate the significance of CD63 for TIMP-1 binding on the cell surface, we established CD63-knockdown MCF10A cells using a vector-based small hairpin RNA (shRNA) strategy. These studies were conducted in MCF10A cells in which the endogenous TIMP-1 expression was previously downregulated by antisense technology to increase the sensitivity of the cells to the exogenous rTIMP-1 (Liu et al, 2003). The antisense TIMP-1-MCF10A cells (referred to as AS TIMP-1-MCF10A) were stably transfected with the pSilencer vector containing CD63 target sequences or scrambled sequences. Immunoblot analyses confirmed significant downregulation of CD63 expression in cells receiving specific CD63 shRNA target sequences (Figure 1E). To examine the role of CD63 in mediating the surface binding of TIMP-1, CD63-knockdown and control AS-TIMP-1-MCF10A cells were incubated at 37°C with biotinylated rTIMP-1 protein. After extensive washes, the bound rTIMP-1 was detected by immunoblot analysis of cell lysates. As shown in Figure 1F, TIMP-1 was readily detected in control cells, but far less bound TIMP-1 was detected in CD63-knockdown AS-TIMP-1-MCF10A cells especially at 60 min postincubation, supporting a notion that CD63 is a cell surface interacting protein for exogenous TIMP-1.
At the plasma membrane, the members of the tetraspanin family, including CD63, are known to interact with cell adhesion molecules such as integrins and regulate intracellular signal transduction pathways. We previously showed that TIMP-1 is a potent inhibitor of apoptosis induced by a variety of apoptotic stimuli including growth factor withdrawal, staurosporine, tumor necrosis factor-related apoptosis-inducing ligand and anoikis, and the antiapoptotic activity of TIMP-1 is mediated by activation of the cell survival pathways including focal adhesion kinase (FAK) and extracellular signal-regulated kinase (ERK) pathway (Airola et al, 1999; Liu et al, 2003, 2005). Here, we asked whether TIMP-1 interacts with CD63/integrin complex, and if so, whether these interactions are critical for TIMP-1 regulation of cell survival. Since integrin β1 is thought to be one of the main tetraspanin-interacting integrins (Berditchevski et al, 1995, 1996, 1997), we first examined CD63 interactions with integrin β1. As shown in Figure 2A, anti-CD63 antibodies co-immunoprecipitated integrin β1 and endogenous TIMP-1 from lysates of MCF10A cells, suggesting that these proteins are forming a complex. Using a confocal microscopic analysis of nonpermeabilized cells, we further examined TIMP-1/CD63/integrin β1 interactions on the cell surface (Figure 2B). While a minimal level of endogenous TIMP-1 staining was detected on the cell surface of control AS-TIMP-1-MCF10A cells as expected from TIMP-1 downregulated cells, incubation of rTIMP-1 with these cells resulted in an enhanced signal consistent with binding of TIMP-1 protein to the cell surface (Figure 2B). Importantly, exogenously added rTIMP-1 protein was co-localized with integrin β1 on the cell surface of AS-shCont#5. When CD63 expression is downregulated by shRNA, cell surface localization of endogenous TIMP-1 was further reduced and these CD63-knockdown cells failed to bind exogenously administered rTIMP-1 (Figure 2B). Consequently, CD63 downregulation abrogated the co-localization of TIMP-1 with integrin β1. Taken together, these results demonstrate a critical role for CD63 in mediating TIMP-1 association with the cell surface and TIMP-1 co-localization with integrin β1.
Figure 2.
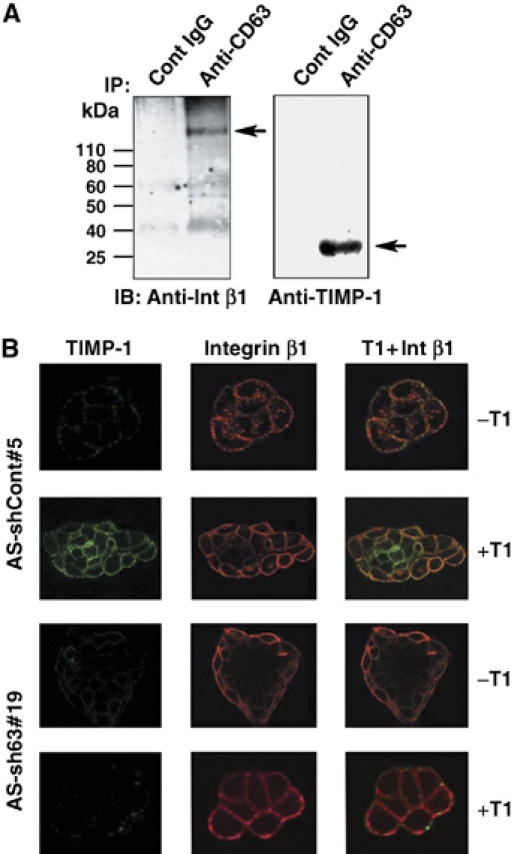
CD63 mediates TIMP-1 binding to cell surface and TIMP-1 co-localization with integrin β1 in human breast epithelial cells. (A) MCF10A cell lysates were immunoprecipitated with anti-CD63 antibody, followed by immunoblotting with anti-integrin β1 or anti-TIMP-1 monoclonal antibody. (B) AS-shCont#5 and AS-shCD63#19 cells were cultured on the coverslips overnight and incubated with or without 500 ng/ml TIMP-1 for 30 min. Live cells were co-stained with anti-integrin β1 Ab/Texas red conjugated secondary Ab (red staining) and anti-TIMP-1 Ab/FITC conjugated secondary Ab (green staining). Colocalization of TIMP-1 and integrin β1 is shown as yellow staining.
Next, we investigated whether CD63 is required for TIMP-1-mediated ERK activation using exogenous administration of rTIMP-1. As shown in Figure 3A, administration of rTIMP-1 to AS-shCont#5 cells resulted in significant ERK activation in agreement with our previous reports (Liu et al, 2003, 2005). In contrast, downregulation of CD63 expression inhibited this effect of rTIMP-1. Consistently, downregulation of CD63 expression in MCF10A cells inhibited the ability of rTIMP-1 to enhance cell survival following serum starvation when compared to control cells (Figure 3B). In accordance, rTIMP-1-mediated inhibition of caspase-3 like activity was reduced in CD63-knockdown cells (Figure 3C). These results strongly suggest that CD63 plays a critical role in TIMP-1-mediated cell survival signaling and apoptosis inhibition.
Figure 3.
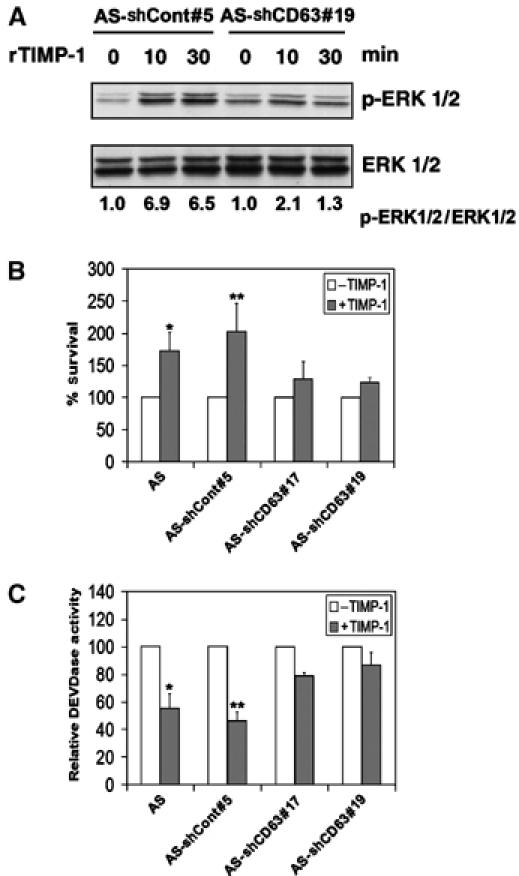
CD63 is required for exogenous TIMP-1-mediated ERKs activation and inhibition of apoptosis. (A) AS-shCont#5 and AS-shCD63#19 cells were cultured in serum-free medium for 48 h and then incubated with 500 ng/ml TIMP-1 for 10 and 30 min. Cell lysates (20 μg/lane) were subjected to immunoblot analysis with anti-active ERKs (pERK1/2) and anti-ERK1/2 antibodies. Densitometry analysis of the signals between pERK1/2 and ERK1/2 were presented by normalizing to the signal ratio in AS-shCont#5 or AS-shCD63#19 cells at 0 min. (B) AS MCF10A, AS-shCont#5, AS-shCD63#17, and AS-shCD63#19 were cultured in serum-free medium for 48 h in the presence and absence of 500 ng/ml TIMP-1. The percentage of cell survival was determined by WST-1 assay and normalized to the respective cells cultured in serum containing medium. Shown are the means±s.e. of the sextuple experiments. *P<0.05 versus AS MCF10A cells without TIMP-1 treatment (t-test); **P<0.05 versus AS-shCont#5 cells without TIMP-1 treatment (t-test). (C) AS MCF10A, AS-shCont#5, AS-shCD63#17, and AS-shCD63#19 were cultured in serum-free medium for 24 h in the presence and absence of 500 ng/ml TIMP-1. The cells were then subjected to DEVDase activity assay. Shown are the means±s.e. of the sextuple experiments. *P<0.05 versus AS cells without TIMP-1 treatment (t-test); **P<0.001 versus AS-shCont#5 cells without TIMP-1 treatment (t-test).
To examine whether CD63 is also required for ectopically expressed TIMP-1-mediated cell survival, we downregulated CD63 in TIMP-1 overexpressing MCF10A cells (referred to as T29 cells) using an shRNA approach. Immunoblot analysis confirmed significant downregulation of CD63 expression in the pooled population of T29 cells transfected with CD63 shRNA vector compared to the control vector transfected cells (Figure 4A). The level of CD63 had little effect on TIMP-1 secretion as shown by comparable levels of extracellular TIMP-1 between control and CD63-knockdown T29 cells, while the cellular TIMP-1 levels decreased, presumably due to lack of TIMP-1 association with the cell surface of CD63-knockdown cells compared to the control cells (Figure 4B). Similar to the results obtained with MCF10A cells treated with exogenous rTIMP-1 shown above, downregulation of CD63 reduced TIMP-1-mediated constitutive phosphorylation of ERK and FAK (∼60 and 50% inhibition, respectively, over control cells) upon growth factor withdrawal (Figure 4C and D). Also, TIMP-1-induced cell survival significantly decreased and caspase-3 like activity was partially recovered in T29 cells with downregulated CD63 expression upon growth factor withdrawal (Figure 4E and F). Similarly, TIMP-1 failed to prevent staurosporine- and anoikis-induced caspase-3 like activity when CD63 expression was downregulated (Figure 4G and H), further demonstrating a critical role for CD63 in TIMP-1 inhibition of apoptosis in human breast epithelial cells.
Figure 4.
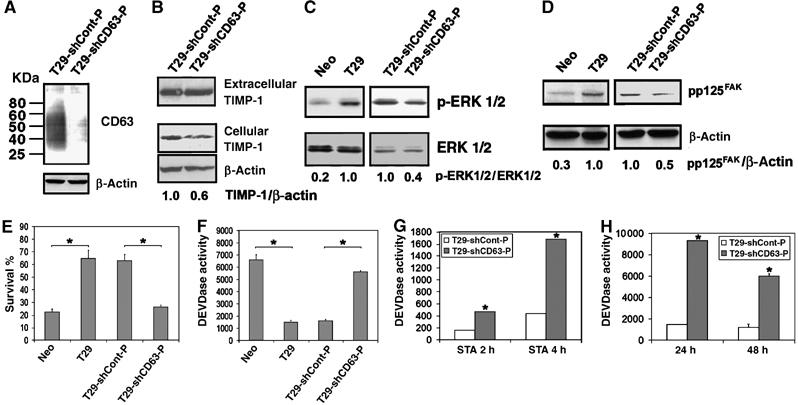
Downregulation of CD63 expression reverses TIMP-1mediated antiapoptotic activity. (A) T29-shCont-P and T29-shCD63-P cells were subjected to immunoblot analysis with an anti-CD63 antibody in a nonreducing condition. The bottom panel shows the β-actin levels of the same blot reprobed with an anti-human β-actin antibody. (B) Conditioned medium and cell lysates of T29-shCont-P and T29-shCD63-P cells were subjected to immunoblot analysis with an anti-TIMP-1 antibody. The bottom panel shows the β-actin levels of the same blot reprobed with an anti-human β-actin antibody. Densitometry analysis of the signals between cellular TIMP-1 and β-actin was presented by normalizing to the signal ratio in T29-ShCont-P cells. (C) Cell lysates (40 μg/lane) of 48 h serum-starved MCF10Aneo (Neo), T29, T29-shCont-P, and T29-shCD63-P cells were subjected to immunoblot analysis with anti-active ERKs (pERK1/2) and anti-ERK1/2 antibodies. Densitometry analysis of the signals between pERK1/2 and ERK1/2 were presented by normalizing to the signal ratio in T29 (left panel) or T29-shCont-P cells (right panel). (D) Cell lysates (40 μg/lane) of 48 h serum-starved Neo, T29, T29-shCont-P, and T29-shCD63-P cells were subjected to immunoblot analysis with anti-FAK (pY397) and anti-FAK antibodies. Densitometry analysis of the signals between anti-FAK (pY397) and anti-FAK were presented by normalizing to the signal ratio in T29 (left panel) or T29-shCont-P cells (right panel). (E) MCF10Aneo (Neo), T29, T29-shCont-P, and T29-shCD63-P cells were cultured in serum-free medium for 48 h. The percentage of cell survival was determined by WST-1 assay, and normalized to the respective cells cultured in serum containing medium. Three independent experiments were performed and the error bars represent standard deviation of the mean of sextuplicates. Asterisks depict statistically significant differences between Neo and T29 cells and between T29-shCont-P and T29-shCD63-P cells, by an unpaired, unequal t-test (*P<0.001). (F) MCF10Aneo (Neo), T29, T29-shCont-P, and T29-shCD63-P cells were cultured in serum-free medium for 48 h. The cells were then washed with PBS and lysed with 200 μl caspase lysis buffer as previously described (Liu et al, 2003, 2005). After lysates were centrifuged at 16 000 g for 10 min, DEVDase activity in 50 μl cytosol was assayed and the activity was normalized per μg protein. Three independent experiments were performed and the error bars represent standard deviation of the mean of sextuplicates. Asterisks depict statistically significant differences between Neo and T29 cells and between T29-shCont-P and T29-shCD63-P cells, by an unpaired, unequal t-test (*P<0.001). (G, H) Apoptosis was induced in T29-shCont-P and T29-shCD63-P cells by treatment with 0.5 μM staurosporine (G) or by culturing on polyHEMA-coated dishes (H). At indicated time points, the cells were washed with PBS and lysed with 200 μl caspase lysis buffer and DEVDase activity was measured. Three independent experiments were performed and the error bars represent standard deviation of the mean of triplicates. *P<0.05 versus T29-shCont-P cells at the respective time points (t-test).
Our study indicates that TIMP-1 binding to CD63 on the cell surface regulates cell survival signaling pathways likely involving the tetraspanin/integrin complex. Next, we asked whether TIMP-1/CD63/integrin β1 interactions modulate the integrin activity independent of cell adhesion. When the activation states of integrins were examined in suspension culture using antibodies that recognize the activated form of integrins (Ginsberg et al, 1990), TIMP-1 levels correlated with the levels of active integrins on the cell surface (compare among Neo, AS, and T29 cells in Figure 5). This is consistent with our previous finding that TIMP-1 constitutively activates FAK-mediated cell survival signaling pathway, independent of cell anchorage (Airola et al, 1999; Liu et al, 2003, 2005). Importantly, CD63 downregulation abolished TIMP-1-mediated integrin activation in T29 cells (T29-shCD63 in Figure 5).
Figure 5.
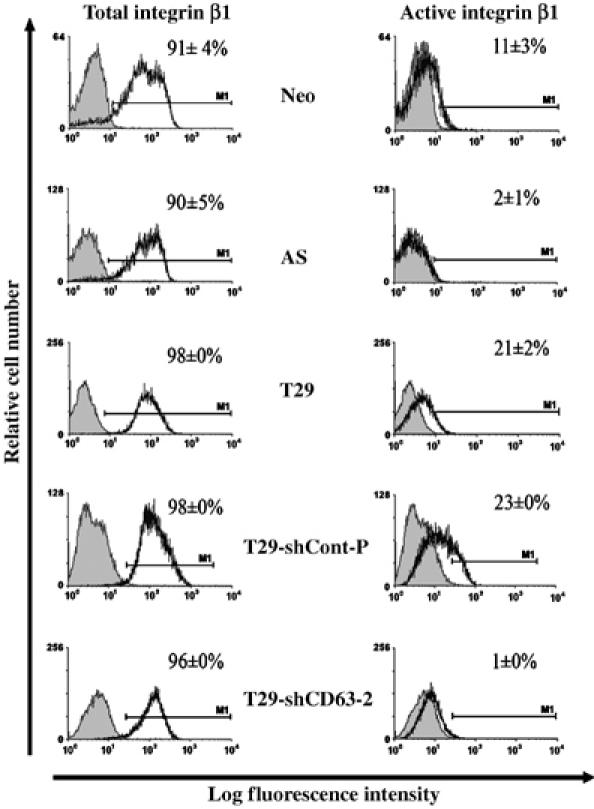
TIMP-1 enhances the level of integrin β1 in activated conformation on the cell surface. MCF10Aneo (Neo), AS TIMP-1 MCF10A (AS), TIMP-1 overexpressing MCF10A (T29), T29-shCont-P and T29-shCD63 cells were plated on polyHEMA coated tissue culture plates for 24 h and stained indirectly with either anti integrin β1 antibody (left panels) or an LIBS antibody recognizing only the active integrin β1 (right panels) followed by incubation with a FITC conjugated secondary antibody. Fluorescence was measured using a FACSCalibur machine. The percentage of gated cells stained for active or total integrin β1 (solid black line) was normalized to the percentage of gated cells stained with the FITC-secondary antibody only (negative control, shaded area). About 20 000 cells per experimental condition were analyzed in duplicates and the means±s.d. were shown.
Integrins regulate diverse cellular processes including cell adhesion, motility, proliferation, and survival (Schwartz and Baron, 1999). In addition, the distribution of cell surface integrins and activation of intracellular signaling is thought to regulate cell polarity and differentiation. In nonmalignant breast epithelium, integrin α6β4 is localized at contact sites with the basement membrane and is associated with establishment of epithelial cell polarity and survival, which is critical for the formation and maintenance of the hollow glandular architecture. When these cells become transformed, their tissue polarity is lost with a greater expression of total and cell surface integrin β1 (Fish and Molitoris, 1994; Reichmann, 1994; Lelievre et al, 1996). The significance of integrin β1 and its downstream signaling involving the FAK/PI3-K pathway in this process was demonstrated when treatment with an integrin β1 functional blocking antibody or pharmacological inhibitors of the FAK/PI3-K pathway restored polarization and apoptosis of transformed cells (Lelievre et al, 1996; Boulday et al, 2004; Xia et al, 2004). In vitro 3D culture of MCF10A cells has shown that the outer layer of polarized MCF10A cells in contact with the basement membrane survives due to activation of integrin-mediated cell adhesion survival signaling pathways, whereas the centrally located cells undergo apoptotic cell death involving caspase-3 activation (Debnath et al, 2002; Mills et al, 2004). When the role for TIMP-1 in forming a hollow acini-like structure was examined in 3D culture, TIMP-1 overexpression prevented both polarization and apoptosis of MCF10A cells growing within and consequently inhibited formation of acini-like structure (Figure 6A), in agreement with our previous report (Liu et al, 2005). Since CD63 mediates TIMP-1 binding to the cell surface, co-localization with integrin β1 and activation of integrin β1/FAK pathway, we asked whether TIMP-1 interaction with CD63 is responsible for its modulation of MCF10A cell polarization and/or inhibition of apoptosis in a 3D culture. Control shRNA transfected TIMP-1 overexpressing T29 cells failed to form a luminal structure as evidenced by weak and disorganized staining of integrin α6, as well as the absence of active caspase-3 in the center layer (Figure 6B). Interestingly, MCF10A T29 cells with CD63 downregulation by shRNA were capable of generating acini-like structures similar to those formed by the parental MCF10A cells (Figure 6B), as demonstrated by a single outer layer of integrin α6 staining and polarized epithelial cells with apoptotic cells in the center of the acini. These studies show that CD63 downregulation reverses the nonpolarization and cell survival induced by TIMP-1 overexpression in breast epithelial MCF10A cells. Approximately half of the T29-shCD63-P acini contained active caspase-3, whereas only ∼10% of T29-shCont-P had detectable levels of active caspase-3. Similarly, when cell polarization was quantitated, more than 80% of T29-shCD63-P acini displayed well-organized DAPI staining and polarized distribution of basal integrin α6, while less than 20% of T29-shCont-P acini formed polarized spheroids (Figure 6C). It should be noted that modulation of CD63 expression had little effect on TIMP-1 secretion, as shown by immunoblot analysis of TIMP-1 using conditioned medium collected from T29-shCont-P and T29-shCD63-P cells (Figure 4B). This demonstrates that TIMP-1 regulation of cell survival and polarization of nonmalignant breast epithelial cells is mediated, at least in part, by its interaction with CD63 on the cell surface leading to modulation of cell signaling, rather than extracellular MMP inhibitory activity of soluble TIMP-1.
Figure 6.
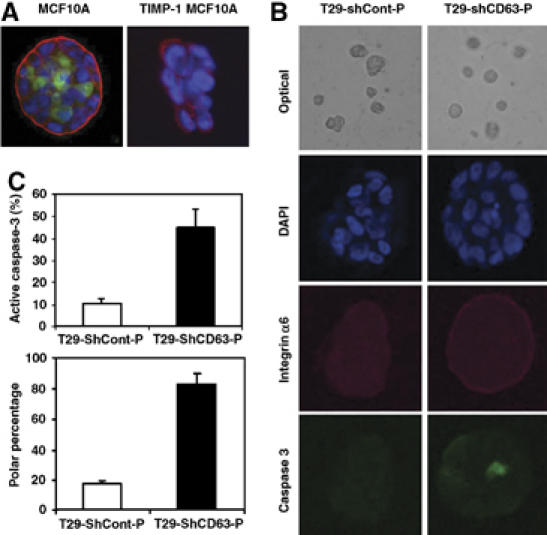
Downregulation of CD63 expression results in phenotypic reversion of TIMP-1 overexpressing MCF10A cells to the parental MCF10A cells in 3D culture. (A, B) MCF10A and TIMP-1 overexpressing MCF10A (T29) cells (A) and T29-shCont-P and T29-shCD63-P cells (B) were cultured in GFR matrigel as described in the Materials and methods (see Supplemental data). At 8 days, confocal microscopic images of cross-sections through the middle of developing acini are shown. Cells were stained with anti-integrin α6 Ab/Texas red conjugated secondary Ab (red staining) to delineate the basement membrane, anti-active caspase-3 Ab/FITC conjugated secondary Ab (green staining) to detect apoptotic cells, and with DAPI (blue) to counterstain the nuclei. (C) Quantitative analysis of the percentage of spheroids including active caspase-3 and polarity are shown in the right panel. (About 30 spheroids were analyzed for each condition from three independent experiments and the means±s.d. were shown).
Discussion
The present study identified CD63 as a cell surface binding partner for TIMP-1 in nonmalignant human breast epithelial cells, regulating cell survival, and polarization of the breast epithelial architecture. CD63 is an established component of the late endosomal and lysosomal membranes, also known as lysosomal associated membrane protein (LAMP-3) (Metzelaar and Nieuwenhuis, 1991). It was first identified as a 40-kDa platelet-activating glycoprotein (Pltgp40) (Hildreth et al, 1991) and was found to be identical to ME491, an antigen associated with human malignant melanoma (Hotta et al, 1988). A recent study reported increased CD63 expression on activated lymphocytes, especially on the cell surface where CD63 was suggested to function as a costimulatory signal to activate T cells and transmit cell survival signals when stimulated by anti-CD63 antibody (Pfistershammer et al, 2004). Our finding raises a question as to whether TIMP-1 is a physiological ligand for CD63 in hematopoietic cells for the activation of specific cell lineage, differentiation, and/or cell survival.
TIMP-1 regulates the integrin signaling complex via its interaction with CD63, a member of the tetraspanins, whereas TIMP-2 directly interacts with α3β1 integrin. Tetraspanins, which consist of 28 members in humans, can modulate integrin signaling pathways in both positive and negative manners. CD82 was shown to function as a metastasis suppressor gene involving downregulation of integrin signaling, whereas CD151 enhances the ligand-binding activity of integrins by maintaining their activated conformation of the integrins (Hemler, 2001; Kovalenko et al, 2005; Nishiuchi et al, 2005; Tonoli and Barrett, 2005). It remains to be fully investigated as to how CD63 regulates the integrin activity at the molecular level, and how TIMP-1 affects CD63's regulation of integrin signaling in a cell-type specific manner. TIMP-1 interaction with the tetraspanin member CD63 implies potentially diverse effects of TIMP-1 signaling on many cellular processes. Tetraspanins not only interact with integrins but also interact with many Ig superfamily proteins, complement regulatory proteins, growth factors, growth factor receptors, and signaling enzymes (Hemler, 2001). Although the potential significance and complexity of tetraspanin-regulated signal transduction and protein trafficking is well appreciated, their molecular actions are poorly defined at present. The intracellular cytoplasmic tail of CD63 interacts with signaling molecules including phosphatidylinositol 4-kinases (PI4-k) and Src (Berditchevski et al, 1997; Yauch and Hemler, 2000; Heijnen et al, 2003). Increasing evidence suggests that CD63 regulates FAK, Src, Gab2, PI3K, Akt, and PI4K signaling pathways as shown by modulation of these pathways upon anti-CD63 antibody binding to CD63 in breast carcinoma cells and immune cells (Berditchevski and Odintsova, 1999; Sugiura and Berditchevski, 1999; Pfistershammer et al, 2004; Kraft et al, 2005). By binding to CD63, TIMP-1 may alter CD63 interactions with integrins within the tetraspanin microdomain and in turn influence integrin signaling. Alternatively, TIMP-1 binding to CD63 may modulate recycling and redistribution of the integrin complex and/or regulate their activity, resulting in changes in signal transduction pathways.
CD63 was shown to interact with integrin β1, but not with the integrin β4 subunit, complexed with its integrin α partner α3, α4, α5, or α6 (Yunta and Lazo, 2003). A TIMP-1/CD63/ integrin β1 complex appears to provide constitutively activated survival signaling involving FAK and ERK pathways regardless of cell contacts with the basement membrane, mimicking transformed cell survival signaling and disrupting normal breast epithelial architecture. Interestingly, we noticed that TIMP-1 overexpression reduces the rate of MCF10A cell proliferation, which is associated with a prolonged G1 phase (Taube et al, 2006). Thus, TIMP-1-mediated potential ‘oncogenic' activity appears to differ from other oncogenes that dysregulate signaling such as ras, cyclin D1, and ErbB2 (Debnath et al, 2002, 2003), which also disrupt normal breast epithelial structure, but are accompanied with cell growth stimulation (Boulday et al, 2004). The present study provides insights as to how extracellular TIMP-1 regulates intracellular signaling pathways critical for cell survival and polarization through its binding to CD63, a member of the tetraspanin family of proteins. Further elucidation of TIMP-1 actions at the molecular levels warrants continued investigation, especially in the light of the clinical studies that TIMP-1 overexpression is associated with poor prognosis of many human cancers (Kossakowska et al, 1991; Zeng et al, 1995; Fong et al, 1996; Yoshiji et al, 1996; Caterina et al, 1997; McCarthy et al, 1999; Schrohl et al, 2004), an unexpected finding considering its MMP inhibitory activity. Since loss of cell polarization and filling of the luminal space in the glandular epithelial structure is one of the hallmarks of breast cancer development/early progression, TIMP-1 regulation of cell signaling in this regard may be of particular importance in understanding breast cancer development and progression.
The functions of TIMP-1 during embryogenesis and branching morphogenesis have been extensively studied using animal models (reviewed in Fata et al, 2001). Downregulation of TIMP-1 expression by transgenic antisense expression resulted in excessive ductal branches with increased mammary epithelial cell proliferation. Conversely, slow-release pellet of rTIMP-1 protein lead to inhibition of ductal elongation with decreased mammary epithelial cell proliferation (Fata et al, 2001). In contrast, overexpression of TIMP-1 enhanced bifurcation in the salivary gland and increased the number of branches in lung development (Nakanishi et al, 1986; Ganser et al, 1991). Although branching morphogenesis was not greatly affected in TIMP-1 null mice, TIMP-1 deficiency resulted in changes in estrous cycles, uterine morphology, and systemic steroid concentrations suggesting a role for TIMP-1 in reproductive processes (Nothnick, 2000). These, sometimes conflicting, results of TIMP-1 upregulation or downregulation were mostly interpreted by its regulation of MMP-mediated ECM turnover and MMP-processing of growth factors, growth factor receptors, and cell surface adhesion molecules. In light of our finding, multifunctions of TIMP-1 may need to be re-evaluated in its relation to cellular microenvironment, the levels of MMPs, and availability of TIMP-1's cell surface binding protein CD63.
Materials and methods
Yeast two-hybrid screening
The TIMP-1 cDNA was inserted into pGBKT7 and transformed into the bait strain AH109 (BD Biosciences Clonetech). The correct orientation and in-frame fusion were confirmed by DNA sequencing. Expression of the fusion proteins was confirmed by immunoblot analysis using anti-TIMP-1 and anti-cMyc antibodies. Before screening, the toxicity of the bait protein on the host strain was tested by comparing the growth rate of cells transformed with the empty DNA-BD vector with the rate of cells transformed with DNA-BD/bait plasmid. We also tested whether the bait alone would activate reporter genes by growth of bait transformants on SD/−Trp/X-α-gal, SD/−His/−Trp/ X-α-gal, and SD/−Trp/−Ade/X-α-gal. TIMP-1 bait strain AH109 was mated with the Y187/pGADT7 human placenta cDNA library as instructed by the Clontech protocol. After three rounds of screening of 1.2 × 107 diploid colony-forming units, 88 colonies were grown on SD/−Ade/−His/−Leu/−Trp (QDO) plates and showed positive phenotypes on QDO/X-α-gal plates. Plasmids were rescued from the QDO/X-α-gal positive colonies by yeast mini-prep method. PCR amplification was performed using primers the T7 sequencing primer (5′-TAATACGACTCACTATAGGGC-3′) and 3′AD sequencing primer (5′-CTGTGCATGGTGCACCATCT-3′) to amplify the inserts. Sequencing analysis of the amplified inserts and BLAST search revealed that 13 independent gene products were reproducibly shown positive. Among those, CD63, protein-tyrosine phosphatase and solute carrier protein 2 were TIMP-1 interacting membrane proteins. Protein-tyrosine phosphatase was eliminated for further study, since the insert encoded the cytoplasmic domain.
To confirm CD63 interaction with TIMP-1, the purified CD63 prey plasmid was transformed back into the Y187 yeast strain and mated with TIMP-1 or TIMP-2 bait in the AH109 strain.
Establishing CD63-knockdown cell lines
Plasmids carrying shRNA targeted to CD63 were constructed following Ambion's web-based protocol. CD63 ShRNA Target 1: 5′-GGAGAACTATTGTCTTATG-3′; Target 2: 5′-AATCCCTTCCATGTCGAAG-3′; Target 3: 5′-TTTCAACGAGAAGGCGATC-3′; Target 4: 5′-TTGCTTTTGTCGAGGTTTT-3′.
Detection of active integrin β1 in suspension culture
Cells were cultured on polyHEMA (polyhydroxyethylmethacrylate) coated tissue culture plates for 24 h. Cells were incubated with anti-integrin β1 antibody (Clone P5D2, Chemicon, MAB1959) or with anti-active integrin β1 antibody (Clone HUTS-4, Chemicon, MAB2079Z) in HEPES/NaCl buffer for 30 min on ice, followed by incubation for 30 min on ice with fluorescein isothiocyanate-conjugated (FITC) donkey anti-mouse IgG secondary antibody (Jackson Laboratories) in Flow PBS (1 × PBS, 2% horse serum, 0.1% sodium azide). Fluorescence was measured using a FACSCalibur machine (Becton Dickinson, San Jose, CA) and the percentage of gated cells (approximately 20 000 cells) stained with active or total integrin β1 (solid black lines) were normalized to the percentage of gated cells stained with FITC- secondary antibody only (shaded area).
See Supplemental data for Materials and methods in detail.
Supplementary Material
Supplementary data
Acknowledgments
We thank Dr Kamiar Moin for helping with confocal microscopic analysis. This work is supported by NIH/National Cancer Institute Grant CA89113 (to H-RCK). Microscopy and Imaging Resources Laboratory is supported in part by center Grants P30 ES06639 from the National Institutes of Environmental Health Sciences and P30CA22453 from the National Cancer Institute.
References
- Airola K, Karonen T, Vaalamo M, Lehti K, Lohi J, Kariniemi AL, Keski-Oja J, Saarialho-Kere UK (1999) Expression of collagenases-1 and -3 and their inhibitors TIMP-1 and -3 correlates with the level of invasion in malignant melanomas. Br J Cancer 80: 733–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos BR, Kaufman SE, Tomonaga M, Williams RE, Golde DW, Gasson JC (1988) K562 cells produce and respond to human erythroid-potentiating activity. Blood 71: 1720–1725 [PubMed] [Google Scholar]
- Berditchevski F (2001) Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci 114: 4143–4151 [DOI] [PubMed] [Google Scholar]
- Berditchevski F, Bazzoni G, Hemler ME (1995) Specific association of CD63 with the VLA-3 and VLA-6 integrins. J Biol Chem 270: 17784–17790 [DOI] [PubMed] [Google Scholar]
- Berditchevski F, Odintsova E (1999) Characterization of integrin–tetraspanin adhesion complexes: role of tetraspanins in integrin signaling. J Cell Biol 146: 477–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berditchevski F, Tolias KF, Wong K, Carpenter CL, Hemler ME (1997) A novel link between integrins, transmembrane-4 superfamily proteins (CD63 and CD81), and phosphatidylinositol 4-kinase. J Biol Chem 272: 2595–2598 [DOI] [PubMed] [Google Scholar]
- Berditchevski F, Zutter MM, Hemler ME (1996) Characterization of novel complexes on the cell surface between integrins and proteins with 4 transmembrane domains (TM4 proteins). Mol Biol Cell 7: 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulday G, Fitau J, Coupel S, Soulillou JP, Charreau B (2004) Exogenous tissue inhibitor of metalloproteinase-1 promotes endothelial cell survival through activation of the phosphatidylinositol 3-kinase/Akt pathway. Ann NY Acad Sci 1030: 28–36 [DOI] [PubMed] [Google Scholar]
- Caterina NC, Windsor LJ, Yermovsky AE, Bodden MK, Taylor KB, Birkedal-Hansen H, Engler JA (1997) Replacement of conserved cysteines in human tissue inhibitor of metalloproteinases-1. J Biol Chem 272: 32141–32149 [DOI] [PubMed] [Google Scholar]
- Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS (2002) The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111: 29–40 [DOI] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS (2003) Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30: 256–268 [DOI] [PubMed] [Google Scholar]
- Fata JE, Leco KJ, Voura EB, Yu HY, Waterhouse P, Murphy G, Moorehead RA, Khokha R (2001) Accelerated apoptosis in the Timp-3-deficient mammary gland. J Clin Invest 108: 831–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish EM, Molitoris BA (1994) Alterations in epithelial polarity and the pathogenesis of disease states. N Engl J Med 330: 1580–1588 [DOI] [PubMed] [Google Scholar]
- Fong KM, Kida Y, Zimmerman PV, Smith PJ (1996) TIMP1 and adverse prognosis in non-small cell lung cancer. Clin Cancer Res 2: 1369–1372 [PubMed] [Google Scholar]
- Ganser GL, Stricklin GP, Matrisian LM (1991) EGF and TGF alpha influence in vitro lung development by the induction of matrix-degrading metalloproteinases. Int J Dev Biol 35: 453–461 [PubMed] [Google Scholar]
- Ginsberg MH, Frelinger AL, Lam SC, Forsyth J, McMillan R, Plow EF, Shattil SJ (1990) Analysis of platelet aggregation disorders based on flow cytometric analysis of membrane glycoprotein IIb-IIIa with conformation-specific monoclonal antibodies. Blood 76: 2017–2023 [PubMed] [Google Scholar]
- Guedez L, Courtemanch L, Stetler-Stevenson M (1998) Tissue inhibitor of metalloproteinase (TIMP)-1 induces differentiation and an antiapoptotic phenotype in germinal center B cells. Blood 92: 1342–1349 [PubMed] [Google Scholar]
- Heijnen HF, Van Lier M, Waaijenborg S, Ohno-Iwashita Y, Waheed AA, Inomata M, Gorter G, Mobius W, Akkerman JW, Slot JW (2003) Concentration of rafts in platelet filopodia correlates with recruitment of c-Src and CD63 to these domains. J Thromb Haemost 1: 1161–1173 [DOI] [PubMed] [Google Scholar]
- Hemler ME (2001) Specific tetraspanin functions. J Cell Biol 155: 1103–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth JE, Derr D, Azorsa DO (1991) Characterization of a novel self-associating Mr 40,000 platelet glycoprotein. Blood 77: 121–132 [PubMed] [Google Scholar]
- Hotta H, Ross AH, Huebner K, Isobe M, Wendeborn S, Chao MV, Ricciardi RP, Tsujimoto Y, Croce CM, Koprowski H (1988) Molecular cloning and characterization of an antigen associated with early stages of melanoma tumor progression. Cancer Res 48: 2955–2962 [PubMed] [Google Scholar]
- Kossakowska AE, Urbanski SJ, Edwards DR (1991) Tissue inhibitor of metalloproteinases-1 (TIMP-1) RNA is expressed at elevated levels in malignant non-Hodgkin's lymphomas. Blood 77: 2475–2481 [PubMed] [Google Scholar]
- Kovalenko OV, Metcalf DG, DeGrado WF, Hemler ME (2005) Structural organization and interactions of transmembrane domains in tetraspanin proteins. BMC Struct Biol 5: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft S, Fleming T, Billingsley JM, Lin SY, Jouvin MH, Storz P, Kinet JP (2005) Anti-CD63 antibodies suppress IgE-dependent allergic reactions in vitro and in vivo. J Exp Med 201: 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre S, Weaver VM, Bissell MJ (1996) Extracellular matrix signaling from the cellular membrane skeleton to the nuclear skeleton: a model of gene regulation. Recent Prog Horm Res 51: 417–432 [PMC free article] [PubMed] [Google Scholar]
- Liu XW, Bernardo MM, Fridman R, Kim HR (2003) Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells against intrinsic apoptotic cell death via the focal adhesion kinase/phosphatidylinositol 3-kinase and MAPK signaling pathway. J Biol Chem 278: 40364–40372 [DOI] [PubMed] [Google Scholar]
- Liu XW, Taube ME, Jung KK, Dong Z, Lee YJ, Roshy S, Sloane BF, Fridman R, Kim HR (2005) Tissue inhibitor of metalloproteinase-1 protects human breast epithelial cells from extrinsic cell death: a potential oncogenic activity of tissue inhibitor of metalloproteinase-1. Cancer Res 65: 898–906 [PubMed] [Google Scholar]
- McCarthy K, Maguire T, McGreal G, McDermott E, O'Higgins N, Duffy MJ (1999) High levels of tissue inhibitor of metalloproteinase-1 predict poor outcome in patients with breast cancer. Int J Cancer 84: 44–48 [DOI] [PubMed] [Google Scholar]
- Metzelaar MJ, Nieuwenhuis HK (1991) Identity of Pltgp40 and lysomal integral membrane protein-CD63. Blood 78: 534–535 [PubMed] [Google Scholar]
- Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS (2004) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci USA 101: 3438–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi Y, Sugiura F, Kishi J, Hayakawa T (1986) Collagenase inhibitor stimulates cleft formation during early morphogenesis of mouse salivary gland. Dev Biol 113: 201–206 [DOI] [PubMed] [Google Scholar]
- Nguyen Q, Willenbrock F, Cockett MI, O'Shea M, Docherty AJ, Murphy G (1994) Different domain interactions are involved in the binding of tissue inhibitors of metalloproteinases to stromelysin-1 and gelatinase A. Biochemistry 33: 2089–2095 [DOI] [PubMed] [Google Scholar]
- Nishiuchi R, Sanzen N, Nada S, Sumida Y, Wada Y, Okada M, Takagi J, Hasegawa H, Sekiguchi K (2005) Potentiation of the ligand-binding activity of integrin alpha3beta1 via association with tetraspanin CD151. Proc Natl Acad Sci USA 102: 1939–1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothnick WB (2000) Disruption of the tissue inhibitor of metalloproteinase-1 gene results in altered reproductive cyclicity and uterine morphology in reproductive-age female mice. Biol Reprod 63: 905–912 [DOI] [PubMed] [Google Scholar]
- Pfistershammer K, Majdic O, Stockl J, Zlabinger G, Kirchberger S, Steinberger P, Knapp W (2004) CD63 as an activation-linked T cell costimulatory element. J Immunol 173: 6000–6008 [DOI] [PubMed] [Google Scholar]
- Qi JH, Ebrahem Q, Moore N, Murphy G, Claesson-Welsh L, Bond M, Baker A, Anand-Apte B (2003) A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med 9: 407–415 [DOI] [PubMed] [Google Scholar]
- Reichmann E (1994) Oncogenes and epithelial cell transformation. Semin Cancer Biol 5: 157–165 [PubMed] [Google Scholar]
- Schrohl AS, Holten-Andersen MN, Peters HA, Look MP, Meijer-van Gelder ME, Klijn JG, Brunner N, Foekens JA (2004) Tumor tissue levels of tissue inhibitor of metalloproteinase-1 as a prognostic marker in primary breast cancer. Clin Cancer Res 10: 2289–2298 [DOI] [PubMed] [Google Scholar]
- Schwartz MA, Baron V (1999) Interactions between mitogenic stimuli, or, a thousand and one connections. Curr Opin Cell Biol 11: 197–202 [DOI] [PubMed] [Google Scholar]
- Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ (1999) Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9- dependent manner. J Cell Biol 144: 281–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipp CS, Kolesnikova TV, Hemler ME (2003) Functional domains in tetraspanin proteins. Trends Biochem Sci 28: 106–112 [DOI] [PubMed] [Google Scholar]
- Sugiura T, Berditchevski F (1999) Function of alpha3beta1-tetraspanin protein complexes in tumor cell invasion. Evidence for the role of the complexes in production of matrix metalloproteinase 2 (MMP-2). J Cell Biol 146: 1375–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube ME, Liu XW, Fridman R, Kim HR (2006) TIMP-1 regulation of cell cycle in human breast epithelial cells via stabilization of p27(KIP1) protein. Oncogene 25: 3041–3048 [DOI] [PubMed] [Google Scholar]
- Tonoli H, Barrett JC (2005) CD82 metastasis suppressor gene: a potential target for new therapeutics? Trends Mol Med 11: 563–570 [DOI] [PubMed] [Google Scholar]
- Xia H, Nho RS, Kahm J, Kleidon J, Henke CA (2004) Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem 279: 33024–33034 [DOI] [PubMed] [Google Scholar]
- Yauch RL, Hemler ME (2000) Specific interactions among transmembrane 4 superfamily (TM4SF) proteins and phosphoinositide 4-kinase. Biochem J 351 (Part 3): 629–637 [PMC free article] [PubMed] [Google Scholar]
- Yoshiji H, Gomez DE, Thorgeirsson UP (1996) Enhanced RNA expression of tissue inhibitor of metalloproteinases-1 (TIMP-1) in human breast cancer. Int J Cancer 69: 131–134 [DOI] [PubMed] [Google Scholar]
- Yunta M, Lazo PA (2003) Tetraspanin proteins as organisers of membrane microdomains and signalling complexes. Cell Signal 15: 559–564 [DOI] [PubMed] [Google Scholar]
- Zeng ZS, Cohen AM, Zhang ZF, Stetler-Stevenson W, Guillem JG (1995) Elevated tissue inhibitor of metalloproteinase 1 RNA in colorectal cancer stroma correlates with lymph node and distant metastases. Clin Cancer Res 1: 899–906 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data