

Abstract
Although mammalian SWI/SNF chromatin remodeling complexes have been well established to play important role in transcription, their role in DNA repair has remained largely unexplored. Here we show that inactivation of the SWI/SNF complexes and downregulation of the catalytic core subunits of the complexes both result in inefficient DNA double-strand break (DSB) repair and increased DNA damage sensitivity as well as a large defect in H2AX phosphorylation (γ-H2AX) and nuclear focus formation after DNA damage. The expression of most DSB repair genes remains unaffected and DNA damage checkpoints are grossly intact in the cells inactivated for the SWI/SNF complexes. Although the SWI/SNF complexes do not affect the expression of ATM, DNA-PK and ATR, or their activation and/or recruitment to DSBs, they rapidly bind to DSB-surrounding chromatin via interaction with γ-H2AX in the manner that is dependent on the amount of DNA damage. Given the crucial role for γ-H2AX in efficient DSB repair, these results suggest that the SWI/SNF complexes facilitate DSB repair, at least in part, by promoting H2AX phosphorylation by directly acting on chromatin.
Keywords: ATP-dependent chromatin remodeling, DNA double-strand break repair, γ-H2AX, radiosensitivity, SWI/SNF complex
Introduction
Eukaryotic DNA is organized into nucleosomes and higher-order structure that presents obstacles to the access of cellular proteins to their natural chromatin substrates. The process of modulating chromatin structure therefore must constitute a pivotal step in the regulation of the nuclear processes such as DNA repair (Kornberg and Lorch, 1999; Fyodorov and Kadonaga, 2001). One of two major mechanisms by which chromatin modulation is achieved involves reversible addition of an acetyl group to the lysine residues of the histones that comprise the nucleosomes. The other one is mediated by the family of ATP-dependent chromatin-remodeling complexes that utilize the energy of ATP hydrolysis to disrupt the histone–DNA interactions. While these two mechanisms have been well established to play crucial roles in the regulation of transcription (Narlikar et al, 2002), their role in chromosomal DNA repair is just beginning to be elucidated (Allard et al, 2004; Peterson and Cote, 2004; van Attikum and Gasser, 2005).
Double-strand breaks (DSBs) of chromosomal DNA, the most destructive form of DNA damage, can be generated by exposure to ionizing radiation (IR), and also naturally occur during the nuclear processes such as DNA replication and V(D)J recombination. Unless accurately and efficiently repaired, DNA DSBs can result in chromosomal instability and cancer development (Hoeijmakers, 2001; Jackson, 2002). Upon generation of DSBs, cells activate the pathways leading to DNA repair as well as propagate the signals for checkpoint activation to arrest the cell cycle until the repair is completed. The three phosphatidylinositol-3 kinase-like kinases (PIKKs), ATM, ATR and DNA-PK, play a key role in initiating these intracellular signaling pathways of DSB responses (Shiloh, 2003; Bakkenist and Kastan, 2004).
Immediately after DSB generation, histone H2AX is phosphorylated at Ser-139 at the conserved SQ motif on the C-terminal tail, and the three PIKKs are all responsible for this phosphorylation. The phosphorylation of H2AX (termed γ-H2AX) occurs specifically at the DSB-surrounding chromatin encompassing hundreds of thousands of base pairs, resulting in organization into a poorly understood subnuclear chromosomal structure which can be visualized by immunostaining, termed as IR-induced nuclear foci (IRIF) or frequently referred to as repair foci. γ-H2AX is thought to serve as binding sites for repair and checkpoint proteins such as Nbs1 as well as to influence chromatin structure in such a way that DNA repair events are facilitated (Bassing and Alt, 2004; Fernandez-Capetillo et al, 2004). Studies using H2AX-deleted cells demonstrated that γ-H2AX is crucial for efficient repair of chromosomal DSBs and hence the maintenance of genome integrity (Downs et al, 2000; Bassing et al, 2002, 2003; Celeste et al, 2002, 2003a).
Mammalian SWI/SNF complexes, belonging to the family of swi2/snf2-based ATP-dependent chromatin remodeling complexes, are capable of facilitating alterations of nucleosome structure to control the accessibility of chromatin substrates. The SWI/SNF complexes consist of at least eight subunits containing either BRG-1 or Brm as the catalytic core subunits with ATPase activity. It has been well established that the SWI/SNF complexes play an important role in transcription both in reconstituted in vitro system and within the cells (Narlikar et al, 2002). Studies also have implicated the SWI/SNF complexes in DNA processes other than transcription such as DNA replication (Flanagan and Peterson, 1999), V(D)J recombination (Kwon et al, 2000) and viral integration (Yung et al, 2001). In vitro studies have shown that yeast SWI/SNF complex can stimulate the nucleotide excision repair on reconstituted nucleosomal substrates (Hara and Sancar, 2002; Gaillard et al, 2003). However, the in vivo function of the SWI/SNF complexes in DNA repair has remained elusive. Here, we report a role for the mammalian SWI/SNF complexes in H2AX phosphorylation, DSB repair and cell survival after DNA damage.
Results
Inactivation of the SWI/SNF complexes results in increased DNA damage sensitivity and decreased DSB repair
To investigate a potential role for the mammalian SWI/SNF complexes in DNA repair, we initially employed the NIH-3T3 cells, named B05-1, in which the SWI/SNF complexes can be conditionally inactivated by the expression of ATPase-defective dominant negative versions of BRG-1 under the control of tet-off system (de la Serna et al, 2000). Cells were cultured under the conditions with (as control) or without tetracycline (to induce the flag-tagged dominant negative BRG-1) for 4 days before being subjected to the experiments described below, and, for the sake of convenience, we often denote such cells as B05-1(+tet) and B05-1(−tet), respectively.
In the first, we examined the effects of SWI/SNF inactivation on cell viability after DNA damage by colony formation assays. As expected, the expression of flag-tagged proteins was nicely induced by tetracycline depletion from B05-1 cells but not from tet-VP16 cells, the control NIH-3T3 cells expressing no flag-tagged proteins (de la Serna et al, 2000) (Figure 1A, top). When we exposed these cells to IR, we found that the viability of B05-1(−tet) cells was markedly decreased compared to B05-1(+tet) cells, whereas tet-VP16 cells showed the similar levels of viability regardless of tetracycline depletion (Figure 1A, bottom), indicating that the SWI/SNF complexes are important for cell survival after DNA damage. The cellular levels of flag-BRG-1 were not affected by irradiation (data not shown). It should be noted that SWI/SNF inactivation reduces clonogenic ability even in undamaged cells (data not shown), suggesting that the SWI/SNF complexes are important for maintaining cell health.
Figure 1.
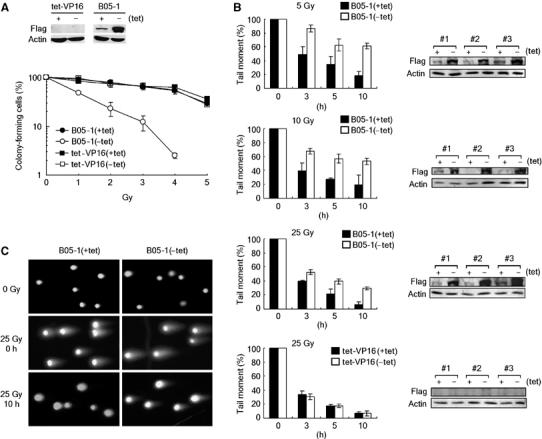
Inactivation of the SWI/SNF complexes results in increased DNA damage sensitivity and decreased DSB repair. (A) (top) tet-VP16 and B05-1 cells were incubated with or without tetracycline for 4 days, and cell lysates were analyzed for the expression of flag-BRG-1 by immunoblottings. The expression of actin was also analyzed as internal control. (Bottom) Inactivation of the SWI/SNF complexes renders cells hypersensitive to IR. Cells grown as per in (A) were irradiated by various doses before the viability was determined by colony formation assays. Data are presented as mean±standard deviation (s.d.) from triplicates. (B) Neutral comet assays show that SWI/SNF inactivation leads to inefficient DSB repair. B05-1 (the first three graphs) and tet-VP16 cells (the last graph) grown as in (A) were irradiated by indicated doses, and the cells were collected immediately (0 h) or at the indicated time points after irradiation for comet assays. Each graph is depicted as mean±s.d. from three independent experiments. Immunoblot analysis for flag-BRG-1 expression in three independent experiments is shown next to the corresponding graphs. (C) The pictures show representative comet images of B05-1(+tet) and B05-1(−tet) cells untreated (0 Gy), or immediately (0 h) and 10 h after irradiation by 25 Gy.
We then asked whether the SWI/SNF complexes are involved in DNA repair. We performed single-cell electrophoresis analysis under the neutral conditions (neutral comet assay) that specifically measures DNA DSBs. Immediately after irradiation (0 h) by 5, 10 and 25 Gy, approximately the same amounts of DNA fragments were generated from B05-1(+tet) and B05-1(−tet) cells at each dose (Figure 1B and C, and data not shown). When remaining unrepaired DNA fragments were monitored at various hours after irradiation, B05-1(−tet) cells exhibited significantly lower repair efficiency than B05-1(+tet) cells for all three doses tested, whereas tet-VP16 cells showed the same repair efficiency regardless of tetracycline depletion (Figure 1B and C). These results show that the SWI/SNF complexes are required for efficient DBS repair as well as cell survival after DNA damage.
Analysis of the genes whose expression is affected by SWI/SNF inactivation
To gain insight into whether the SWI/SNF complexes directly participate in DSB repair or contribute indirectly by transcription, we analyzed the genes that are differentially expressed between B05-1(+tet) and B05-1(−tet) cells using the mouse 11K microarray consisting of 11 376 genes that include 110 DNA repair genes. Out of the total 11 376 genes, 371 genes were induced or repressed at the significant levels (>1.8-fold) by SWI/SNF inactivation (Supplementary Table S1). Among them, only three genes were categorized into DNA repair, with two genes implicated in nucleotide excision repair and one in base excision repair (Supplementary Table S2). The human genome is estimated to contain about 150 genes encoding DNA repair enzymes and some proteins associated with cellular responses to DNA damage, among which 24 genes are directly implicated in DSB repair (Wood et al, 2005). The microarray data and RT–PCR experiments showed that none of the 24 DSB repair genes were significantly affected by SWI/SNF inactivation (Figure 2). These results raised a possibility that the SWI/SNF complexes may play a direct role in DSB repair.
Figure 2.
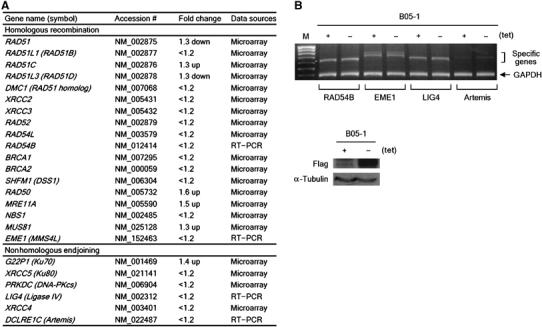
The expression of the known DSB repair genes is not significantly affected by SWI/SNF inactivation. (A) The fold changes of the expression of thus far known 24 DBS repair genes by SWI/SNF inactivation are summarized (these are all below the cutoff value). The data sources for the fold change of each gene are indicated in the last column. Note that the majority of the genes (17 genes) were changed by less than 1.2-fold by SWI/SNF inactivation, and that Rad51 and Rad51D were decreased by only 1.3-fold and the remaining five genes were rather increased by SWI/SNF inactivation. (B) (Top) The effects of SWI/SNF inactivation on the expression of the DSB repair genes that were not included in the mouse 11K gene chip. RT–PCR was performed using total RNA isolated from B05-1(+tet) and B05-1(−tet) cells. The mRNA expression of GAPDH was analyzed as internal control. The predicted sizes of the PCR products are as follow: RAD54B, 614 bp; EME1, 681 bp; LIG4, 641 bp; Artemis, 655 bp; GAPDH, 361 bp. The first lane (M) is 100-bp standard size marker (the most bottom band is 400 bp). (Bottom) The expression of flag-BRG-1 and α-tubulin (internal control) from the cells used in the top panel was analyzed by immunoblottings.
Inactivation of the SWI/SNF complexes compromises the induction of γ-H2AX after DNA damage
γ-H2AX is known to be essential for efficient repair of chromosomal DSBs (Bassing and Alt, 2004; Fernandez-Capetillo et al, 2004). The above results therefore prompted us to test whether the SWI/SNF complexes regulate γ-H2AX as one of the potential mechanisms to facilitate DBS repair. For this, we irradiated tet-VP16 and B05-1 cells that had been cultured under the conditions with or without tetracycline to induce the expression of flag-BRG-1 (Figure 3A, top). To our surprise, immunoblot analysis using the specific antibodies showed that the phosphorylation of H2AX after irradiation (25 Gy) was severely compromised in B05-1(−tet) cells, whereas B05-1(+tet) cells exhibited a typical kinetics of γ-H2AX with its levels maximized at 30–60 min and declined afterwards, reflecting DSB generation and repair (Figure 3A, middle). tet-VP16 cells showed the similar kinetics as B05-1(+tet) cells regardless of tetracycline depletion (Figure 3A, bottom). The defect of H2AX phosphorylation in B05-1(−tet) cells was detected when the cells were irradiated by various doses from 0.5 to 100 Gy (Figure 3B and see below). The levels of H2AX proteins were not different between B05-1(+tet) and B05-1(−tet) cells (Figure 3B and also see below), indicating that the SWI/SNF complexes are critical for the efficient induction of H2AX phosphorylation after DNA damage.
Figure 3.
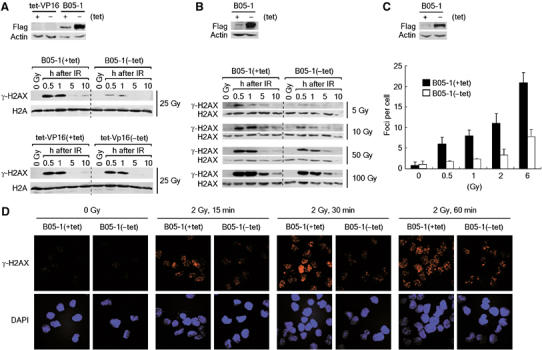
Cells inactivated for the SWI/SNF complexes are compromised in the induction of γ-H2AX after DNA damage. (A) (Top) Immunoblot analysis for the expression of flag-BRG-1 and actin (internal control) from the cells used below. (Middle and bottom) Indicated cells were untreated (0 Gy) or irradiated by 25 Gy, and histones were acid-extracted at various time points for analysis of the levels of γ-H2AX and H2A (loading control) by immunoblottings. (B) (Top) Immunoblot analysis for the expression of flag-BRG-1 and actin (internal control) from the cells used below. (Bottom) The effects of SWI/SNF inactivation on the induction of γ-H2AX after irradiation by various doses. The experiments were carried out as in (A) except that the expression of H2AX was analyzed as loading control. (C) (Top) Immunoblot analysis for the expression of flag-BRG-1 and actin (internal control) from the cells used below. (Bottom) The effects of SWI/SNF inactivation on the formation of γ-H2AX foci were analyzed by immunofluorescence microscopy. Indicated cells were untreated (0 Gy) or irradiated by 0.5, 1, 2 and 6 Gy, and the cells were fixed after 1 h for immunostaining with γ-H2AX antibodies. The graph shows the average number of γ-H2AX foci determined by counting about 50 nuclei per sample. The error bar indicates mean±s.d. from three independent experiments. (D) Representative confocal images of γ-H2AX foci taken 15, 30 and 60 min after irradiation by 2 Gy. The nuclei were visualized by DAPI staining.
Next, we examined whether the SWI/SNF complexes also would affect the formation of γ-H2AX foci after DNA damage by immunofluorescence microscopy. For quantitative analysis of the foci, we irradiated cells by low-dose IR (0.5–6 Gy). When analyzed at 1 h after irradiation, the number of γ-H2AX foci of B05-1(−tet) cells was approximately 30% of that of the control cells (Figure 3C). The defect of γ-H2AX focus formation in B05-1(−tet) cells was manifested as early as 15 min postirradiation and detected throughout the tested time course up to 4 h (Figure 3D and data not shown). In addition, the majority of γ-H2AX foci detected from B05-1(−tet) cells appeared to be much smaller in size relative to those of control cells (Figure 3D). These results, together with the immunoblot data, suggest that the SWI/SNF complexes are critical for the formation of γ-H2AX foci as well as the phosphorylation of H2AX after DNA damage.
Downregulation of BRG-1 and hBrm results in γ-H2AX defect, inefficient DSB repair and increased DNA damage sensitivity
To further demonstrate specifically the role of the SWI/SNF complexes in γ-H2AX induction and DSB repair, we used small interference RNA (siRNA) approaches. When BRG-1 and human Brm (hBrm), the catalytic core subunits of the human SWI/SNF complexes, were downregulated by cotransfecting HeLa cells with the corresponding siRNAs, both H2AX phosphorylation (Figure 4A and 4B) and γ-H2AX focus formation (Figure 4C) following DNA damage were largely compromised. The cells downregulated for BRG-1 and hBrm also showed increased DNA damage sensitivity (Figure 4D) and decreased DSB repair (Figure 4E). These results, confirming the data from the dominant negative experiments, demonstrate that the SWI/SNF complexes are specifically responsible for the optimal induction of γ-H2AX, efficient DSB repair and cell survival after DNA damage. It is noted that, as individual knockdown of either BRG-1 or hBrm appears to influence each other in their cellular levels (HL and JK, unpublished observations), we currently do not understand what relative extent to which each subunit would contribute to these functions.
Figure 4.
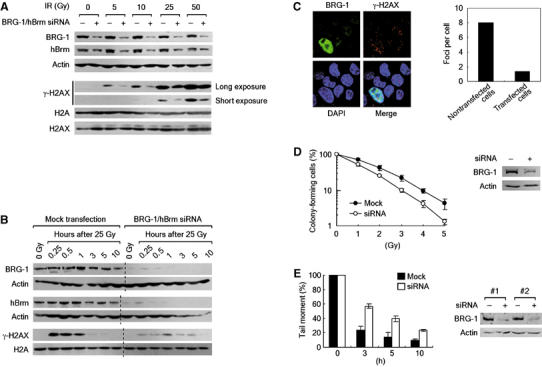
Downregulation of both BRG-1 and hBrm results in γ-H2AX defect, inefficient DSB repair and increased DNA damage sensitivity. (A) The effects of BRG-1/hBrm downregulation on the γ-H2AX induction after irradiation. HeLa cells were mock-transfected (−) or cotransfected with BRG-1 and hBrm siRNAs (+) for 48 h before irradiation by indicated doses. After 1 h, cells were collected and divided into two for preparation of whole-cell lysates and acid extracts of histones. The whole-cell lysates were analyzed for the expression of BRG-1, hBrm and actin (internal control) by immunoblottings. The acid extracts were divided into two for analyzing the levels of γ-H2AX and the expression of H2A and H2AX by immunoblottings in separate gels. The γ-H2AX blots show both short and long exposures for better comparison of γ-H2AX levels. A representative of five independent experiments is shown. (B) The effects of BRG-1/hBrm downregulation on the kinetics of γ-H2AX induction. HeLa cells transfected as in (A) were irradiated by 25 Gy and collected at various time points for analyzing the expression of indicated proteins. A representative of five independent experiments is shown. (C) The effects of BRG-1/hBrm downregulation on the formation of γ-H2AX foci. (Left) HeLa cells cotransfected with BRG-1 and hBrm siRNAs were irradiated by 2 Gy, and after 1 h cells were fixed and double stained with antibodies for BRG-1 and γ-H2AX. Confocal images were taken so as to capture both nontransfected and transfected cells in the same picture. The nuclei were visualized by DAPI staining. (Right) γ-H2AX foci were counted using the confocal image in the left panel and depicted as a graph. (D) The effects of BRG-1/hBrm downregulation on the viability after DNA damage. HeLa cells transfected as in (A) were irradiated by indicated doses before viability was evaluated by colony formation assays. Data are presented as mean±s.d. from triplicates. Immunoblot analysis of siRNA knockdown of BRG-1 is shown next to the graph. (E) The effects of BRG-1/hBrm downregulation on DSB repair. HeLa cells were transfected as in (A) and irradiated by 50 Gy and the cells were collected at the indicated time points before subjecting to neutral comet assays. Each graph is depicted as mean±s.d. from two independent experiments. Immunoblot analysis of siRNA knockdown of BRG-1 for the two independent experiments is shown next to the graph.
The effects of the SWI/SNF complexes on γ-H2AX are independent of ATM, DNA-PK and ATR
We then wished to understand how the SWI/SNF complexes regulate γ-H2AX. As a simple possibility, the SWI/SNF complexes could stimulate the phosphorylation of H2AX by regulating the expression of the kinase genes responsible for H2A phosphorylation. We found that this was not the case; the levels of ATM, DNA-PKcs and ATR proteins were not affected by inactivation of the SWI/SNF complexes (data not shown) or downregulation of BRG-1 and hBrm (Figure 5A). Next, we examined whether the SWI/SNF complexes are involved in the activation of ATM, the major player for the phosphorylation of H2AX after exposure to IR (Burma et al, 2001; Fernandez-Capetillo et al, 2002). ATM exists as an inactive dimer form before DNA damage, and, after DNA damage, ATM is autophosphorylated on Ser-1981 (p-ATM) and dissociated into active monomers (Bakkenist and Kastan, 2003). Using the specific antibodies recognizing this diagnostic autophosphorylation, we found that ATM was normally activated after irradiation in the SWI/SNF-inactivated cells (Figure 5B) and in the cells downregulated for BRG-1 and hBrm (Figure 5C). Further confirming the ATM activation, Nbs1, one of the major substrates of ATM, was phosphorylated in those cells after irradiation (Figure 5D and data not shown). These results suggest that the SWI/SNF complexes do not participate in the process of activating ATM following DNA damage.
Figure 5.
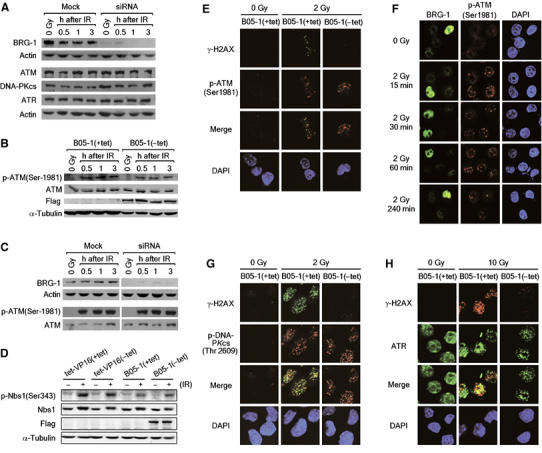
The effects of the SWI/SNF complexes on γ-H2AX are independent of ATM, DNA-PK and ATR. (A) siRNA knockdown of BRG-1 and hBrm has no effect on the expression of ATM, DNA-PKcs and ATR. HeLa cells mock transfected or cotransfected with BRG-1 and hBrm siRNA for 48 h were untreated (0 Gy) or irradiated by 10 Gy. After 1 h, cell lysates were analyzed for the expression of the indicated proteins by immunoblottings. (B) ATM activation after DNA damage normally occurs in SWI/SNF-inactivated cells. B05-1(+tet) and B05-1(−tet) cells were untreated (0 Gy) or irradiated by 10 Gy, and the cell lysates were prepared at the indicated time points for analysis of the diagnostic autophosphorylation of ATM at Ser-1981 as well as the expression of ATM by immunoblottings. The expression of flag-tagged proteins was analyzed to ensure the induction of the dominant negative BRG-1, and the expression of α-tubulin was analyzed as internal control. (C) siRNA knockdown of BRG-1 and hBrm has no effect on the activation of ATM after DNA damage. HeLa cell lysates prepared as in (A) were analyzed for the expression of the indicated proteins. (D) Nbs1 is phosphorylated in SWI/SNF-inactivated cells after DNA damage. The expression of phospho-Nbs1(Ser-343) and Nbs1 was analyzed by immunoblottings at 1 h postirradiation (30 Gy). The expression of flag-BRG-1 and α-tubulin (internal control) was also analyzed. (E) ATM foci can be formed in SWI/SNF-inactivated cells after DNA damage. B05-1(+tet) and B05-1(−tet) cells were fixed at 1 h after irradiation by 2 Gy, and double stained by antibodies for γ-H2AX and p-ATM(Ser-1981) before confocal images were captured. The nuclei were visualized by DAPI staining. (F) siRNA knockdown of BRG-1/hBrm has no effect on the formation of ATM foci after DNA damage. HeLa cells cotransfected with BRG-1 and hBrm siRNAs for 48 h were untreated (0 Gy) or irradiated by 2 Gy. Irradiated cells were collected after 15, 30, 60 and 240 min for double staining with antibodies for BRG-1 and p-ATM(Ser-1981). Confocal images were taken so as to capture both nontransfected and transfected cells in the same picture for each sample. The nuclei were visualized by DAPI staining. (G, H) DNA-PKcs and ATR can form foci in SWI/SNF-defected cells after DNA damage. B05-1(+tet) and B05-1(−tet) cells were irradiated by 2 Gy (G) or 10 Gy (H) followed by incubation for 2 h. Cells were collected for double staining with antibodies for γ-H2AX and p-DNA-PKcs(Thr-2609) (G), or with antibodies for γ-H2AX and ATR (H). The nuclei were visualized by DAPI staining.
A current model predicts that activated ATM is initially recruited to a DSB to phosphorylate nearby H2AX and that the phosphorylated H2AX recruit more ATM to the DSB site in a positive activation loop (Bakkenist and Kastan, 2004; Stavridi and Halazonetis, 2005). We therefore asked if the SWI/SNF complexes promote H2AX phosphorylation by regulating ATM recruitment. Immunofluorescence microscopy showed that p-ATM foci after irradiation were normally formed in B05-1(−tet) cells (Figure 5E and Supplementary Figure S1A) and in the cells downregulated for BRG-1 and hBrm (Figure 5F); their levels and time-course induction are not distinguishable from those of control cells, in keeping with the above results showing normal ATM activation in irradiated SWI/SNF-defective cells. We note that, as previously shown, γ-H2AX foci were barely detected from those SWI/SNF-defective cells (Figure 5E and Supplementary Figure S1A, and data not shown). These results were somewhat unexpected, however, given the number of studies suggesting the requirement of H2AX for ATM recruitment to DBS sites; cells lacking H2AX fail to form Nbs1 foci (Celeste et al, 2002, 2003b), and Nbs1 is required for ATM activation and recruitment to DSBs (Uziel et al, 2003; Kitagawa et al, 2004; Lee and Paull, 2005; Difilippantonio et al, 2005; Falck et al, 2005). We reasoned that the relative γ-H2AX levels (null versus downregulation) could make such differences. Indeed, after irradiation, cells lacking H2AX exhibited a pan-nuclear staining pattern of p-ATM in contrast to the control cells showing distinct p-ATM foci (Supplementary Figure S1B, and also shown by Stucki et al, 2005). The levels of γ-H2AX in SWI/SNF-defected cells, although largely decreased, might be still sufficient for supporting the accumulation of ATM into foci. Further, the recruitment of the phosphorylated DNA-PKcs on Thr-2609 (Chan et al, 2002) and ATR to DSBs was not affected by SWI/SNF inactivation (Figure 5G and H). These results suggest that the optimal phosphorylation of H2AX after DNA damage is not guaranteed just by ATM (and other PIKKs) activation and recruitment, but requires additional steps that likely involve the activity of the SWI/SNF complexes.
The SWI/SNF complexes rapidly bind to DSB-surrounding chromatin via interaction with γ-H2AX
The results thus far led us to hypothesize that the SWI/SNF complexes might promote H2AX phosphorylation by directly acting on chromatin. To investigate this hypothesis, we first examined whether the SWI/SNF complexes would bind to chromatin in response to DNA damage using detergent extraction chromatin retention assays (Andegeko et al, 2001). When we analyzed the detergent extracts of the nuclei isolated from control and irradiated NIH-3T3 cells, we found that both BRG-1 and mouse Brm (mBrm) became more resistant to detergent extraction after irradiation (Figure 6A, see Fraction III), indicative of increased binding to chromatin upon DNA damage. Nbs1 as positive control also more bound to chromatin after irradiation (Figure 6A, see Fraction III). Time-course analysis showed that the increased chromatin binding of BRG-1 occurred as early as 10 min after irradiation and continued until 2 h, and decreased afterwards to a certain level that was sustained throughout the tested time course up to 8 h (Figure 6B). The levels of the chromatin binding of BRG-1 increased in proportion to the exposed doses up to 50 Gy (Figure 6C). mBrm also showed similar kinetics and dose dependency as BRG-1 in the chromatin binding following IR exposure (data not shown). Interestingly, while BRG-1 and Nbs1 showed some difference in the pattern of dose-dependent chromatin binding (Figure 6C), the kinetics of the chromatin binding of these two proteins was almost identical (Figure 6B).
Figure 6.
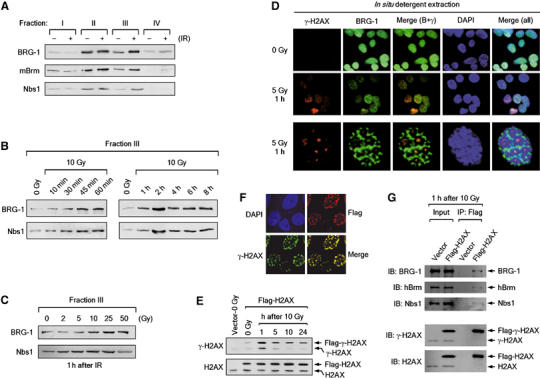
The SWI/NF complexes bind to DSB-surrounding chromatin via interaction with γ-H2AX. (A) BRG-1 and mouse Brm (mBrm) bind to chromatin after DNA damage. NIH-3T3 cells were untreated or irradiated by 10 Gy, and after 1 h cells were collected and subjected to detergent extraction. Sequentially fractionated extracts (Fractions I–III) and pellet (Fraction IV) were subjected to immunoblottings with the antibodies against BRG-1, mBrm and Nbs1. A representative of four independent experiments is shown. (B) Time-course analysis for the chromatin binding of BRG-1 and mBrm (data not shown) after DNA damage. NIH-3T3 cells were untreated (0 Gy) or irradiated by 10 Gy, and at various time points after irradiation, cells were collected and subjected to detergent extraction chromatin retention assays as in (A). Equal protein loading was ensured by Ponceau staining (data not shown). Immunoblots for fraction III are shown. A representative of four independent experiments is shown. (C) Binding of BRG-1 and mBrm (data not shown) to chromatin increases in proportion to irradiated IR doses. NIH-3T3 cells were untreated (0 Gy) or irradiated by 2, 5, 10, 25 and 50 Gy. After 1 h, cells were collected and subjected to detergent extraction chromatin retention assays as in (A). Equal protein loading was ensured by Ponceau staining (data not shown). Immunoblots for fraction III are shown. A representative of three independent experiments is shown. (D) BRG-1 is specifically retained in the chromatin overlapping with γ-H2AX after detergent extraction. NIH-3T3 cells untreated (0 Gy) or irradiated by 5 Gy, and after 1 h, the cells were detergent fractionated in situ. Cells were then fixed and double stained with the antibodies for γ-H2AX and BRG-1 with the nuclei labeled with DAPI. The third row shows a representative staining pattern of the irradiated cells shown in the second row. (E) The phosphorylation of Flag-H2AX at Ser-139 (Flag-γ-H2AX) was analyzed using the histones extracted from 293T cells that stably express N-terminal Flag-tagged H2AX after irradiation as indicated. The size differences permitted a detection of Flag-tagged proteins and their endogenous counterparts on the same immunoblot gel using the antibodies for γ-H2AX or H2AX. The first lane is nonirradiated 293T cells harboring empty vector. (F) Flag-H2AX expressing 293T cells were examined for nuclear focus formation by double staining with the Flag and γ-H2AX antibodies 1 h after irradiation by 2 Gy. (G) 293T cells expressing an empty vector or Flag-H2AX were subjected to ChIP at 1 h after irradiation by 10 Gy. After formaldehyde crosslinking and sonication, chromatin was immunoprecipitated by anti-Flag antibodies and co-precipitation of BRG-1, hBrm and Nbs1 was analyzed by immunoblottings with the indicated antibodies.
To determine where the SWI/SNF complexes would bind to the chromatin after DNA damage, we performed in situ detergent extraction experiments combined with immunofluorescence microscopy (Andegeko et al, 2001). Nuclei were isolated from the cells after irradiation and subjected to detergent extraction before staining with the antibodies against BRG-1 and γ-H2AX. Strikingly, while BRG-1 was detergent extracted from the large subnuclear regions surrounding γ-H2AX, it was retained specifically at the chromatin overlapping with γ-H2AX foci, leaving a doughnut-shaped BRG-1-free region (dark area, Figure 6D, third row). Under our experimental conditions, this pattern of BRG-1 staining was detected from the major population of irradiated cells (>70%) but not from untreated cells (Figure 6D, second and first rows, respectively). These results suggest that the SWI/SNF complexes bind to the chromatin surrounding a DSB but dissociate from a large chromatin domain beyond that region. Intriguingly, BRG-1 and DAPI were mutually exclusively stained; the doughnut-shaped BRG-1-free regions were stained intensely with DAPI (heterochromatin), whereas the rest of the nucleus and the region of γ-H2AX foci were BRG-1 positive but stained poorly with DAPI (euchromatin) (Figure 6D, third row), consistent with the previous report that the SWI/SNF complexes are preferentially associated with active chromatin (Reyes et al, 1997). Although the functional meanings underlying this peculiar BRG-1 staining pattern remain to be further investigated, a possible interpretation would be that, upon DSB generation, a large chromosomal domain undergoes reorganization into highly ordered structure at the distal region of the DSB whereas the DSB-proximal chromatin is maintained open structure by the SWI/SNF complexes. In support of this interpretation, a recent work of laser microirradiation experiments showed that the regions containing DSBs displayed less condensed 10–30 nm chromatin fibers relative to the neighboring regions adopting the heterochromatin-like structure (Kruhlak et al, 2006).
Next, we performed chromatin immunoprecipitation (ChIP) experiments to examine whether the SWI/SNF complexes interact with γ-H2AX. We generated human embryonic kidney 293T cells that stably express N-terminal Flag-tagged H2AX (Flag-H2AX) as well as control cells that harbor an empty vector. After irradiation, Flag-H2AX was phosphorylated at Ser-139 (Flag-γ-H2AX) with a similar kinetics as the endogenous H2AX (Figure 6E) and properly formed into the nuclear foci undistinguishable from γ-H2AX foci (Figure 6F). When the lysates of irradiated Flag-H2AX cells were immunoprecipitated using the anti-Flag antibodies, both BRG-1 and hBrm were co-precipitated (Figure 6G). The co-precipitation of BRG-1 and hBrm was not detected from the lysates of irradiated control cells (Figure 6G) and detected at lower levels from the lysates of unirradiated Flag-H2AX cells (data not shown), indicating that the SWI/SNF complexes directly or indirectly interact with γ-H2AX in a specific manner. We also confirmed the interaction of Nbs1 and γ-H2AX as previously shown (Figure 6G) (Kobayashi et al, 2002).
In the meantime, we asked whether we could detect colocalization of the SWI/SNF complexes and γ-H2AX by immunofluorescence microscopy without detergent extraction. While present evenly throughout the nucleus except the nucleoli before DNA damage, BRG-1 was not concentrated in speckles even after DNA damage; the staining pattern was not apparently changed by exposure to various doses of IR at various time points (Supplementary Figure S2). Thus, unlike many repair and checkpoint proteins, the SWI/SNF complexes may not migrate at the global levels towards the DNA lesion following DNA damage.
DNA damage checkpoints are grossly intact in SWI/SNF-defective cells
The increased DNA damage sensitivity of SWI/SNF-defective cells could be owing to DNA damage checkpoint defect as well as inefficient DSB repair. We thus wanted to ask whether the SWI/SNF complexes affect the DNA damage checkpoints. We found that, while marginally defective in the arrest at G2 after irradiation by 0.5 and 1 Gy, B05-1(−tet) cells exhibited no G2/M checkpoint defect in response to 2 Gy and higher doses (Figure 7A). When BRG-1 and hBrm were downregulated in HeLa cells by siRNA, the G2 arrest was only slightly defective in response to 0.5 Gy but not in response to 1 Gy and higher doses (Figure 7B). To further examine the effects of the SWI/SNF complexes on the G2/M DNA damage checkpoint, we treated the cells with adriamycin, a DSB-generating chemotherapeutic drug. We detected no G2/M checkpoint defect from B05-1(−tet) cells after adriamycin treatment (Figure 7C). Thus, the G2/M DNA damage checkpoint appears to be intact in SWI/SNF-defective cells in general, and the slight defect of G2/M checkpoint of these cells in response to low-dose IR might be due to decreased levels of γ-H2AX as previously reported (Fernandez-Capetillo et al, 2002). Next, we analyzed the effects of the SWI/SNF complexes on S-phase checkpoint. B05-1(−tet) cells showed normal S checkpoint in response to DSB-generating agents such as IR and adriamycin (Figure 7D). Interestingly, consistent with the previous report (Strobeck et al, 2000), B05-1(−tet) cells exhibited S checkpoint defect in response to the DNA crosslinker cisplatin (Figure 7D), reflecting a difference in requirement of the SWI/SNF complexes for the S checkpoint pathways depending on the types of genotoxic stresses.
Figure 7.
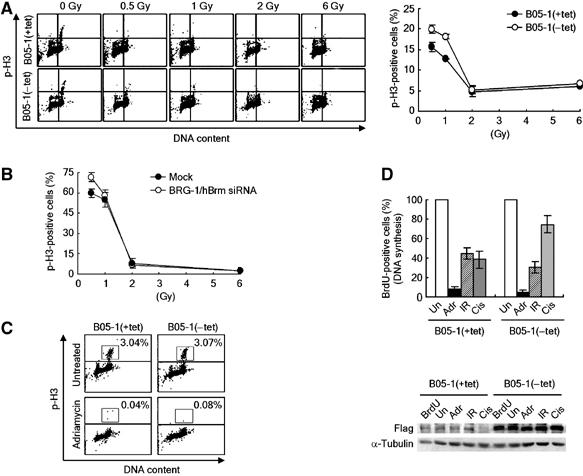
The effects of SWI/SNF inactivation on DNA damage checkpoint. (A) (Left) Analysis of G2/M checkpoint in B05-1 cells. At 1 h after irradiation by indicated doses, cells were fixed for double staining with PI and the anti-phospho-H3 antibodies followed by FACS analysis. A representative FACS result is shown. (Right) The frequency of phospho-H3-positive cells (mitotic cells) at 1 h after irradiation was represented by a percentage (irradiated/nonirradiated). An average from three independent experiments is plotted as graph with mean±s.d. (B) The effects of siRNA downregulation of BRG-1 and hBrm on DNA damage G2/M checkpoints. HeLa cells were mock transfected or transfected with BRG-1 and hBrm siRNA for 48 h, and irradiated by the indicated doses. The mitotic cells were analyzed as in (B). An average from three independent experiments is plotted as graph with mean±s.d. Note that the overall levels of G2 arrest of HeLa cells in response to 0.5 and 1 Gy are much lower than those of NIH-3T3 cells. (C) SWI/SNF inactivation has no effect on the G2/M checkpoint in response to adriamycin. Cells were treated with 0.5 μM of adriamycin for 1 h, and harvested for analysis of mitotic cells as in (A). Percentages of phospho-H3-positive mitotic cells are indicated. (D) S-phase checkpoint analysis. (Top) The levels of new DNA synthesis were determined by BrdU incorporation method after cells were untreated (Un), treated with 0.5 μM of adriamycin (Adr), irradiated by 10-Gy IR, or 32 μM of cisplatin (Cis). The number of BrdU-positive nonirradiated cells was set to 100%, and an average percentage from three independent experiments was plotted as graph with mean±s.d. (Bottom) Immunoblot analysis shows that the expression of flag-BRG-1 was properly induced by tetracycline depletion. BrdU indicates the control reactions without BrdU and other abbreviations are same as the top panel.
Discussion
In the present study, we reveal the role for the mammalian SWI/SNF complexes in DSB repair and cell survival after DNA damage. Inactivation of the SWI/SNF complexes and downregulation of their catalytic subunits both results in inefficient DBS repair and increased DNA damage sensitivity, with DNA damage checkpoint grossly intact and the expression of most DSB repair genes largely unaffected. We find that the SWI/SNF complexes are critical for the phosphorylation of H2AX and the formation of γ-H2AX foci following DNA damage. Given the crucial role for γ-H2AX in chromosomal DSB repair and cell survival after DNA damage, these results suggest that the SWI/SNF complexes facilitate DSB repair and hence increase the resistance to DNA damage, at least in part, by promoting γ-H2AX induction. Our data, showing that BRG-1/Brm bind to DSB-surrounding chromatin via interaction with γ-H2AX, suggest that the SWI/SNF complexes directly act on the chromatin to facilitate the phosphorylation of H2AX. These results provide the first example, to our knowledge, for direct implication of ATP-dependent chromatin remodeling in DNA repair in mammalian cells.
Our findings have raised a number of interesting questions to be addressed. Most importantly, whether the SWI/SNF complexes and γ-H2AX function in a common pathway of DSB repair, in other words, whether the SWI/SNF complexes facilitate DSB repair mainly through the γ-H2AX pathway remains to be investigated. Therefore, we cannot formally exclude the possibility that the SWI/SNF complexes also contribute to DSB repair by regulating the expression of yet unidentified DSB repair genes. For the same reason, it is also possible that the SWI/SNF complexes contribute to DSB repair in parallel by altering the chromatin structure surrounding the DNA lesion in such a way that subsequent repair events are facilitated. In raising this possibility, the SWI/SNF complexes appear to remain associated with chromatin for an extended period of time after reaching the maximum levels of binding at 2 h after irradiation (Figure 6B). We are currently investigating the molecular mechanisms underlying the DNA damage-induced chromatin binding of the SWI/SNF complexes as well as its functional significance in the γ-H2AX induction and DSB repair.
How might the SWI/SNF complexes promote the phosphorylation of H2AX following DNA damage? Our results support direct mechanisms for this process. In the first, the data clearly excluded the possibilities that the SWI/SNF complexes promote H2AX phosphorylation indirectly by regulating gene expression or affecting the upstream kinase pathways. The SWI/SNF complexes do not affect the expression of H2AX itself (Figures 3B and 4A) or the expression of the kinases, ATM, DNA-PKcs and ATR, which are all responsible for the phosphorylation of H2AX (Figure 5A). In addition, ATM, the major player for H2AX phosphorylation after IR (Burma et al, 2001; Fernandez-Capetillo et al, 2002), can be fully activated and normally recruited to DSB sites in SWI/SNF-defective cells following exposure to IR (Figure 5B–F). Moreover, the other two kinases are also properly recruited to DSB sites in irradiated SWI/SNF-defective cells (Figure 5G and H). Second, the detergent extraction experiments show that the SWI/SNF complexes rapidly bind to chromatin in response to IR in the manner such that the levels of their binding are proportional to the amount of DNA damage (Figure 6A–C). In addition, these experiments also show that the SWI/SNF complexes specifically bind to the chromatin at the sites of γ-H2AX foci (Figure 6D). Finally, the ChIP experiments demonstrated that the SWI/SNF complexes specifically interact with γ-H2AX (Figure 6E–G). All these data strongly suggest that the SWI/SNF complexes directly act on the chromatin to facilitate H2AX phosphorylation. Alterations of the structure of individual nucleosomes by SWI/SNF remodeling could directly affect the accessibility of H2AX at the sites of DSBs. In support of this possibility, it has been shown that DNA-PK-mediated H2AX phosphorylation in the context of nucleosomal structure can be modulated by histone acetylation, an important mechanism to regulate the accessibility of nucleosomes (Park et al, 2003). Alternatively, but not mutually exclusively, the SWI/SNF complexes might facilitate H2AX phosphorylation by influencing the higher order chromatin structure in such a way as to increase the accessibility of the H2AX-containing nucleosomes.
Recent studies showed that several members of the SWI2/SNF2 superfamily of ATP-dependent chromatin remodeling complexes are directly implicated in DSB repair in yeast. Using the system that permits generation of a single DSB at a defined chromosomal locus, the INO80 complex have been shown to be recruited to the DSB site by specifically interacting with γ-H2AX (Downs et al, 2004; Morrison et al, 2004; van Attikum et al, 2004). Similarly, the yeast SWI/SNF and related RSC complexes have been shown to be recruited to the DSB site by unknown mechanisms and by the mechanisms involving the repair proteins such as Mre11 and yKu70, respectively (Chai et al, 2005; Shim et al, 2005). Our work shows that mammalian SWI/SNF complexes are also implicated in DBS repair, emphasizing the evolutionally conserved functions of this family of chromatin remodeling complexes in DNA repair. Interestingly, however, these results also suggest that different members of the family may adopt distinct mechanisms for facilitating DSB repair. The INO80 complex, albeit it interacts with γ-H2AX, is not required for the induction of γ-H2AX following DNA damage (Morrison et al, 2004), whereas mammalian SWI/SNF complexes are critical for the optimal induction of γ-H2AX (this report). Therefore, it is tempting to propose that the SWI/SNF complexes can function upstream of γ-H2AX, whereas the INO80 complex rather contribute to the downstream repair events. Indeed, a recent work showed that the INO80 complex, after being recruited to a DSB, disrupts the nucleosome structure around the DSB in the manner such that the broken DNA becomes accessible to repair proteins (Tsukuda et al, 2005). In this respect, it is of great interest to ask whether yeast SWI/SNF complex would promote γ-H2AX induction, and whether mammalian INO80 complex binds to γ-H2AX but is dispensable for its induction.
Increasing evidence suggests that the SWI/SNF complexes function as a tumor suppressor (Roberts and Orkin, 2004). Most of the experimental evidence supporting the tumor suppressor function of the SWI/SNF complexes relies on their activities to regulate the expression of the genes involved in cell cycle and proliferation. In this report, we show that the SWI/SNF complexes can function as a positive regulator of γ-H2AX which has recently been defined as a haploinsufficiency tumor suppressor (Bassing et al, 2003; Celeste et al, 2003a). Our findings therefore provide an important insight into the understanding of the tumor suppressor function of these evolutionally conserved ATP-dependent chromatin remodeling complexes in the context of the function in the chromosomal DSB repair as well as their well-established role for transcription.
Materials and methods
Cell culture and transfection
tet-VP16 and B05-1 cells and their culture conditions have been previously described (de la Serna et al, 2000). Cells were cultured under the conditions with (+tet) or without tetracycline (−tet) for 4 days before being subjected to all the experiments described in this work. Human BRG-1 (sc-29827) and hBrm siRNAs (sc-29831) were purchased from Santa Cruz biotechnology (CA, USA). HeLa cells were cotransfected with BRG-1 and hBrm siRNAs using lipofectamine for 48 h before being subjected to the experiments described.
Colony formation assays
After irradiation by 1–5 Gy of γ-ray (137Cs; Cell Irradiation System, GC 3000 Elan-Model β; MDS Nordion, Ontario, Canada), 5 × 102 cells were seeded onto a 60 mm dish in triplicate and incubated for 10 or more days. Visible colonies of more than 50 cells were counted.
Comet assays
Neutral comet assays were performed using the Trevigen's CometAssay kit (4250-050-K) according to the manufacturer's instruction. DNA was stained with Trevigen SYBR green as provided in the kit. Comet images were captured by fluorescence microscopy before tail moments were analyzed by TriTek CometScore Freeware program. Average value of tail moments was determined by counting at least 100 cells per sample.
Chromatin retention assays
Biochemical fractionation of chromatin-associated proteins and in situ detergent extraction experiments were performed as previously described (Andegeko et al, 2001). In brief, approximately 2 × 106 NIH-3T3 cells were suspended in 50 μl of the fractionation buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, protease inhibitors (5 μg/ml each pepstatin, leupeptin and aprotinin), and 1 × phsophatase inhibitor cocktail 1 (Sigma)) containing 0.2% NP-40. After incubation for 5 min on ice, supernatant (Fraction I) was collected by centrifugation at 1000 g for 5 min. Pellet was resuspended in 50 μl of the same buffer and immediately centrifuged as before to collect the second supernatant (Fraction II). The nuclear pellet was further extracted with 50 μl of the fractionation buffer containing 0.5% NP-40 for 40 min on ice followed by centrifugation at 16 000 g for 15 min to separate supernatant (Fraction III) and insoluble pellet (Fraction IV). For in situ detergent extraction, NIH-3T3 cells grown on 22-mm2 glass coverslips were in situ extracted by incubating the coverslips in the fractionation buffer containing 0.2% NP-40 for 20 min on ice. Cells were then fixed for immunocytochemistry.
ChIP
Approximately 6 × 107 cells were fixed with 1% formaldehyde on ice for 10 min followed by incubation with 0.1 M glycine for 5 min. After PBS wash, cells were collected and resuspended in 3.5 ml of sonication buffer containing 50 mM HEPES pH 7.5, 150 mM NaCl, 0.1% SDS, protease-phosphatase inhibitors. Cell suspension was sonicated 12 times for 10 s at 30% setting using Cole-Parmer 400 Watt Ultrasonic Homogenizer (these conditions give mostly mononucleosome-size DNA fragments). The lysate was then clarified by centrifuge at 1500 g for 5 min. The supernatant was added by sonicated herring sperm DNA (final concentration at 1 μg/ml) and BSA (final concentration at 1 mg/ml), and precleared with protein G sepharose at 4°C for 2 h followed by incubation with 5 μl of anti-Flag M2 affinity gel (Sigma) at 4°C for overnight. After four time washing with the sonication buffer, pellet was suspended in SDS–PAGE sample buffer and boiled for 10 min before being subjected to SDS–PAGE and immunoblot analysis.
Microarray experiments
A full list of the genes differentially expressed between B05-1(+tet) and B05-1(−tet) cells are available at The EMBO Journal Online. The microarray data have been deposited in the ArrayExpress database (Accession # A-MEXP-256, E-MEXP-434).
Immunofluorescence microscopy, checkpoint analysis, immunoblot analysis, plasmid construction and RT–PCR
Detailed experimental procedures are available at The EMBO Journal Online.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Materials
Acknowledgments
We thank Fred Alt for providing H2AX−/− mouse ES cells and Daekee Lee for helping ES cell culture. This work was supported by Korea Science and Engineering Foundation (KOSEF) grant to JK (R01-2004-000-10047-0), and the Nuclear Research and Development Program from the Ministry of Science and Technology of Korea granted to JK (M2-0408-00-0028), and by the Korea Science and Engineering Foundation (KOSEF) grant to the Center for Cell Signaling Research (Ewha Woman's University) and the Brain Korea 21 Scholars Program.
References
- Allard S, Masson JY, Cote J (2004) Chromatin remodeling and the maintenance of genome integrity. Biochim Biophys Acta 1677: 158–164 [DOI] [PubMed] [Google Scholar]
- Andegeko Y, Moyal L, Mittelman L, Tsarfaty I, Shiloh Y, Rotman G (2001) Nuclear retention of ATM at sites of DNA double strand breaks. J Biol Chem 276: 38224–38230 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2004) Initiating cellular stress responses. Cell 118: 9–17 [DOI] [PubMed] [Google Scholar]
- Bassing CH, Alt FW (2004) H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle 3: 149–153 [DOI] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC (2002) Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc Natl Acad Sci USA 99: 8173–8178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW (2003) Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 114: 359–370 [DOI] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276: 42462–42467 [DOI] [PubMed] [Google Scholar]
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A (2003a) H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 114: 371–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A (2003b) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675–679 [DOI] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA (2002) Genomic instability in mice lacking histone H2AX. Science 296: 922–927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai B, Huang J, Cairns BR, Laurent BC (2005) Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 19: 1656–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DW, Chen BP, Prithivirajsingh S, Kurimasa A, Story MD, Qin J, Chen DJ (2002) Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 16: 2333–2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Serna IL, Carlson KA, Hill DA, Guidi CJ, Stephenson RO, Sif S, Kingston RE, Imbalzano AN (2000) Mammalian SWI–SNF complexes contribute to activation of the hsp70 gene. Mol Cell Biol 20: 2839–2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S, Celeste A, Fernandez-Capetillo O, Chen HT, Reina-San-Martin B, Van Laethem F, Yang YP, Petukhova GV, Eckhaus M, Feigenbaum L, Manova K, Kruhlak M, Camerini-Otero RD, Sharan S, Nussenzweig M, Nussenzweig A (2005) Role of Nbs1 in the activation of the ATM kinase revealed in humanized mouse models. Nat Cell Biol 7: 675–685 [DOI] [PubMed] [Google Scholar]
- Downs JA, Allard S, Jobin-Robitaille O, Javaheri A, Auger A, Bouchard N, Kron SJ, Jackson SP, Cote J (2004) Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol Cell 16: 979–990 [DOI] [PubMed] [Google Scholar]
- Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408: 1001–1004 [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A (2002) DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat Cell Biol 4: 993–997 [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo O, Lee A, Nussenzweig M, Nussenzweig A (2004) H2AX: the histone guardian of the genome. DNA Repair 3: 959–967 [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Peterson CL (1999) A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res 27: 2022–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyodorov DV, Kadonaga JT (2001) The many faces of chromatin remodeling: switching beyond transcription. Cell 106: 523–525 [DOI] [PubMed] [Google Scholar]
- Gaillard H, Fitzgerald DJ, Smith CL, Peterson CL, Richmond TJ, Thoma F (2003) Chromatin remodeling activities act on UV-damaged nucleosomes and modulate DNA damage accessibility to photolyase. J Biol Chem 278: 17655–17663 [DOI] [PubMed] [Google Scholar]
- Hara R, Sancar A (2002) The SWI/SNF chromatin-remodeling factor stimulates repair by human excision nuclease in the mononucleosome core particle. Mol Cell Biol 22: 6779–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH (2001) Genome maintenance mechanisms for preventing cancer. Nature 411: 366–374 [DOI] [PubMed] [Google Scholar]
- Jackson SP (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis 23: 687–696 [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB (2004) Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev 18: 1423–1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi J, Tauchi H, Sakamoto S, Nakamura S, Morishima K-I, Matsuura S, Kobayashi T, Tamai K, Tanimoto K, Komatsu K (2002) NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr Biol 12: 1846–1851 [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y (1999) Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 98: 285–294 [DOI] [PubMed] [Google Scholar]
- Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Müller WG, McNally JG, Bazett-Jones DP, Nussenzweig A (2006) Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 172: 823–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon J, Morshead KB, Guyon JR, Kingston RE, Oettinger MA (2000) Histone acetylation and hSWI/SNF remodeling act in concert to stimulate V(D)J cleavage of nucleosomal DNA. Mol Cell 6: 1037–1048 [DOI] [PubMed] [Google Scholar]
- Lee J, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science 308: 551–554 [DOI] [PubMed] [Google Scholar]
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X (2004) INO80 and γ-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 119: 767–775 [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE (2002) Cooperation between complexes that regulate chromatin structure and transcription. Cell 108: 475–487 [DOI] [PubMed] [Google Scholar]
- Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J (2003) DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res 31: 6819–6827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson CL, Cote J (2004) Cellular machineries for chromosomal DNA repair. Genes Dev 18: 602–616 [DOI] [PubMed] [Google Scholar]
- Reyes JC, Muchardt C, Yaniv M (1997) Components of the human SWI/SNF complex are enriched in active chromatin and are associated with the nuclear matrix. J Cell Biol 137: 263–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Orkin SH (2004) The SWI/SNF complex—chromatin and cancer. Nat Rev Cancer 4: 133–142 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Shim EY, Ma JL, Oum JH, Yanez Y, Lee SE (2005) The yeast chromatin remodeler RSC complex facilitates end joining repair of DNA double-strand breaks. Mol Cell Biol 25: 3934–3944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavridi ES, Halazonetis TD (2005) Nbs1 moving up in the world. Nat Cell Biol 7: 648–650 [DOI] [PubMed] [Google Scholar]
- Strobeck MW, Knudsen KE, Fribourg AF, DeCristoforo MF, Weissman BE, Imbalzano AN, Knudsen ES (2000) BRG-1 is required for RB mediated cell cycle arrest. Proc Natl Acad Sci USA 97: 7748–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. (2005) MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123: 1213–1226 [DOI] [PubMed] [Google Scholar]
- Tsukuda T, Fleming AB, Nickoloff JA, Osley MA. (2005) Chromatin remodelling at a DNA double-strand break site in Saccharomyces cerevisiae. Nature 438: 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel T, Lerenthal Y, Moyal L, Andegeko Y, Mittelman L, Shiloh Y (2003) Requirement of the MRN complex for ATM activation by DNA damage. EMBO J 22: 5612–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Attikum H, Fritsch O, Hohn B, Gasser SM (2004) Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 119: 777–788 [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM (2005) The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 6: 757–765 [DOI] [PubMed] [Google Scholar]
- Wood RG, Mitchell M, Lindahl T (2005) Human DNA repair genes (2005). Mutat Res 577: 275–283, (http://www.cgal.icnet.uk/DNARepairG enes.html) [DOI] [PubMed] [Google Scholar]
- Yung E, Sorin M, Pal A, Craig E, Morozov A, Delattre O, Kappes J, Ott D, Kalpana GV (2001) Inhibition of HIV-1 virion production by a transdominant mutant of integrase interactor 1. Nat Med 7: 920–926 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Table S1
Supplementary Table S2
Supplementary Materials