

Abstract
The ribonucleoprotein enzyme RNase P processes all pre-tRNAs, yet some substrates apparently lack consensus elements for recognition. Here, we compare binding affinities and cleavage rates of Escherichia coli pre-tRNAs that exhibit the largest variation from consensus recognition sequences. These results reveal that the affinities of both consensus and nonconsensus substrates for the RNase P holoenzyme are essentially uniform. Comparative analyses of pre-tRNA and tRNA binding to the RNase P holoenzyme and P RNA alone reveal differential contributions of the protein subunit to 5′ leader and tRNA affinity. Additionally, these studies reveal that uniform binding results from variations in the energetic contribution of the 5′ leader, which serve to compensate for weaker tRNA interactions. Furthermore, kinetic analyses reveal uniformity in the rates of substrate cleavage that result from dramatic (>900-fold) contributions of the protein subunit to catalysis for some nonconsensus pre-tRNAs. Together, these data suggest that an important biological function of RNase P protein is to offset differences in pre-tRNA structure such that binding and catalysis are uniform.
Keywords: C5 protein, pre-tRNA, ribonucleoprotein, RNase P, RNA processing
Introduction
In bacteria, a single RNase P enzyme, consisting of a ca. 400 nucleotide RNA subunit (P RNA) and a smaller, ca. 100 amino-acid protein, processes all pre-tRNAs (Harris and Christian, 2003; Hsieh et al, 2004). Biochemical studies show that RNase P recognizes sequences and structures at the site of cleavage that are common, but not universal among pre-tRNAs. A fundamental unresolved question is what impact, if any, such natural structural variation has on this essential step in pre-tRNA processing. The majority of the substrate binding interface resides in P RNA including the active site; however, the RNase P protein subunit enhances affinity and confers a broader range of substrate specificity (Peck-Miller and Altman, 1991; Kirsebom and Svard, 1992; Liu and Altman, 1994; Park et al, 2000; Loria and Pan, 2001). Kinetic and thermodynamic studies reveal that the protein contacts 5′ leader sequences proximal to the cleavage site resulting in increased pre-tRNA affinity (Crary et al, 1998; Niranjanakumari et al, 1998; Hsieh et al, 2004). Leader sequences do not appear to be conserved, however, implying that protein contacts are nonspecific. Recent analyses show that Escherichia coli RNase P protein (termed C5) also strengthens interactions with tRNA by stabilizing the native structure of P RNA (Buck et al, 2005). Thus, although the protein subunit contributes significantly to binding affinity and appears to modulate binding specificity in some way, the key determinants of specificity reside in interactions between P RNA and pre-tRNA.
In vitro structure–function studies reveal that P RNA recognizes functional groups flanking the pre-tRNA cleavage site and in the T-stem and loop (Christian et al, 2002; Hsieh et al, 2004). The molecular details of substrate recognition are based largely on studies of P RNA alone under high monovalent ion concentrations that stabilize binding in the absence of the protein subunit. These studies demonstrate that, with respect to determinants at the cleavage site, P RNA contacts a U residue at N(−1) (Conventional tRNA nucleotide numbering (Lowe and Eddy, 1997; Sprinzl and Vassilenko, 2005). N(−1) is one nucleotide 5′ of the mature tRNA 5′ end.), the closing G(1)–C(72) base pair of the acceptor stem, and the sequence RCC at the tRNA 3′ end. The 3′ RCC sequence and N(−1) have been shown to form direct contacts with residues in the P RNA subunit (Kirsebom and Svard, 1994; Busch et al, 2000; Brannvall et al, 2002; Zahler et al, 2003, 2005), whereas the specific interaction with the G(1)–C(72) pair is unclear. Additionally, experiments with Bacillus subtilis RNase P demonstrate that P RNA contacts C(58) and 2′ hydroxyls in the T-stem and loop (Pan et al, 1995; Loria and Pan, 1997, 1999). Indeed, RNase P cleaves a variety of non-pre-tRNA substrates as long as these determinants are present (e.g. Peck-Miller and Altman, 1991; Hartmann et al, 1995).
Nonetheless, inspection of E. coli pre-tRNA sequences reveals that some lack recognizable RNase P recognition elements at the cleavage site (Lowe and Eddy, 1997; Sprinzl and Vassilenko, 2005). Several pre-tRNAs have nonconsensus base pairs 3′ to the cleavage site (see below, Figure 1), which can alter affinity and specificity of cleavage by P RNA alone when engineered into model substrates (Svard and Kirsebom, 1992). Mutation of N(−1), G(1), C(72) or the 3′RCC results in significant decreases in binding affinity and catalytic rate, whereas disruption of two or more results in miscleavage (Brannvall et al, 1998, 2004; Kikovska et al, 2005; Zahler et al, 2005). Thus, the effect of structural variation among different pre-tRNA substrates that do not match the consensus may be large. However, comparative analyses of different substrates have been limited, and as a result, current models of substrate recognition do not account for the obvious structural differences between pre-tRNA substrates. To address how natural structural variation affects RNase P processing, we measured the precursor and product binding affinity, as well as the cleavage specificity and rate for a set of E. coli substrates that embody the greatest variation from consensus RNase P recognition elements. Together, the results reveal significant substrate-specific effects of RNase P protein on both molecular recognition and catalysis, and suggest that these differential effects are important for maintaining uniformity in substrate binding affinity and cleavage rate.
Figure 1.
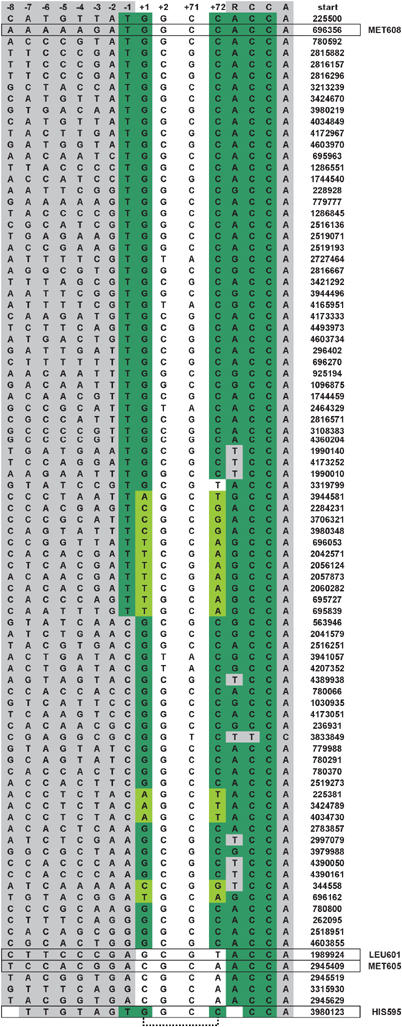
E. coli pre-tRNA gene sequences proximal to the RNase P cleavage site. Aligned sequences include −8 to +2 relative to the RNase P cleavage site at N(1) and the last six nucleotides (71–76), which together encompass the 5′ leader sequence, the first two base pairs of the acceptor stem and the 3′ RCC motif. Nucleotides 5′ and 3′ to the acceptor stem are shown in gray. Nucleotides that match the RNase P recognition consensus are shown in green. Watson–Crick base pairs other than G–C at (+1) to (+72) are shown in light green. The nucleotide position of the tRNA 5′ ends in the E. coli genome are indicated at right. The pre-tRNAs used in this study are boxed.
Results
Uniform binding affinity of examples of consensus and nonconsensus pre-tRNA substrates for E. coli RNase P
A compilation of E. coli pre-tRNA sequences adjacent to the site of RNase P cleavage is shown in Figure 1. Inspection reveals that the majority contains consensus recognition elements at the cleavage site that interact with P RNA. A significant number (ca. 30%) lack a consensus U necessary for interaction at the N(−1) position, but nonetheless retain a G(1)–C(72) or other consensus Watson–Crick base pair at the cleavage site. About 20% have Watson–Crick base pairs at the N(1)–N(72) position other than the consensus G–C, but the majority of these retain a consensus U at N(−1). As introduced above, such single differences from consensus recognition elements have measurable effects on processing in vitro. However, several E. coli pre-tRNAs have an even greater degree of variation in the consensus recognition elements and would be predicted, based on the current model for RNAse P recognition, to be very poor substrates for the enzyme. These substrates include pre-tRNAPHIS595, which retains a closing G–C pair and U(−1) contacts, but has an extra base pair in its acceptor stem. Additionally, substrates represented by pre-tRNAPMET605 and pre-tRNALEU601 have nonconsensus C–A or G–U pairs, respectively, at the cleavage site and also lack a U at N(−1). In order to gain a deeper understanding of RNase P substrate recognition, we examined the binding and cleavage of pre-tRNAMET605, pre-tRNAPLEU601 and pre-tRNAHIS595, as examples of pre-tRNAs that show the greatest deviation from consensus structure, by the RNase P holoenzyme and by P RNA alone. As examples of consensus substrates we also analyzed B. subtilis pre-tRNAASP, which is commonly used in structure–function studies, and E. coli pre-tRNAPMET608.
Computational analyses of E. coli tRNA gene sequences indicates that the length of leader sequences of different pre-tRNAs can vary significantly (Fredrik Pettersson et al, 2005); however, the precise leader lengths encountered by RNase P in vivo are not defined. Long leader sequences could result in alternative conformations or competing structures that could complicate comparative analytical studies. For the substrates used in these studies, specific leaders include two G residues at the 5′ terminus for optimal transcription in vitro followed by the eight nucleotide sequence found upstream of the individual E. coli tRNA genes (seven for the pre-tRNAPHIS595 leader). This design includes the entire region of the leader known to interact with the protein subunit, N(−3) to N(−7), without introducing significant extraneous sequences. The sequences and secondary structures of the pre-tRNAs including the leader sequences are depicted in Figure 2.
Figure 2.
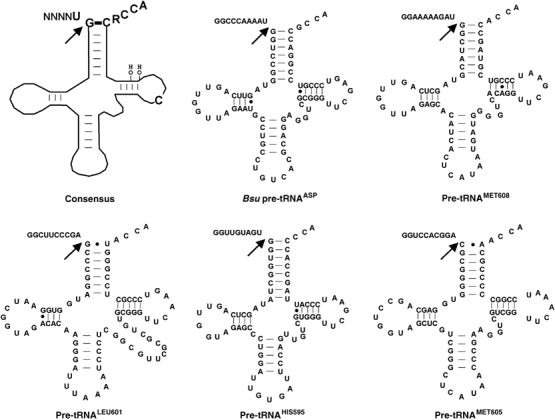
Consensus recognition elements for bacterial RNase P and secondary structures of the pre-tRNAs examined in this study. A cartoon of pre-tRNA is depicted at the upper left; nucleobases and 2′ OH groups important for RNase P recognition are indicated. B. subtilis pre-tRNAASP and E. coli pre-tRNAMET608, pre-tRNALEU601, pre-tRNAHIS595 and pre-tRNAMET605 are shown. Base pairs are represented by a dash and non-Watson–Crick pairs by dots. Arrows indicate RNase P cleavage sites.
First, we determined the apparent dissociation constant (Kd) for binding to the RNase P holoenzyme under standard conditions of 100 mM NaCl, 17.5 mM M2+P (Figure 3A, Table I). Remarkably, all of the pre-tRNAs bind with essentially equivalent high affinity (ca. 1 nM) regardless of the presence or absence of consensus recognition elements at the cleavage site. Given previous studies, the tight binding of pre-tRNAs lacking consensus elements, like pre-tRNAMET605 and pre-tRNALEU601, was unexpected. Accordingly, we considered factors that would artificially give rise to the observed uniformity in pre-tRNA binding affinity. The above binding experiments were performed in Ca2+ rather than the optimal metal ion, Mg2+, in order to suppress substrate cleavage; thus, the results might be idiosyncratic to Ca2+. However, estimation of Kd from kinetic analysis of substrate competition experiments performed in Mg2+ also indicates equivalent substrate affinity (Supplementary data). Another possibility is that the intrinsic differences in affinities for different substrates are reduced owing to saturating concentrations of divalent metal ions (17.5 mM) present in the assay. To test this possibility, we repeated the analysis at 5 mM Ca2+. Under these conditions, binding affinity is reduced 10–100-fold, yet there is still only a three-fold difference between the apparent binding affinities of these substrates (Table I). This result also indicates that the narrow range of Kd observed at higher metal ion concentrations is not due to a loss of linearity at high substrate affinity. Importantly, previous studies demonstrate that these divalent ion concentrations are sufficient for folding of E. coli P RNA (Zarrinkar et al, 1996). Furthermore, control reactions probing P RNA folding using oligo-directed RNase H cleavage demonstrate that P RNA is folded under the reactions conditions used here (Supplementary data).
Figure 3.
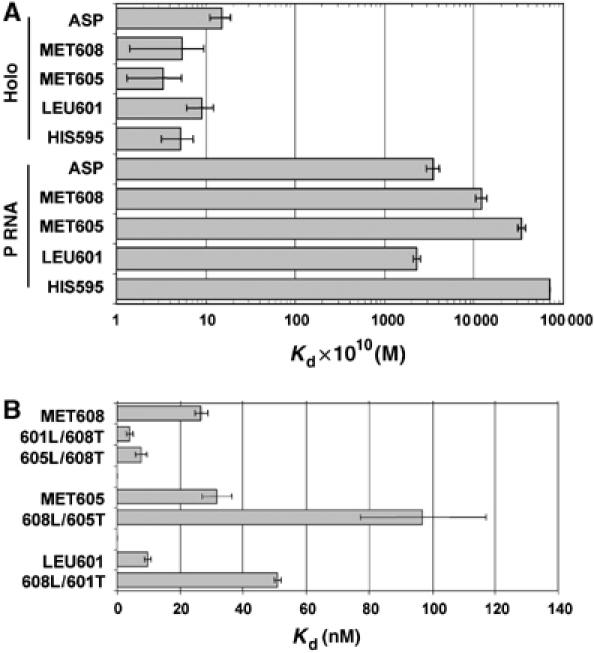
Equilibrium binding analysis of E. coli pre-tRNAs. (A) The average apparent dissociation constants (Kd) measured at 100 mM NaCl and 17.5 mM CaCl2 (from Table I) of E. coli pre-tRNAs for the holoenzyme (top) and P RNA (bottom) are shown. (B) Dissociation (Kd) constants for leader swapped substrates measured at 5 mM CaCl2 are shown. The data are derived from the average dissociation constants reported in Table I.
Table 1.
Apparent dissociation constants for the binding of pre-tRNAs and tRNAs to the E. coli RNase P RNA and holoenzymea,b
| P RNA |
RNase P |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Kdpre-tRNA (nM) |
ΔGpre-tRNA (kcal/mol) |
Kdpre-tRNA (nM) |
KdtRNA (nM) |
ΔGtRNA (kcal/mol) |
Kdpre-tRNA (nM) |
ΔGpre-tRNA (kcal/mol) |
KdtRNA (nM) |
ΔGtRNA (kcal/mol) |
Kdpre-tRNA (nM) |
ΔGpre-tRNA (kcal/mol) |
|
| 100 mM NaCl | 1 M NaCl | 100 mM NaCl | 100 mM NaCl | 100 mM NaCl | 100 mM NaCl | ||||||
| 17.5 mM CaCl2 | 17.5 mM CaCl2 | 17.5 mM CaCl2 | 17.5 mM CaCl2 | 17.5 mM CaCl2 | 5 mM CaCl2 | ||||||
| Bsu tRNAASP | 352±57 | −9.2 | 85±7 | 560±70 | −8.9 | 1.5±0.4 | −12.5 | 7.5±1.5 | −11.5 | ||
| Eco tRNAMET608 | 1200±170 | −8.4 | 540±80 | 150±40 | −9.7 | 0.5±0.4 | −13.2 | 10.7±3.0 | −11.3 | 27±2 | −10.8 |
| Eco tRNAMET605 | 3390±340 | −7.8 | 1950±70 | 190±50 | −9.5 | 0.3±0.2 | −13.5 | 200±20 | −9.5 | 32±5 | −10.6 |
| Eco tRNALEU601 | 230±20 | −9.4 | 110±30 | 350±95 | −9.2 | 0.9±0.3 | −12.8 | >500 | −8.9 | 10±1 | −11.4 |
| Eco tRNAHIS595 | >7000 | −7.3 | >5000 | 1100±90 | −8.5 | 0.5±0.2 | −13.2 | >500 | −8.9 | 16±2 | −11.1 |
| 601L/608T | 170±20 | −9.6 | <0.2 | −13.8 | 4±1 | −11.9 | |||||
| 608L/601T | 1100±300 | −8.5 | 0.4±0.2 | −13.3 | 51±1 | −10.4 | |||||
| 605L/608T | 310±80 | −9.2 | 0.3±0.1 | −13.5 | 8±2 | −11.5 | |||||
| 608L/605T | 1750±70 | −8.2 | 1.0±0.2 | −12.8 | 97±20 | −10.0 | |||||
| aAverage values and standard deviations from three or more independent trials are shown. | |||||||||||
| bMeasured at 50 mM MES, pH 5.75, 100 mM or 1 M NaCl and 5 or 17.5 mM CaCl2. | |||||||||||
Next, we determined the affinity of the same substrates for binding to P RNA alone under identical reaction conditions (100 mM NaCl, 17.5 mM CaCl2). Consistent with previous studies, P RNA has relatively low affinity for the pre-tRNAs under conditions of low ionic strength (Gardiner et al, 1985; Crary et al, 1998). However, in contrast to the results obtained with the RNase P holoenzyme, a range of affinities is observed (230 to >7000 nM) for P RNA with the highest affinity for pre-tRNAASP and pre-tRNALEU601 (Kd=352 and 230 nM, respectively). The pre-tRNAMET608 and pre-tRNAMET605 substrates were bound with lower affinity (Kd=1200 and 3390 nM, respectively), whereas pre-tRNAHIS595 was bound with lowest affinity. Comparison of the P RNA and holoenzyme binding affinities reveals that C5 has a ca. 250-fold effect on substrate binding affinity for both pre-tRNAASP and pre-tRNALEU601 substrates; however, greater contributions are observed for pre-tRNAMET608, pre-tRNAMET605 and pre-tRNAHIS595 (∼2000, ∼10 000 and >13 000-fold, respectively).
These differential contributions of the RNase P protein subunit to overall binding free energy can be estimated by comparing the free energy of binding to P RNA alone and to the RNase P holoenzyme (i.e, ΔΔGprotein(pre-tRNA)=ΔGpre-tRNA(Holo)–ΔGpre-tRNA(P RNA); ΔG=−RT ln(1/Kd)). This analysis reveals that C5 increases binding affinity for pre-tRNALEU601 by −3.4 kcal/mol and for pre-tRNAMET608 by −4.8 kcal/mol, whereas the contributions for pre-tRNAMET605 and pre-tRNAHIS595 are larger (−5.7 and −5.9 kcal/mol, respectively) (Table I and Figure 4). It has been reported that the protein subunit offsets electrostatic repulsion between P RNA and pre-tRNA (Gardiner et al, 1985; Smith et al, 1992). Thus, it is possible that higher monovalent ion concentrations could mimic the effect of C5 in establishing uniform binding. To test this possibility, we repeated the analysis of binding affinity with P RNA alone at 1 M NaCl. Although these conditions result in somewhat tighter binding for all substrates (Table I), importantly, it neither eliminates nor narrows the differences in pre-tRNA binding affinity. We conclude, therefore, that C5 has variable positive contributions to binding thermodynamics for these pre-tRNAs resulting in uniform affinity for both consensus and nonconsensus substrates.
Figure 4.
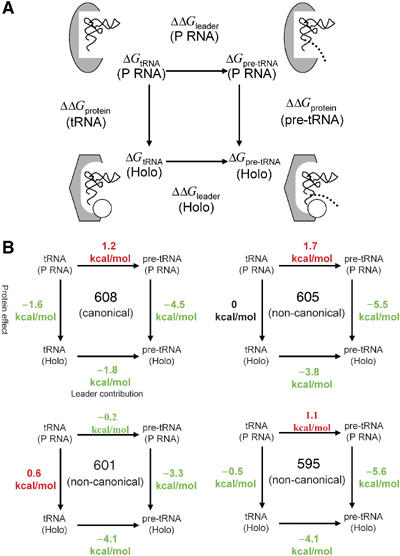
Differential effects of C5 protein on pre-tRNA, tRNA and leader sequence binding thermodynamics. (A) The experimentally determined binding free energies of pre-tRNA (ΔGpre-tRNA) and tRNA (ΔGtRNA) for the E. coli RNase P holoenzyme (Holo) and P RNA subunit alone (P RNA) are used to construct a thermodynamic box relating the effects of the protein on tRNA and pre-tRNA binding affinities (ΔΔGprotein(tRNA) and ΔΔGprotein(pre-tRNA), respectively) in the vertical dimension and the contributions of the 5′ leader sequence to P RNA and holoenzyme binding affinities (ΔΔGleader(P RNA) and ΔΔGleader(Holo), respectively) in the horizontal dimension. In the diagrams, the P RNA subunit alone is shown as a gray oval whereas the P RNA in complex with C5 protein (depicted as an white circle) is shown as a gray hexagon to symbolize indirect effects of the protein subunit on P RNA structure. tRNA is represented as a thin, black ribbon, and the leader sequence is shown as a dotted line. Arrows between complexes indicate the means by which ΔΔG values are calculated. For example, ΔΔGleader is computed by subtracting ΔGtRNA from ΔGpre-tRNA, and ΔΔGprotein is computed by subtracting ΔG(P RNA) from ΔG(Holo) as described in the text. (B) Thermodynamic boxes and ΔΔG values for pre-tRNAMET608, pre-tRNAMET605, pre-tRNALEU601 and pre-tRNAHIS595, organized according to panel A, are shown. ΔΔG values that increase binding affinity are shown in green whereas values that decrease affinity are shown in red.
Analysis of differential tRNA and leader sequence contributions to pre-tRNA binding affinity
Given that all of the tested substrates have essentially the same free energy of binding (Table I), we considered whether there are differences between the substrates in the way in which that fixed quantity of binding energy is distributed between the leader and tRNA portions of the substrate. To address this issue, we compared the affinities of tRNA domains corresponding to each of the pre-tRNA substrates for RNase P holoenzyme. In contrast to holoenzyme binding to pre-tRNA, a much broader range of binding affinities is observed for the corresponding tRNA domains. As shown in Table I, the tRNA portion of consensus substrates such as pre-tRNAMET608 and B. subtilis pre-tRNAASP bind with relatively high affinity (ca. 10 nM) to the RNase P holoenzyme. However, the three tRNAs derived from nonconsensus substrates bind 20-fold to >50-fold weaker. The differential affinities of the tRNAs for the RNase P holoenzyme were confirmed by inhibition experiments (Supplementary data). Assuming that tRNA binding reflects the contribution of the tRNA portion of pre-tRNA in substrate binding, pre-tRNA and tRNA equilibrium binding measurements allow the thermodynamic contribution of the leader sequence to be estimated for the holoenzyme (ΔΔGleader(Holo))=ΔGpre-tRNA(Holo)–ΔGtRNA(Holo). Comparison of the thermodynamic contributions of the tRNA (ΔGtRNA(Holo)) and the leader sequence (ΔΔGleader(Holo)) indicates that whereas the tRNA domain of the substrate makes the greatest contribution to overall binding affinity for all of the substrates (−8.9 to −11.3 kcal/mol), the presence of leader sequence significantly increases binding affinity of all substrates to the holoenzyme ranging from −1.9 kcal/mol for pre-tRNAMET608 (∼20-fold increase in Kd) up to ca. −4.0 kcal/mol for pre-tRNAHIS595, pre-tRNALEU601 and pre-tRNAMET605 (>600-fold increase in Kd) (Figure 4).
An inspection of these data reveals that all of the tRNAs derived from nonconsensus substrates have a free energy of binding that is ca. 2 kcal/mol lower than that of the tRNA domain of the consensus substrate. This relatively weaker contribution of these tRNAs to binding for nonconsensus substrates is offset by compensating increases in the thermodynamic contribution of the 5′ leader sequences (ΔΔGleader(Holo)) of ca. 2 kcal/mol relative to the leader from the consensus substrate (e.g. −1.9 kcal/mol, pre-tRNAMET608 versus −3.9 kcal/mol, pre-tRNALEU601).
The different apparent thermodynamic contributions of 5′ leader sequences to pre-tRNA binding to the RNase P holoenzyme (ΔΔGleader(Holo)) are surprising given the prevailing model that interactions with the leader are nonspecific. Such differences might be due to as yet unidentified sequence-dependent contacts, or may depend on the tRNA context. To assess these two possibilities, we swapped the leader sequences between substrates with weak and strong binding tRNAs. If there are leader-specific contributions, then swapping leaders should modulate the observed pre-tRNA affinity; however, if the effect is idiosyncratic to the tRNA used, then uniform binding is expected independent of leader sequence.
For these analyses, the leader sequence from pre-tRNAMET608, which has a relatively small apparent contribution to affinity in that context (−1.9 kcal/mol), was attached to the weak binding tRNAMET605 and tRNALEU601 to generate the hybrid substrates 608L/605T and 608L/601T. Similarly, leader sequences from pre-tRNAMET605 and pre-tRNALEU601, both of which have a much greater apparent contribution to affinity in their native context (−4.0 and −3.9 kcal/mol, respectively), were attached to tRNAMET608, which already binds with relatively high affinity in the absence of a leader sequence, to generate substrates 605L/608T and 601L/608T. The pre-tRNAHIS595 leader is one nucleotide shorter than the other pre-tRNA leaders and was excluded from the analysis to control for effects of sequence length.
At 17.5 mM Ca2+, we observe little apparent difference in the affinities of the leader swapped substrates. However, the necessity of maintaining substrate concentrations significantly lower than Kd limits the precision of binding analyses at very high affinity (<0.5 nM Kd). In fact, most of the measured affinities are below 0.5 nM. Consequently, we performed the same experiments at 5 mM Ca2+, which results in a ca. 10–100-fold decrease in binding affinity (see above). Indeed, in 5 mM Ca2+ we observe that replacement of the pre-tRNAMET608 leader sequence with the leaders sequences that have greater apparent contributions to binding (601L/608T and 605L/608T) results in even tighter binding compared to native pre-tRNAMET608 (Figure 3B and Table I). In contrast, replacement of the leaders from pre-tRNAMET605 and pre-tRNALEU601 with the leader derived from pre-tRNAMET608, which has a much smaller apparent contribution to affinity (608L/605T and 608L/601T), results in significantly weaker binding relative to pre-tRNAMET605 and pre-tRNALEU601, respectively. Thus, we conclude that different leader sequences can modulate holoenzyme affinity for pre-tRNA consistent with their differential contributions observed from comparison of pre-tRNA and tRNA affinity. Interestingly, the sensitivity to leader sequence changes is not unique to the RNase P holoenzyme, because 601L/608T and 608L/605T bind stronger than the parent pre-tRNAs as predicted based on relative affinities of tRNA and pre-tRNA to P RNA alone. Similarly, 608L/601T binds weaker than pre-tRNA601 as anticipated. However, the leader sequence of pre-tRNA605 fails to weaken binding of pre-tRNA608 to P RNA alone as would be expected. Yet, it is possible that there are differences in the sensitivity of these substrates to changes in leader sequence owing to tRNA contextual effects arising from differences in binding mechanism.
Nonetheless, these results suggest that, contrary to the current model for substrate recognition, RNase P as well as P RNA can have significantly different affinities for different 5′ leaders. Previous direct analyses of leader product binding to B. subtilis RNase P demonstrated very low affinity (Kd>1 μM) (Crary et al, 1998), yet no comparative analysis has been made. This led us to consider whether the different leader products have similar low affinity to the E. coli RNase P holoenzyme, or whether specific interactions result in different leader product binding affinities consistent with the trends observed in the leader swapping experiments. Indeed, leaders from pre-tRNAMET605 and pre-tRNALEU601 bound with appreciable affinity (Kd=10–100 nM) (data not shown). In contrast, binding of the leader from pre-tRNAMET608 was essentially undetectable (Kd>1000 nM). These results correlate in a qualitative manner with the apparent differences in leader contributions to pre-tRNA binding affinity and are consistent with the idea that pre-tRNA leader sequences may make specific, differential contributions to affinity as observed with the hybrid pre-tRNAs. It should be noted, however, that the effects of swapping leader sequences are relatively small (3–7-fold) compared to the total thermodynamic contribution of the individual leaders to pre-tRNA affinity (i.e. pre-tRNA versus tRNA, 20–1000-fold). Additionally, it is important to consider that quantitative analysis of leader sequence contribution is complicated by the potential for different modes of binding for pre-tRNA, tRNA and leader products (see below, Discussion). Nonetheless, whereas contextual effects of the tRNA portion of the substrate are apparent, the observations that swapping leader sequences influences pre-tRNA affinity and that different leader sequence products have different affinities support the interpretation that the apparent uniformity in pre-tRNA affinity is due in part to differential contributions of leader sequences to binding.
Differential effects of C5 on the apparent thermodynamic contributions of tRNA and leader sequence to pre-tRNA binding affinity
Previous studies using a consensus substrate have shown that the protein subunit can increase the affinity of both tRNA and pre-tRNA, and that part of the contribution to pre-tRNA binding is likely to result from direct interactions with the 5′ leader sequences (Crary et al, 1998; Buck et al, 2005). This raises the question of whether the effects of C5 on pre-tRNA affinity are attributable to increases in the thermodynamic contribution of tRNA, the leader sequence or both. Construction of thermodynamic boxes relating the effects of the protein subunit on tRNA and pre-tRNA binding (vertical dimension) and the contribution of the leader sequence to binding (horizontal dimension) provide a more complete view of the manner in which the additional binding energy conferred on substrates by C5 protein (ΔΔGprotein(pre-tRNA)) is distributed between the tRNA and leader portions of the substrate. To this end, a comparison of the affinity of tRNA binding to the holoenzyme and P RNA permits an estimate of the effect of the protein subunit on the thermodynamic contribution of tRNA to binding (ΔΔGprotein(tRNA)=ΔGtRNA(Holo)−ΔGtRNA(P RNA)). As shown in Figure 4 and Table I, the protein makes a significant contribution to tRNAMET608 affinity (−1.6 kcal/mol) whereas it makes only minor contributions to the binding of tRNAHIS595 and tRNAMET605 (−0.4 and 0 kcal/mol, respectively) and results in somewhat weaker binding of tRNALEU601 (a decrease of 0.3 kcal/mol). Thus, the protein subunit is observed to contribute differentially to tRNA domain affinity with minimal effects on tRNAs derived from nonconsensus substrates, but with a significantly higher contribution for the tRNA domain from the consensus substrate. Likewise, the protein effect on the thermodynamic contribution of the leader comes from comparing the leader contribution to P RNA and holoenzyme binding affinity (ΔΔGleader(Holo)−ΔΔGleader(P RNA)). These analyses reveal large protein effects on the thermodynamic contributions of the leader for all substrates, with the largest for pre-tRNAMET605 and pre-tRNAHIS595 (−5.7 and −5.5 kcal/mol, respectively). Smaller effects are observed for pre-tRNALEU601 and pre-tRNAMET608 (−3.7 and −3.2 kcal/mol, respectively).
A comparison of the protein effect on the free energy of binding to tRNA (ΔΔGprotein(tRNA)) and the corresponding protein effect on the leader for all substrates reveals differences in the way that the additional binding energy conferred on substrates by C5 protein (ΔΔGprotein(pre-tRNA)) is distributed (Figure 4). For the consensus substrate, this binding energy is distributed to both tRNA and leader domains. However, for the nonconsensus substrates virtually all of the additional binding energy is the result of effects of the protein on the thermodynamic contribution of the leader.
C5 protein contributes to uniformity in pre-tRNA cleavage rates
In addition to binding affinity, we also examined the effect of variation from consesnsus pre-tRNA structure on the apparent rate of substrate cleavage (kobs) in single turnover reactions catalyzed by the RNA alone and the holoenzyme (Table II, Figure 5). As the observed single turnover rate may not necessarily reflect the intrinsic chemical reaction rate (kc) owing to other rate limiting steps or conformational changes, the term kobs is used. Here, ‘effects on catalysis' includes not only effects on the intrinsic chemical rate, but also changes in kinetic mechanism or enzyme structure that affect rate enhancement. Both RNase P and P RNA alone process these substrates accurately (Supplementary data).
Table 2.
Single turnover rate constants for cleavage of pre-tRNAs by the RNase P holoenzyme, kobs (Holo), and P RNA alone, kobs (P RNA)a
| 17.5 mM MgCl2 |
5 mM MgCl2b |
|||
|---|---|---|---|---|
| kobs (P RNA) (min−1) | kobs (Holo) (min−1) | kobs (Holo)/kobs (P RNA)c | kobs (Holo) (min−1) | |
| Bsu pre-tRNAASP | 0.83±0.06 | 2.92±0.56 | 3.5 | |
| Eco pre-tRNAMET608 | 0.14±0.05 | 1.30±0.18 | 9.3 | 0.41±0.04 |
| Eco pre-tRNAMET605 | 0.0011±0.0001 | 1.00±0.03 | 909 | 0.49±0.04 |
| Eco pre-tRNALEU601 | 0.031±0.005 | 2.44±0.19 | 79 | 0.96±0.03 |
| Eco pre-tRNAHIS595 | <0.001d | 0.70±0.03 | >700 | 0.39±0.08 |
| 601L/608T | 0.38±0.08 | |||
| 608L/601T | 0.57±0.06 | |||
| 605L/608T | 0.39±0.04 | |||
| 608L/605T | 0.74±0.15 | |||
| aMeasured at 50 mM MES, pH 5.75, 100 mM NaCl, and the indicated concentration of MgCl2 for both P RNA and holoenzyme. Average values and standard deviations from three or more independent trials are shown. | ||||
| bThe rates for leader swapped substrates as well as their native counterparts were determined at 5 mM MgCl2 as the differences in binding affinities for these substrates are well documented in the presence of 5 mM divalent cation (Table I). | ||||
| cRatio of kobs (P RNA) and kobs (Holo) determined at 100 mM NaCl, 17.5 mM MgCl2. | ||||
| dOwing to limitations arising from RNA substrate degradation over long time periods, only an estimate of the maximal rate is reported. | ||||
Figure 5.
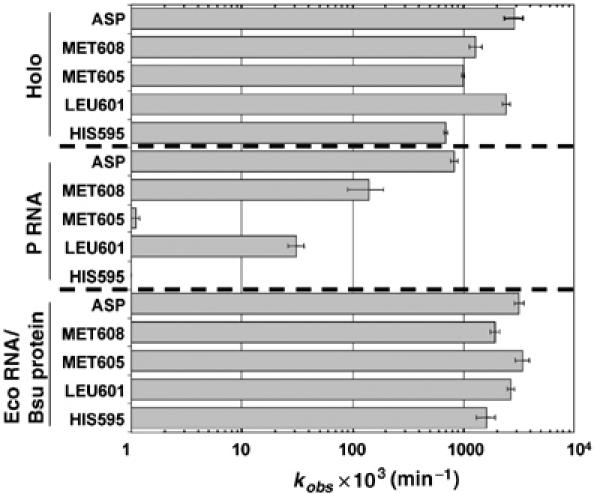
Substrate-specific contributions of C5 to catalysis. The kobs for cleavage of pre-tRNAASP ASP), pre-tRNAMET608 (MET608), pre-tRNAMET605 (MET605), pre-tRNALEU601 (LEU601) and pre-tRNAHIS595 (HIS595) by either E. coli P RNA, E. coli holoenzyme or by a hybrid holoenzyme containing the E. coli P RNA and the B. subtilis protein subunit (Eco RNA/Bsu protein) as indicated on the left are shown. Rate constants were measured at 50 mM MES, pH 5.75, 100 mM NaCl and 17.5 mM MgCl2 for both P RNA and holoenzyme. The data are derived from the average rate constants reported in Table II.
As shown previously, the holoenzyme cleaves B. subtilis pre-tRNAASP only slightly faster (three-fold) than P RNA, consistent with the current model for RNase P protein function in which it contributes little to the cleavage step. Remarkably, all of the E. coli substrates are cleaved considerably faster by the holoenzyme than by P RNA. This effect is most dramatic for the nonconsensus pre-tRNAMET605, which is cleaved by the holoenzyme at a >900-fold higher kobs. Likewise, the nonconsensus pre-tRNALEU601 and pre-tRNAHIS595 are cleaved 80-fold and >700-fold faster by holoenzyme, respectively, whereas the consensus E. coli pre-tRNAMET608 has only a nine-fold faster kobs for holoenzyme compared to P RNA. Thus, in contrast to the current model, these data demonstrate that C5 increases kobs by 80 to >900-fold for the three nonconsensus substrates. Hence, the protein contribution to catalysis is substrate specific with significantly greater contributions for substrates that are predicted to have suboptimal interactions with P RNA. Importantly, like substrate binding affinities, there is remarkable uniformity in the rates of holoenzyme cleavage for both consensus and nonconsensus substrates, which show no more than a four-fold range in kobs.
The large contribution of C5 to catalysis runs counter to the current models based on analysis of the B. subtilis protein. Consequently, we asked whether the ability to stimulate catalysis for nonconsensus substrates is a unique property of the E. coli protein. Although the sequence of the B. subtilis protein differs significantly from E. coli C5, previous studies showed that it can functionally assemble with the E. coli P RNA. Consistent with these results, we find that hybrid holoenzymes composed of the E. coli P RNA and the B. subtilis protein have essentially the same single turnover rate constants for this set of substrates as the native E. coli holoenzyme (Figure 5). Thus, the ability to provide differential effects on catalysis appears to be a general feature of the bacterial RNase P protein. Given the differential leader-specific contributions to binding, we also asked whether varying the leader sequence also influences the cleavage step. As indicated in Table II, swapping the leader sequence had little effect on kobs under conditions where an influence on binding is clearly observed (5 mM Mg2+), consistent with little if any sequence-specific contribution of the leader to cleavage rate. It is possible that the protein acts to overcome structural defects related to substrate cleavage by screening electrostatic repulsion. However, comparison of kobs for P RNA in 100 mM versus 1 M NaCl reveals only a ca. two-fold enhancement of the cleavage rate in high salt (data not shown). Thus, the contribution of C5 to catalysis appears to be a general feature of the RNase P protein that goes beyond generic effects on electrostatic repulsion.
Discussion
The first clues that the RNase P protein subunit contributes to substrate-specific recognition were reported in 1983 by Guerrier-Takada et al (1983) who showed that, whereas P RNA alone can cleave pre-tRNA, only the holoenzyme will process pre-4.5S rRNA. Subsequent analyses showed that the protein contribution to pre-4.5S processing, as well as that of some pre-tRNAs, could largely be attributed to differences in Km (Peck-Miller and Altman, 1991; Tallsjo and Kirsebom, 1993). In vitro selection experiments also highlighted differences in the way P RNA alone and the holoenzyme recognize substrates. In the presence of the protein subunit, substrates resembling precursor tRNA were selected as were others with different sequences and structures including some resembling pre-4.5S RNA that were not readily cleaved by the catalytic RNA alone (Liu and Altman, 1994). Subsequent experiments demonstrated that the protein contacts 5′ leader sequences and also appears to act indirectly by enhancing interactions between P RNA and tRNA (Niranjanakumari et al, 1998; Buck et al, 2005). However, the functional and mechanistic basis for protein contributions to multiple substrate recognition has been difficult to pin down.
The differences in the thermodynamic and kinetic properties of P RNA and the holoenzyme reported here suggest that C5 acts to compensate for the lack of recognition elements in pre-tRNA resulting in binding affinity and cleavage rate uniformity. Such uniformity despite structural variation is evocative of molecular recognition of aminoacyl-tRNAs by EF-Tu and the ribosome as described by Uhlenbeck and colleagues (Dale et al, 2004; Fahlman et al, 2004; Fahlman and Uhlenbeck, 2004; Olejniczak et al, 2005). Like RNase P, both the ribosome and EF-Tu interact with all tRNAs. For EF-Tu and the ribosome, weak or strong tRNA interactions are compensated by weaker or stronger interactions with the esterified amino acid. The observed differential thermodynamic contributions of leader sequences of different pre-tRNAs suggest a conceptually similar mechanism for achieving uniformity by RNase P in which weak tRNA interactions are compensated by strong interactions with the leader and vice versa. The inverse correlation between the thermodynamic contributions of the leader sequence and tRNA, as well as the effects of swapping leader sequences provide support for this hypothesis. Nonetheless, there are likely to be important differences between the interactions of RNase P and other factors that bind to tRNAs. For EF-Tu and the ribosome, thermodynamic compensation is a mechanism for maintaining fidelity in translation. For RNase P, cleavage specificity is independent of leader sequence, and compensation would act to maintain overall uniformity in processing rate rather than acting as an error correction mechanism. Addition of the protein subunit may have been an evolutionary mechanism for offsetting drift in tRNA sequences. As compensatory mutations in P RNA would likely affect all pre-tRNAs, a mechanism in which the protein subunit arose to modulate molecular recognition via sequence-specific leader contacts seems reasonable. A broader analysis of tRNA, leader sequence and pre-tRNA affinities is necessary to test these ideas.
Differences in the relative affinity of RNase P for the pre-tRNAs tested and their tRNA products demonstrate differential thermodynamic contributions of leader sequences for different substrates. However, this result alone does not reveal whether this effect is direct, simply involving novel contacts with leader functional groups in substrates with weak binding tRNAs; or indirect, arising in some manner from conformational differences between tRNA and pre-tRNA binding. The data presented here, together with other biochemical studies and comparative sequence analysis, suggests that differences in precursor and product affinities is likely to involve both leader-specific contacts and differences in the binding modes for different tRNAs and pre-tRNAs. Leader swapping experiments clearly show that different leader sequences can influence binding affinity. Also, we observe that leader sequence products can have widely different affinities. Thus, leader-specific contributions are significant and appear to contribute to uniformity; yet, the effects of changing leader sequence are relatively small compared to the overall contribution of the leader. At first glance, the relative affinity of the leader sequence appears to correlate with the purine/pyrimidine content. Comparison of E. coli pre-tRNAs, however, reveals no obvious correlation between leader sequences and the presence or absence of consensus recognition elements. Understanding the precise thermodynamic contribution of the 5′ leader is complicated by the fact that it is not entirely possible to equate the binding of tRNA to the affinity of the enzyme for the tRNA portion of a particular pre-tRNA as crosslinking indicates that different substrates share some contacts but may have distinct binding modes to E. coli P RNA (Pomeranz Krummel and Altman, 1999). Furthermore, modification interference provides evidence for differences in the binding modes of pre-tRNA and tRNA to P RNA (Heide et al, 2001). Thus, the uniform affinity of the pre-tRNAs examined here may arise from a combination of factors including differential contributions of leader functional groups to binding, as well as changes in the conformation of the E–S complex owing to the presence of leader interactions.
An unanticipated development is the observation that C5 makes significant contributions to catalysis for the nonconsensus substrates examined here. The robust catalytic activity of E. coli P RNA with tight binding substrates and the ability of the B. subtilis protein to affect catalysis similar to C5 argue that the protein does not contribute functional groups to active site formation or interact directly with the substrate phosphate. Instead, the large protein effects on both binding and catalysis suggest that the influence on rate is indirect, involving stabilization of the catalytic enzyme-substrate complex rather than affecting the intrinsic rate of catalysis (kc). Such a conclusion is consistent with recent models for pre-tRNA processing, based on kinetic analyses as well as structure probing studies (Loria and Pan, 1998; Pomeranz Krummel et al, 2000; Zahler et al, 2005), that invoke a conformational change involving docking of the substrate into the active site of RNase P. Here, a second E–S complex (E–S*), corresponding to the docked state, is hypothesized to form after binding and before the bond-breaking step (kc). This E–S* complex is in equilibrium, described by Kconf, with the initial E–S complex formed by bimolecular collision (Kdpre-tRNA), and P RNA interactions with the cleavage site are thought to shift Kconf in favor of E–S*. Pan and colleagues proposed that formation of E–S* requires optimal geometry of the two independently folding domains of the P RNA subunit to precisely position crucial functional groups and Mg2+ ions in the active site (Loria and Pan, 1998). Fierke and colleagues have demonstrated that the B. subtilis protein can enhance the affinity of metal ions necessary for binding and catalysis (Kurz and Fierke, 2002), thus, the influence of the protein on the apparent cleavage rate may be due to effects on the metal ion dependence of docking, or conformational change steps for different substrates.
Figure 6 shows a hypothetical scheme for the contribution of E. coli C5 to RNase P holoenzyme function with nonconsensus substrates within the framework of a two-step binding mechanism. Here, tight, direct contacts between the protein and leader, as well as interactions of tRNA with P RNA would act to overcome strain introduced by juxtaposition of RNA functional groups in poor geometric and electrostatic environments for substrates lacking consensus elements, and thus contribute to shifting the equilibrium into the docked state. The proposed mechanism for protein effects on catalysis (stabilization of E–S*) is consistent with the observation that binding and catalysis effects are correlated because measurements of each involve a Kconf component. If the docked and undocked states are in equilibrium, then the observed rate (kobs) is a function of both kc and Kconf (Loria and Pan, 1998; Zahler et al, 2005). The observed binding affinity will depend on both Kdpre-tRNA and Kconf, and the protein can have effects on both, resulting in a large shift to the docked state in the presence of the protein. Identifying the specific metal ion interactions influenced by protein binding to P RNA and to the substrate, as well as defining the structural basis for differential protein contributions to binding and catalysis remain important goals.
Figure 6.
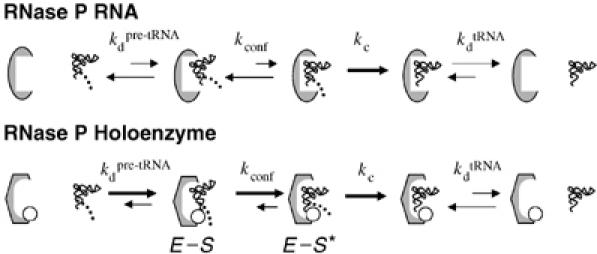
Model for the contributions of E. coli C5 to RNase P holoenzyme function. The P RNA alone or bound to C5 is depicted as a grey oval or hexagon, respectively, and the RNase P protein is shown as an open circle. C5 enhances the affinity of the holoenzyme for both precursor (Kdpre-tRNA) and product (KdtRNA). Precursor binding is nonetheless more favorable owing to additional interactions between the protein subunit and the 5′ leader sequence (depicted as a dotted line). For nonconsensus substrates the protein makes a further contribution to catalysis by promoting a step or steps upstream of catalysis that involve positioning the cleavage site for catalysis. In this model, the effect on catalysis is hypothesized to be on an upstream equilibrium docking step (Kconf). Once the substrate is bound in the appropriate state, the RNA active site promotes the chemical reaction at the rate kc.
Materials and methods
Determination of pre-tRNA and tRNA binding affinity
E. coli P RNA, B. subtilis pre-tRNAASP, tRNAASP and E. coli pre-tRNAs were generated by in vitro transcription from cloned DNA templates as described previously (Zahler et al, 2005). Leader swapped substrates were transcribed directly from linear PCR product DNAs. Mature tRNAs were generated by E. coli holoenzyme multiple turnover cleavage of the corresponding substrate. C5 protein was purified using the Impact expression system (NEB) as described (Guo et al, 2006).
The apparent dissociation constants for pre-tRNA, tRNA and leader sequence were determined by gel filtration spin column as previously described (Zahler et al, 2005) with the following differences. Binding reactions were performed under the following conditions: 50 mM MES, pH 5.75, 100 mM NaCl (or 1 M NaCl as indicated in the text and Table I) and 17.5 mM CaClB2 (or 5 mM as indicated). Holoenzyme reactions also contained 0.005% Triton X-100 which improved enzyme stability. Free and bound substrates were resolved on spin columns (Costar) containing 0.7 ml of packed G100 Sephadex in a reaction buffer. Columns were spun for 15 s at 1200 g. Enzyme–substrate complexes in the flow through and free substrate in the column were recovered, and the amount of bound RNA was determined by Cerenkov scintillation counting. The fractions of bound substrate were fit to a standard binding isotherm: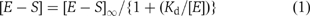 where [E−S] is the concentration of the bound fraction, [E−S]∞ is the maximum fraction bound at saturating enzyme concentration, [E] is the concentration of enzyme in the binding reaction, and Kd is the dissociation constant. Measurements of Kd values less than 5 nM necessitated the use of the expanded binding isotherm, which takes into account the concentration of substrate in the reaction. The following equation was used (Zahler et al, 2003):
where [E−S] is the concentration of the bound fraction, [E−S]∞ is the maximum fraction bound at saturating enzyme concentration, [E] is the concentration of enzyme in the binding reaction, and Kd is the dissociation constant. Measurements of Kd values less than 5 nM necessitated the use of the expanded binding isotherm, which takes into account the concentration of substrate in the reaction. The following equation was used (Zahler et al, 2003):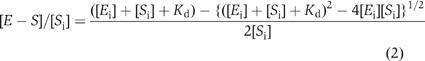 where [E−S] is the concentration of the bound fraction, [Si] is the total substrate concentration and [Ei] is the total enzyme concentration. Equilibrium constants reported are the average of at least three independent determinations, each with <30% error in curve fit to the primary data.
where [E−S] is the concentration of the bound fraction, [Si] is the total substrate concentration and [Ei] is the total enzyme concentration. Equilibrium constants reported are the average of at least three independent determinations, each with <30% error in curve fit to the primary data.
Kinetic analyses
Single turnover kinetic analyses were performed essentially as described (Siew et al, 1999). Rates were measured under the following conditions: 50 mM MES, pH 5.75, 100 mM NaCl, MgCl2 at the concentrations indicated in the text, and 0.005% Triton X-100 (for holoenzyme reactions only). The conversion of substrate to product was quantified by phosphorimaging using a Molecular Dynamics system and ImageQuant software (Amersham). The data were plotted versus time using KaleidaGraph software (Synergy) and fit to a single exponential function: where Fc is the fraction cleaved, A is the extent of the reaction, B is the amplitude of the exponential, k is the observed cleavage rate constant (kobs) and t is the time. Rate constants reported are the average of at least three independent determinations, each with <30% error in the curve fit to the primary data.
where Fc is the fraction cleaved, A is the extent of the reaction, B is the amplitude of the exponential, k is the observed cleavage rate constant (kobs) and t is the time. Rate constants reported are the average of at least three independent determinations, each with <30% error in the curve fit to the primary data.
Supplementary Material
Supplementary Material
Acknowledgments
This work was supported by NIH GM04765 to MEH. The B. subtilis RNase P protein was a generous gift of the Fierke Laboratory. We thank Eric Christian, Xia Guo and Amy Buck for comments on the manuscript, and Amy Buck and Norman Pace for sharing unpublished results.
References
- Brannvall M, Fredrik Pettersson BM, Kirsebom LA (2002) The residue immediately upstream of the RNase P cleavage site is a positive determinant. Biochimie 84: 693–703 [DOI] [PubMed] [Google Scholar]
- Brannvall M, Kikovska E, Kirsebom LA (2004) Crosstalk between the +73/294 interaction and the cleavage site in RNase P RNA mediated cleavage. Nucleic Acids Res 32: 5418–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannvall M, Mattsson JG, Svard SG, Kirsebom LA (1998) RNase P RNA structure and cleavage reflect the primary structure of tRNA genes. J Mol Biol 283: 771–783 [DOI] [PubMed] [Google Scholar]
- Buck AH, Dalby AB, Poole AW, Kazantsev AV, Pace NR (2005) Protein activation of a ribozyme: the role of bacterial RNase P protein. EMBO J 24: 3360–3368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch S, Kirsebom LA, Notbohm H, Hartmann RK (2000) Differential role of the intermolecular base-pairs G292-C(75) and G293-C(74) in the reaction catalyzed by Escherichia coli RNase P RNA. J Mol Biol 299: 941–951 [DOI] [PubMed] [Google Scholar]
- Christian EL, Zahler NH, Kaye NM, Harris ME (2002) Analysis of substrate recognition by the ribonucleoprotein endonuclease RNase P. Methods 28: 307–322 [DOI] [PubMed] [Google Scholar]
- Crary SM, Niranjanakumari S, Fierke CA (1998) The protein component of Bacillus subtilis ribonuclease P increases catalytic efficiency by enhancing interactions with the 5′ leader sequence of pre-tRNAAsp. Biochemistry 37: 9409–9416 [DOI] [PubMed] [Google Scholar]
- Dale T, Sanderson LE, Uhlenbeck OC (2004) The affinity of elongation factor Tu for an aminoacyl-tRNA is modulated by the esterified amino acid. Biochemistry 43: 6159–6166 [DOI] [PubMed] [Google Scholar]
- Fahlman RP, Dale T, Uhlenbeck OC (2004) Uniform binding of aminoacylated transfer RNAs to the ribosomal A and P sites. Mol Cell 16: 799–805 [DOI] [PubMed] [Google Scholar]
- Fahlman RP, Uhlenbeck OC (2004) Contribution of the esterified amino acid to the binding of aminoacylated tRNAs to the ribosomal P- and A-sites. Biochemistry 43: 7575–7583 [DOI] [PubMed] [Google Scholar]
- Fredrik Pettersson BM, Ardell DH, Kirsebom LA (2005) The length of the 5′ leader of Escherichia coli tRNA precursors influences bacterial growth. J Mol Biol 351: 9–15 [DOI] [PubMed] [Google Scholar]
- Gardiner KJ, Marsh TL, Pace NR (1985) Ion dependence of the Bacillus subtilis RNase P reaction. J Biol Chem 260: 5415–5419 [PubMed] [Google Scholar]
- Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 35: 849–857 [DOI] [PubMed] [Google Scholar]
- Guo X, Campbell FE, Sun L, Christian EL, Anderson VE, Harris ME (2006) RNA-dependent folding and stabilization of C5 protein during assembly of the E. coli RNase P holoenzyme. J Mol Biol 360: 190–203 [DOI] [PubMed] [Google Scholar]
- Harris ME, Christian EL (2003) Recent insights into the structure and function of the ribonucleoprotein enzyme ribonuclease P. Curr Opin Struct Biol 13: 325–333 [DOI] [PubMed] [Google Scholar]
- Hartmann RK, Heinrich J, Schlegl J, Schuster H (1995) Precursor of C4 antisense RNA of bacteriophages P1 and P7 is a substrate for RNase P of Escherichia coli. Proc Natl Acad Sci USA 92: 5822–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide C, Busch S, Feltens R, Hartmann RK (2001) Distinct modes of mature and precursor tRNA binding to Escherichia coli RNase P RNA revealed by NAIM analyses. RNA 7: 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh J, Andrews AJ, Fierke CA (2004) Roles of protein subunits in RNA–protein complexes: lessons from ribonuclease P. Biopolymers 73: 79–89 [DOI] [PubMed] [Google Scholar]
- Kikovska E, Brannvall M, Kufel J, Kirsebom LA (2005) Substrate discrimination in RNase P RNA-mediated cleavage: importance of the structural environment of the RNase P cleavage site. Nucleic Acids Res 33: 2012–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsebom LA, Svard SG (1992) The kinetics and specificity of cleavage by RNase P is mainly dependent on the structure of the amino acid acceptor stem. Nucleic Acids Res 20: 425–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsebom LA, Svard SG (1994) Base pairing between Escherichia coli RNase P RNA and its substrate. EMBO J 13: 4870–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz JC, Fierke CA (2002) The affinity of magnesium binding sites in the Bacillus subtilis RNase P-pre-tRNA complex is enhanced by the protein subunit. Biochemistry 41: 9545–9558 [DOI] [PubMed] [Google Scholar]
- Liu F, Altman S (1994) Differential evolution of substrates for an RNA enzyme in the presence and absence of its protein cofactor. Cell 77: 1093–1100 [DOI] [PubMed] [Google Scholar]
- Loria A, Pan T (1997) Recognition of the T stem-loop of a pre-tRNA substrate by the ribozyme from Bacillus subtilis ribonuclease P. Biochemistry 36: 6317–6325 [DOI] [PubMed] [Google Scholar]
- Loria A, Pan T (1998) Recognition of the 5′ leader and the acceptor stem of a pre-tRNA substrate by the ribozyme from Bacillus subtilis RNase P. Biochemistry 37: 10126–10133 [DOI] [PubMed] [Google Scholar]
- Loria A, Pan T (1999) The cleavage step of ribonuclease P catalysis is determined by ribozyme- substrate interactions both distal and proximal to the cleavage site. Biochemistry 38: 8612–8620 [DOI] [PubMed] [Google Scholar]
- Loria A, Pan T (2001) Modular construction for function of a ribonucleoprotein enzyme: the catalytic domain of Bacillus subtilis RNase P complexed with B. subtilis RNase P protein. Nucleic Acids Res 29: 1892–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niranjanakumari S, Stams T, Crary SM, Christianson DW, Fierke CA (1998) Protein component of the ribozyme ribonuclease P alters substrate recognition by directly contacting precursor tRNA. Proc Natl Acad Sci USA 95: 15212–15217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olejniczak M, Dale T, Fahlman RP, Uhlenbeck OC (2005) Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Mol Biol 12: 788–793 [DOI] [PubMed] [Google Scholar]
- Pan T, Loria A, Zhong K (1995) Probing of tertiary interactions in RNA: 2′-hydroxyl-base contacts between the RNase P RNA and pre-tRNA. Proc Natl Acad Sci USA 92: 12510–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BH, Lee JH, Kim M, Lee Y (2000) Effects of C5 protein on Escherichia coli RNase P catalysis with a precursor tRNA(Phe) bearing a single mismatch in the acceptor stem. Biochem Biophys Res Commun 268: 136–140 [DOI] [PubMed] [Google Scholar]
- Peck-Miller KA, Altman S (1991) Kinetics of the processing of the precursor to 4.5S RNA, a naturally occurring substrate for RNase P from Escherichia coli. J Mol Biol 221: 1–5 [DOI] [PubMed] [Google Scholar]
- Pomeranz Krummel DA, Altman S (1999) Multiple binding modes of substrate to the catalytic RNA subunit of RNase P from Escherichia coli. RNA 5: 1021–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomeranz Krummel DA, Kent O, MacMillan AM, Altman S (2000) Evidence for helical unwinding of an RNA substrate by the RNA enzyme RNase P: use of an interstrand disulfide crosslink in substrate. J Mol Biol 295: 1113–1118 [DOI] [PubMed] [Google Scholar]
- Siew D, Zahler NH, Cassano AG, Strobel SA, Harris ME (1999) Identification of adenosine functional groups involved in substrate binding by the ribonuclease P ribozyme. Biochemistry 38: 1873–1883 [DOI] [PubMed] [Google Scholar]
- Smith D, Burgin AB, Haas ES, Pace NR (1992) Influence of metal ions on the ribonuclease P reaction. Distinguishing substrate binding from catalysis. J Biol Chem 267: 2429–2436 [PubMed] [Google Scholar]
- Sprinzl M, Vassilenko KS (2005) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 33: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svard SG, Kirsebom LA (1992) Several regions of a tRNA precursor determine the Escherichia coli RNase P cleavage site. J Mol Biol 227: 1019–1031 [DOI] [PubMed] [Google Scholar]
- Tallsjo A, Kirsebom LA (1993) Product release is a rate-limiting step during cleavage by the catalytic RNA subunit of Escherichia coli RNase P. Nucleic Acids Res 21: 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler NH, Christian EL, Harris ME (2003) Recognition of the 5′ leader of pre-tRNA substrates by the active site of ribonuclease P. RNA 9: 734–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahler NH, Sun L, Christian EL, Harris ME (2005) The pre-tRNA nucleotide base and 2′-hydroxyl at N(−1) contribute to fidelity in tRNA processing by RNase P. J Mol Biol 345: 969–985 [DOI] [PubMed] [Google Scholar]
- Zarrinkar PP, Wang J, Williamson JR (1996) Slow folding kinetics of RNase P RNA. RNA 2: 564–573 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material