

Abstract
Stabilization and maturation of synapses are important for development and function of the nervous system. Previous studies have implicated cholesterol-rich lipid microdomains in synapse stabilization, but the underlying mechanisms remain unclear. We found that cholesterol stabilizes clusters of synaptic acetylcholine receptors (AChRs) in denervated muscle in vivo and in nerve–muscle explants. In paralyzed muscles, cholesterol triggered maturation of nerve sprout-induced AChR clusters into pretzel shape. Cholesterol treatment also rescued a specific defect in AChR cluster stability in cultured src−/−;fyn−/− myotubes. Postsynaptic proteins including AChRs, rapsyn, MuSK and Src-family kinases were strongly enriched in lipid microdomains prepared from wild-type myotubes. Microdomain disruption by cholesterol-sequestering methyl-β-cyclodextrin disassembled AChR clusters and decreased AChR–rapsyn interaction and AChR phosphorylation. Amounts of microdomains and enrichment of postsynaptic proteins into microdomains were decreased in src−/−;fyn−/− myotubes but rescued by cholesterol treatment. These data provide evidence that cholesterol-rich lipid microdomains and SFKs act in a dual mechanism in stabilizing the postsynapse: SFKs enhance microdomain-association of postsynaptic components, whereas microdomains provide the environment for SFKs to maintain interactions and phosphorylation of these components.
Keywords: cholesterol, lipid microdomains, neuromuscular junction, Src-family kinases
Introduction
Synaptogenesis is a key process in the development and function of the nervous system. In a first phase, neurotransmitter receptors and associated proteins accumulate to form a postsynaptic density. Proteins and nonprotein factors trigger synaptic differentiation (Fox and Umemori, 2006). A major factor is glia-derived cholesterol, which induces synaptogenesis in cultured retinal ganglion cells (Mauch et al, 2001). Its roles in vivo and specifically in postsynaptic assembly remain unknown. In a second phase of synaptogenesis, some synapses and postsynaptic densities mature and are stabilized, while others are eliminated. Whereas neural activity is known to regulate this process (Cohen-Cory, 2002), the effector machinery in synapse stabilization is poorly understood.
Cholesterol, along with sphingolipids, is enriched in subcompartments of the cellular membrane system. These lipid microdomains, often isolated due to their detergent-resistance, also include signaling proteins, regulate trafficking and signal transduction processes, and may partly correspond to membrane rafts, dynamic structures that bring together activated receptors and transducer molecules (Brown and London, 1998; Simons and Toomre, 2000; Golub et al, 2004; Pike, 2006). The cholesterol-rich lipid microdomains (CRLMs) are involved in aspects of synaptic function in cultured cells. Depletion of cholesterol leads to loss of surface AMPA receptors and of synapses in hippocampal neurons (Hering et al, 2003). In ciliary neurons, CRLMs are necessary for the maintenance of α7 neuronal nicotinic acetylcholine receptors (AChRs) in synapse-associated clusters (Bruses et al, 2001). At the neuromuscular junction (NMJ), the presence of plasmalemmal cholesterol is necessary for proper AChR gating functions (Barrantes, 1993); AChRs associate with CRLMs in trafficking toward the plasma membrane in transfected heterologous cells (Marchand et al, 2002). However, the relevance of lipid microdomains and cholesterol for synaptogenesis in vivo, and the identity of the signaling pathways operating through the microdomains, has remained unclear.
During NMJ formation, myotubes respond to neural agrin, assembling AChRs at nascent synapses (Gautam et al, 1996). This scaffolding function is assigned to MuSK, the trans-membrane kinase that translates agrin into a clustering signal (Glass et al, 1996). Besides MuSK and AChR, rapsyn is the third core protein for the AChR clustering process (Gautam et al, 1995; Marangi et al, 2001). In response to agrin, the association of rapsyn with AChRs increases and mediates binding to cytoskeletal proteins (Moransard et al, 2003). AChR β subunits become tyrosine-phosphorylated and this modification regulates cytoskeletal linkage and efficient clustering (Borges and Ferns, 2001).
During the maturation of NMJs, plaque-shaped AChR clusters are stabilized and adopt pretzel-shaped configurations, with AChRs located at the crests of postjunctional folds. AChR half-life time is highly increased and synaptic proteins are selectively produced by subsynaptic nuclei (Sanes and Lichtman, 2001). The molecular mechanisms mediating this postnatal NMJ stabilization differ from those involved in NMJ induction, and much less is known about them (Willmann and Fuhrer, 2002). Stability of AChR clusters can also be analyzed in cultured myotubes, by adding and then withdrawing agrin or other factors and studying the half-life time of clusters. Although the time scale is different, this assay reveals many parallels to postnatal NMJ stabilization in vivo. Thus, the utrophin complex with its components dystroglycan and dystrobrevin, and Src-family kinases (SFKs) are important in postsynaptic stabilization both in vivo and in vitro (Grady et al, 2000; Jacobson et al, 2001; Smith et al, 2001; Marangi et al, 2002; Sadasivam et al, 2005). SFKs are activated by agrin (Mittaud et al, 2001) and maintain AChR–rapsyn interaction and AChR β phosphorylation (Sadasivam et al, 2005). In cultured src−/−;fyn−/− myotubes, agrin- or laminin-induced AChR clusters are unstable and disperse rapidly after withdrawal of these factors (Smith et al, 2001; Marangi et al, 2002). Interfering with SFK function in vivo causes postsynaptic disintegration of adult NMJs (Sadasivam et al, 2005). Since SFK functions are specifically associated with CRLMs in other cells (Resh, 1999; Simons and Toomre, 2000), these results raise the possibility that microdomain-dependent processes might be involved in postsynaptic apparatus maintenance through SFKs.
To investigate mechanisms of postsynaptic maturation, we determined whether, and through what signaling molecules, cholesterol and lipid microdomains might stabilize NMJs in vivo and in vitro. We find that cholesterol addition stabilizes NMJs and promotes their maturation from patch- to pretzel-type configurations. Postsynaptic proteins reside in CRLMs, and CRLM dispersion disrupts AChR clusters, AChR–rapsyn interaction and AChR β phosphorylation. In src−/−;fyn−/− myotubes, cholesterol addition normalizes the reduced CRLM association of postsynaptic proteins and stabilizes AChR clusters. These results suggest a dual mechanism for postsynaptic cluster stabilization through SFKs, involving an enhancement of the association of cluster components with CRLMs, and interactions and phosphorylation of these components at the microdomains.
Results
Cholesterol stabilizes AChR clusters in vivo
To investigate a possible role of cholesterol and lipid microdomains in promoting postsynaptic apparatus maintenance in vivo, we analyzed the state of assembly of AChR clusters at denervated NMJs in the absence or presence of exogenous cholesterol. The sciatic nerve was cut in 1-month-old mice, and AChR clusters were visualized 12 days later in two DeSyn muscles (lateral gastrocnemius and medial gastrocnemius), which exhibit substantial postsynaptic cluster disassembly under these experimental conditions (Pun et al, 2002). To visualize denervated synaptic sites, we counterstained muscle sections with an antibody against p75, a protein upregulated in Schwann cells in the absence of nerve contact (Taniuchi et al, 1986). As expected, denervated synaptic sites exhibited only remnants of AChR clusters after 12 days of denervation (Figure 1A; note irregular AChR labeling patterns, with only small regions of intense labeling). In contrast, when cholesterol was applied daily to denervated muscles, starting 5 days after denervation, AChR signals at denervated synaptic sites were much better preserved (Figure 1A; note that p75 signals were not affected by the cholesterol treatment). These AChR signals were comparable to clusters in nondenervated control animals (not shown, but refer to an earlier paper (Pun et al, 2002)). A quantitative analysis of AChR labeling intensities revealed that synaptic sites had lost most of their AChR signal 12 days after denervation, but that the synaptic signal was largely preserved in the presence of exogenous cholesterol (Figure 1C).
Figure 1.
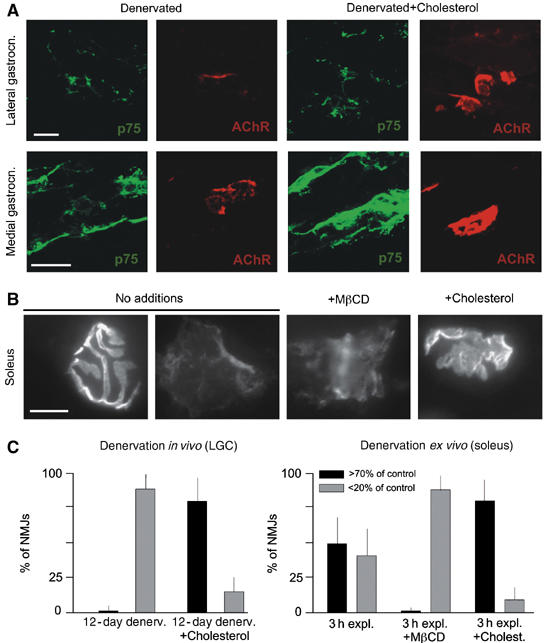
Cholesterol stabilizes AChR clusters in denervated muscles. (A) Appearance of AChR clusters in two DeSyn muscles 12 days after denervation. Sciatic nerves were cut in 1 month mice; the absence of intact axons is confirmed by the expression of p75 in Schwann cells. Where indicated, cholesterol was applied daily, starting 5 days after denervation. The larger p75-positive area in the medial gastrocnemius panel with cholesterol is due to the particular plane of section, and does not reflect a systematic elevation of Schwann cell p75 immunofluorescence signals in denervated muscles treated with cholesterol. (B) Examples of AChR clusters (visualized by rhodamine-α-bungarotoxin; RITC-α-BT) in soleus nerve–muscle explants after 3 h in vitro. (C) Quantitative analysis of data as shown in (A) (left) and (B) (right). AChR labeling intensities (RITC-α-BT) were compared to controls; shown are fractions of NMJs with signal at least 70%, or less than 20% of control values. The 20 and 70% boundaries were selected to highlight the differences among the samples of these experiments. N=300 AChR clusters (from 3 mice each). Bars: 40 (A) and 20 μm (B).
To investigate AChR cluster protection by cholesterol under more challenging experimental conditions, we analyzed nerve–muscle explant preparations of soleus maintained at 37°C in Ringer solution supplemented with calcium. To reliably identify synaptic sites, we carried out these experiments using transgenic mice expressing a synaptophysin-GFP construct in neurons (Thy1-spGFPmu) (De Paola et al, 2003). Under these experimental conditions, many synaptic sites lost most of their AChR signal after 3 h ex vivo, such that about half the synapses appeared normal while others had only low-intensity AChR label (Figure 1B and C). Inclusion of the cholesterol sequestering agent methyl-β-cyclodextrin, which disrupts CRLMs (Simons and Toomre, 2000; Tansey et al, 2000; Ma et al, 2003), accelerated the loss of AChR signal (Figure 1B and C). In contrast, inclusion of cholesterol in the culture medium protected most AChR clusters (Figure 1B and C).
To determine whether cholesterol might also promote the assembly of new AChR clusters in vivo, we used reporter mice expressing membrane-targeted GFP in neurons (Thy1-mGFPs) (De Paola et al, 2003) and carried out cholesterol supplementation experiments in lateral gastrocnemius muscle chronically treated with Botulinum toxin A. These experimental conditions (lateral gastrocnemius in 1-month-old mice; toxin applications every 4th day for a total of 20 days) induce the disassembly of postsynaptic apparatus at NMJs, a massive nerve sprouting response, and induction of small ectopic AChR plaques along the nerve sprouts (Figure 2A and B (left panels); see also Santos and Caroni, 2003). Daily local applications of cholesterol from day 10 of the BotA treatment, that is, at a time when NMJ disassembly and nerve sprouting were not yet pronounced (Santos and Caroni, 2003), led to a suppression of the AChR cluster disassembly process, which was accompanied by a suppression of nerve sprouting and of ectopic AChR plaque induction by sprouts in these paralyzed muscles (Figure 2A, right panels). The resulting AChR signals appeared very similar to those in nontreated control animals (not shown; but refer to Santos and Caroni, 2003). Significantly, initiation of the cholesterol treatment at day 15, when sprouting was well advanced (Santos and Caroni, 2003), led to the assembly of large, pretzel-shaped ectopic AChR clusters along the sprouts (Figure 2B).
Figure 2.
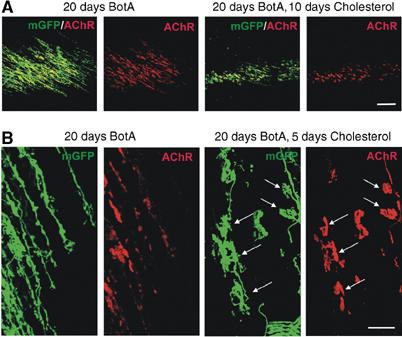
Cholesterol promotes AChR cluster assembly at original and ectopic NMJs in paralyzed DeSyn muscles. Low- (A) and high-magnification (B) views of presynaptic nerves (mGFP) and postsynaptic AChR clusters (RITC-α-BT) in lateral gastrocnemius muscles treated with Botulinum toxin A (BotA). The chronic BotA treatment elicited a massive nerve sprouting response in this DeSyn muscle; cholesterol promoted AChR cluster assembly, and inhibited nerve sprouting. Note pretzel-shaped AChR clusters (arrows, right) induced by sprouts (arrows, left) in the presence of exogenous cholesterol. Bars: 200 (A) and 40 μm (B).
Taken together, these data provide evidence that local applications of exogenous cholesterol in vivo protect AChR clusters against denervation-induced disassembly, and promote the maturation of sprout-induced ectopic AChR clusters in paralyzed muscles from an embryonic-type plaque shape into a pretzel shape. We thus propose that cholesterol is an important factor for the maturation and stabilization of the NMJ in vivo.
Cholesterol stabilizes AChR clusters in cultured src−/−;fyn−/− myotubes
To analyze the mechanism of action of cholesterol in stabilizing AChR clusters, we turned to aggregation assays in cultured myotubes. Furthermore, we took advantage of cells from mice lacking Src and Fyn, where AChR clusters are normally induced by agrin or laminin treatment, but disassemble within a few hours after the removal of these factors from the medium (Smith et al, 2001; Marangi et al, 2002). We treated src−/−;fyn−/− myotubes with agrin to induce maximal AChR clustering, then withdrew agrin and determined whether the addition of cholesterol might stabilize AChR clusters. We found that after 5 h, cholesterol-treated cells showed the same number of AChR clusters as cells from which agrin was not withdrawn (Figure 3). Cells from which agrin was withdrawn for 5 h, without addition of cholesterol, showed a low cluster number, comparable to the level of spontaneous clustering. In wild-type cells, clusters were very stable following removal of agrin, as published previously (Smith et al, 2001; Marangi et al, 2002). This stability prohibited assessing significant effects of cholesterol.
Figure 3.
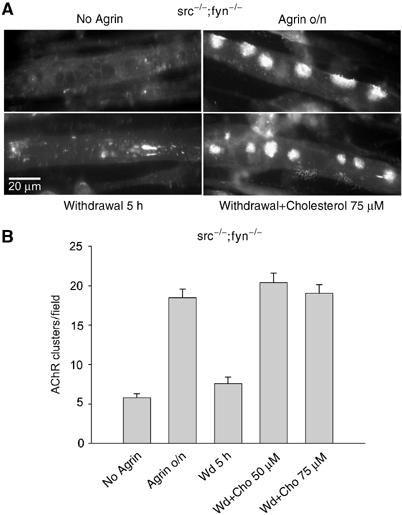
Cholesterol stabilizes AChR clusters in src−/−;fyn−/− myotubes. (A) src−/−;fyn−/− myotubes were not treated or stimulated overnight with 1 nM agrin to induce AChR clusters (top row). Agrin was withdrawn, cells were washed and incubated for 5 h in agrin-free medium lacking (bottom left) or containing (bottom right) 75 μM cholesterol. Myotubes were stained with rhodamine-α-BT to visualize AChR clusters. (B) For cluster quantification, visual fields covering about three times the area of a panel shown in (A) were taken, and only compact clusters with a mimimum size of 5 μm were counted.
We next determined whether cholesterol might compensate for agrin withdrawal by enhancing signaling processes involved in the formation of the NMJ. Cholesterol addition to wild-type myotubes did not induce formation of AChR clusters (Figure 4A) and did not lead to phosphorylation of MuSK or the β-subunit of AChRs, unlike agrin (Figure 4B and C). These data show that cholesterol stabilizes AChR clusters in cultured myotubes but does not activate agrin/MuSK signaling.
Figure 4.
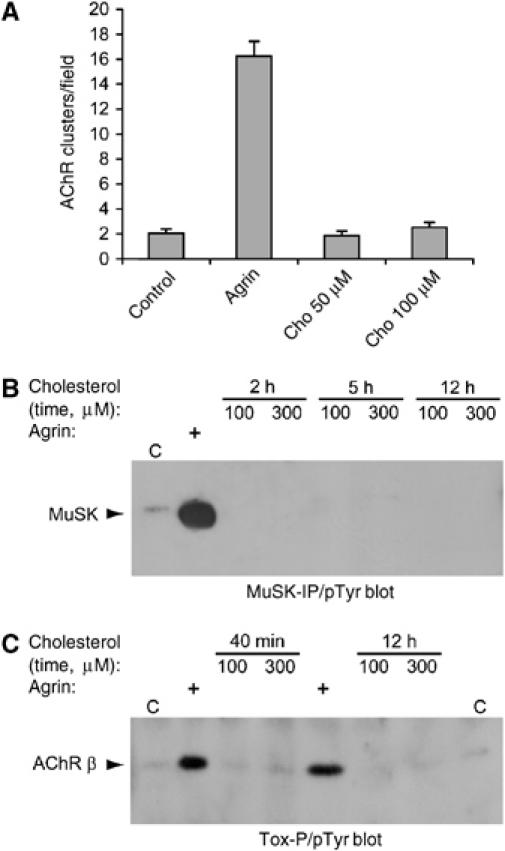
Cholesterol does not induce AChR clustering and phosphorylation of MuSK and AChR β subunits. (A) Cholesterol or 1 nM agrin were added overnight to C2C12 myotubes. Cells were stained with rhodamine-α-BT and AChR clusters quantitated as in Figure 3. (B, C) C2C12 myotubes were treated with different doses of cholesterol, or with 1 nM agrin for 40 min, as indicated; C, untreated control. From cell lysates, MuSK was immunoprecipitated (B) or AChRs were precipitated using biotin-α-BT and streptavidin-agarose (Tox-P; C). Phosphotyrosine immunoblotting detected phosphorylation of MuSK and AChR β subunits. The identity of these phosphoproteins was confirmed by reprobing with MuSK- or AChR β-specific antibodies (not shown).
The proteins involved in AChR cluster stabilization reside in CRLMs
Cholesterol is a key component of lipid microdomains, and its action in cluster stabilization might involve microdomain-dependent processes. We therefore prepared and analyzed CRLMs from cultured wild-type myotubes using a well-established detergent-free protocol (Song et al, 1996a, 1996b; Riddell et al, 2001; Nishio et al, 2004; Rhainds et al, 2004; Zhang et al, 2005). Cell homogenates were floated on discontinuous sucrose gradients, and fractions were analyzed by immunoblots. The CRLM fractions (4–6) were found at the interface between 5 and 35% sucrose and defined by strong enrichment of typical markers such as caveolin-3, flotillin-2, cholesterol and the sphingolipid, ganglioside GM1 (Figure 5A and B). Measurement of protein concentration showed that fractions 4–6 contained little of the overall protein (only 9.5±0.2%; mean±s.e.m., n=5); most protein was found at the bottom of the gradient, in fractions 8–12 which contain free (not microdomain associated) cellular proteins (Figure 5C). Another negative control was α-tubulin, which did not associate with CRLMs and was mostly recovered in the free fractions (Figure 5A). These controls established the validity of the method for preparation of CRLMs from myotubes.
Figure 5.
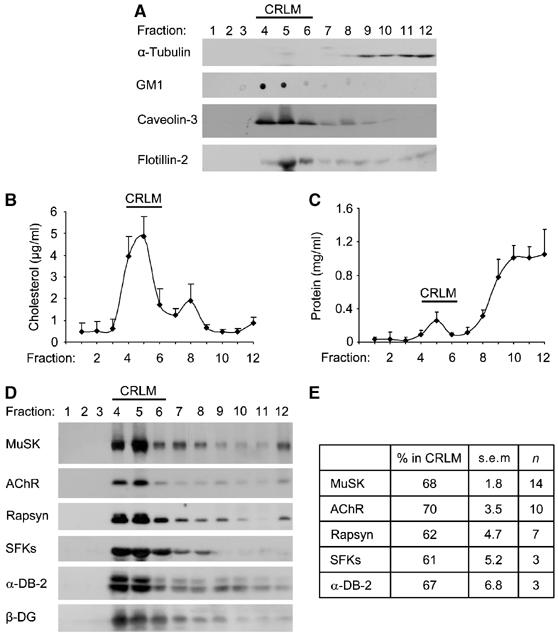
Postsynaptic proteins associate with CRLMs in myotubes. CRLMs were prepared from wild-type myotubes (C2C12 or clones SW10 and SW5), and fractions of the discontinuous sucrose gradients were collected. Fractions 9–12 represent the bottom gradient step (45% sucrose) containing the total cell extract. Fractions 5–8 represent the 35% sucrose layer and fractions 1–4 the top layer (5% sucrose). (A) Fractions were analyzed by immunoblotting (α-tubulin, caveolin-3, flotillin-2) or dot blotting (ganglioside GM1). Fractions 4–6 contain CRLMs and α-tubulin served as a negative control. (B) Fractions were analyzed for the content of cholesterol, showing high enrichment in CRLM fractions 4–6 (n=4). (C) Protein assays of gradient fractions reveal the bulk of protein in the bottom fractions, illustrating the specificity of the CRLM preparation (n=4). (D) Fractions were subjected to immunoblotting, showing that MuSK, AChRs (β-subunit), rapsyn, SFKs, α-dystrobrevin-2 (α-DB-2) and β-dystroglycan (β-DG) all partition efficiently into CRLMs. (E) Blots as shown in (D) were quantitated by densitometric scanning. For each protein, the intensities of bands in CRLM fractions 4–6 were related to the sum of all fractions to quantify the percentage in CRLMs (n=number of experiments).
We probed fractions for the content of postsynaptic proteins. We found the AChR highly enriched in CRLMs, as 70% of the total receptor present in all fractions of the gradient resided in fractions 4–6 (Figure 5D and E). This reflects a 7.3-fold enrichment over bulk protein (Figure 5C). Like AChRs, elements of the agrin signaling pathway such as MuSK and rapsyn were similarly concentrated in CRLMs. The same was true for SFKs (Figure 5D and E), which are known from other cell types to be typical constituents of CRLMs (Resh, 1999; Simons and Toomre, 2000). Finally, β-dystroglycan and α-dystrobrevin-2, members of the utrophin complex important for NMJ stabilization (Grady et al, 2000; Jacobson et al, 2001), were also recovered efficiently in CRLMs. An overnight incubation with agrin, sufficient to produce maximal AChR clustering, did not detectably affect CRLM-association of AChR, rapsyn and MuSK (data not shown).
Dispersion of CRLMs disrupts AChR clusters, AChR–rapsyn interaction and AChR β phosphorylation
Besides microdomain-association of proteins, another standard tool to investigate the role of CRLMs in a given cellular process is to disrupt them by methyl-β-cyclodextrin (MβCD). We treated wild-type myotubes overnight with agrin to induce maximal AChR clusters and then added MβCD for 1–1.5 h. The number of clusters of normal size and morphology was strongly reduced by MβCD (Figure 6). On MβCD treatment, we noticed many smaller clusters and areas containing cluster fragments. The myotube morphology was unaffected, and following removal of MβCD the myotubes lived for extended periods of time and formed normal agrin-induced stable AChR clusters, like untreated controls (data not shown). This indicates that the MβCD effect was specific and not a consequence of impaired myotube health. These data thus show that the integrity of CRLMs is required to maintain the accumulation of large focal AChR clusters at the cell surface.
Figure 6.
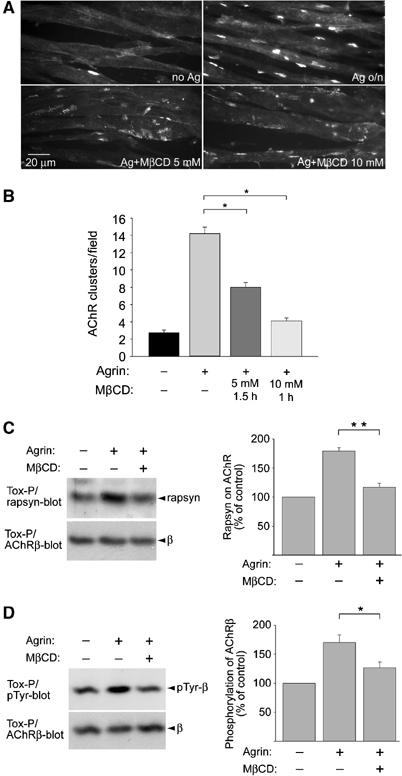
MβCD disrupts AChR clusters, AChR–rapsyn interaction and AChR β phosphorylation in C2C12 myotubes. (A) C2C12 myotubes where first treated overnight with agrin (Ag) to induce AChR clusters. MβCD was then added in the continued presence of agrin, causing AChR clusters to fragment and disappear, as revealed by rhodamine-α-BT staining. (B) Clusters of 5 μM minimal size were quantitated. (C, D) Cells were treated with agrin and MbCD (5 mM, 1.5 h) as in (A). AChRs were precipitated from cell lysates using biotin-α-BT (Tox-P). In immunoblots, AChR-associated rapsyn (C), phosphorylation of AChR β (D) and AChR β itself (C, D; whole protein-control) were detected and quantified by densitometric scanning (C, n=4; D, n=8). *P<0.05, **P<0.01, by two-tailed t-test.
Maintenance of the AChR–rapsyn interaction and of AChR β-phosphorylation depends on SFKs and is crucial in maintaining clusters (Sadasivam et al, 2005). We therefore analyzed the role of CRLMs in these processes. Myotubes were again treated overnight with agrin to induce maximal clustering, followed by addition of 5 mM MβCD for 1.5 h. AChRs were isolated from cell lysates, and associated rapsyn or phosphotyrosine content determined by immunoblotting. We found that the agrin-induced increase in AChR–rapsyn interaction was disrupted by MβCD (Figure 6C). Likewise, agrin-induced phosphorylation of AChR β was reduced to basal levels by MβCD (Figure 6D).
Taken together, these results show that dispersion of CRLMs prevents the maintenance of large focal AChR clusters by disrupting agrin-induced AChR–rapsyn interaction and AChR phosphorylation.
Impaired partitioning of postsynaptic proteins into CRLMs in the absence of Src and Fyn, and rescue by cholesterol
To further define the molecular mechanism through which cholesterol and lipid microdomains stabilize AChR clusters, we analyzed the composition of CRLMs prepared from src−/−;fyn−/− myotubes, in which clusters are unstable. Like those from wild-type cells, CRLMs from mutant myotubes were enriched for ganglioside GM1, caveolin-3 and flotillin-2 (Figure 7A). A cholesterol profile revealed enrichment in CRLMs, but to a lesser extent than in wild-type cells (Figure 7B), as the CRLM peak was smaller (fractions 4–6) whereas non-CRLM fractions (e.g. 8) were increased (see also Figure 7E). CRLMs from src−/−;fyn−/− myotubes contained little overall protein (Figure 7C), as did wild-type CRLMs. Interestingly, significantly less of the total AChR and MuSK were in CRLMs from src−/−;fyn−/− myotubes when compared to wild type, the decrease being 30% for AChRs and 23% for MuSK (Figure 7D).
Figure 7.
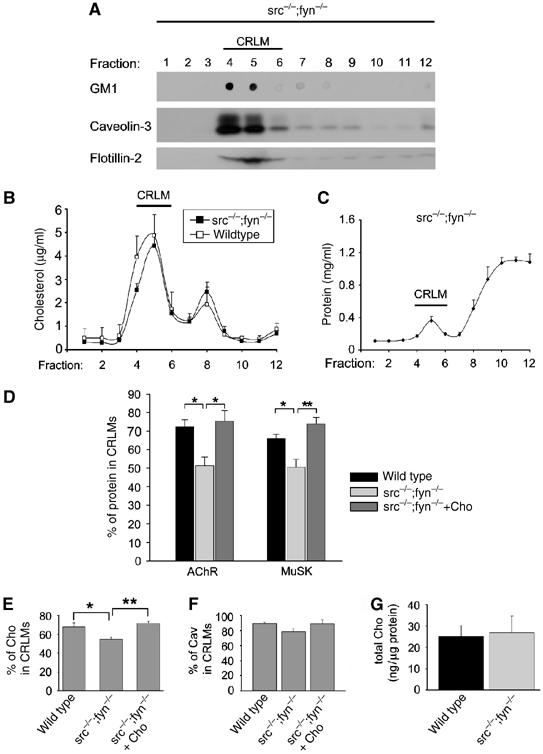
In src−/−;fyn−/− myotubes, CRLM association of postsynaptic proteins is reduced but restored by cholesterol. (A–C) Characterization of CRLMs in src−/−;fyn−/− myotubes. CRLMs were prepared from clones DM11 or DM15, and the content, in gradient fractions, of ganglioside GM1, caveolin-3, flotillin-2 (A), cholesterol (B, n=12) and total protein (C, n=4) was analyzed. Markers are concentrated in CRLM fractions 4–6, with overall protein enriched at the gradient bottom (negative control). Cholesterol is less enriched in CRLMs than in wild-type cells (B; we show the profile from Figure 5B for comparison). (D) Gradient fractions were analyzed for the content of AChR and MuSK, and the percentage of these proteins in CRLM fractions 4–6 was quantified as in Figure 5E. Wild-type cells (C2C12 or clones SW5 and SW10; n=3–7), src−/−;fyn−/− myotubes (n=4–6) and src−/−;fyn−/− myotubes treated with cholesterol (n=4–5) were used. src−/−;fyn−/− myotubes have significantly lower percentages of AChRs and MuSK in CRLMs, and cholesterol restores this. (E) Cells were treated as in (D) and the percentage of cholesterol in CRLM fractions 4–6 was quantitated. CRLM association of cholesterol is lower in src−/−;fyn−/− myotubes (n=12) than in wild-type myotubes (n=8). Addition of cholesterol to the cell culture medium restores the amount of cholesterol in the CRLM fractions to the levels of wild-type myotubes (n=6). (F) Analysis as in (E), examining caveolin-3 (Cav). (G) The total amount of cholesterol, detected in total cell extracts, is the same in wild-type and src−/−;fyn−/− myotubes (n=8). *P<0.05, **P<0.01, by two-tailed t-test.
The overproportionally decreased CRLM association of AChRs in src−/−;fyn−/− myotubes could have two reasons: Src and Fyn may maintain normal numbers of CRLMs in a myotube and/or act as a recruitment signal that brings postsynaptic proteins (such as AChRs) into CRLMs. To investigate these possibilities, we quantitated CRLM partitioning of typical CRLM markers. 19% less of total cholesterol were found in the CRLM fractions 4–6 in src−/−;fyn−/− myotubes when compared to wild type (Figure 7E) and similar observations were made for caveolin-3 (Figure 7F). Overall cellular levels of cholesterol, quantified per microgram of cellular protein, were normal in the mutants, excluding overall nonspecific effects from the lack of Src and Fyn (Figure 7G). These data suggest that src−/−;fyn−/− myotubes have less CRLMs than wild-type cells. The reduction in CRLMs however appears smaller than the reduction in CRLM association of AChRs. Thus, Src and Fyn most likely also act as a recruitment factor for AChRs (and MuSK) into CRLMs.
Importantly, cholesterol addition to src−/−;fyn−/− myotubes not only stabilized AChR clusters (Figure 3), but restored the CRLM partitioning of AChRs and MuSK back to normal (Figure 7D). Likewise, the CRLM enrichment of cholesterol itself and of caveolin were normalized by cholesterol treatment (Figure 7E and F). Thus, while the absence of Src and Fyn decreases the number of CRLMs and the recruitment of postsynaptic proteins into these microdomains, cholesterol addition overcomes this, normalizing the enrichment of AChR and MuSK in CRLMs. Taken together, these loss- and gain-of-function data strongly implicate a role of intact CRLMs, through SFKs, in AChR cluster stabilization.
As the half-life time of AChRs is highly increased at postnatal NMJs, we investigated if changes in AChR turnover may underlie some of our CRLM effects on cluster stability. The half-life time of surface AChRs was the same in wild-type, src−/−;fyn−/−, and src−/−;fyn−/− myotubes treated with cholesterol (Supplementary Figure 1). Furthermore, our MβCD and cholesterol treatments, or the absence of Src and Fyn, did not affect the overall distribution of AChRs between surface and intracellular pools (Supplementary Figure 2). Thus, our observed effect of CRLMs on cluster stability is unlikely to originate from changes in AChR degradative or synthetic pathways.
Discussion
We have shown that cholesterol is an important factor for the stabilization and maturation of the NMJ in vivo and in vitro. This involvement of cholesterol does not stem from activation of the agrin/MuSK signaling pathway, but from promoting the incorporation of postsynaptic proteins into CRLMs. We provide evidence that CRLM integrity is important for the maintenance of AChR clusters, and that SFKs trigger postsynapse stabilization by enhancing the association of critical postsynaptic proteins with these microdomains. CRLMs, in turn, allow SFKs to phosphorylate the AChR and to maintain AChR–rapsyn interaction. These data show that concerted action of cholesterol with lipid microdomains and SFKs forms a mechanism for stabilization of the postsynapse of NMJs.
Cholesterol promotes postsynaptic stabilization
We provide the first evidence that cholesterol promotes synaptic stability in vivo. Cholesterol addition to denervated adult DeSyn gastrocnemius muscle during 12 days prevented the postsynaptic disassembly that would normally occur. Furthermore, soleus nerve–muscle explants exhibited substantial AChR pretzel disassembly within 3 h, which was largely prevented by cholesterol treatment. Finally, Botulinum toxin A caused massive nerve sprouting and induction of plaque-shaped AChR clusters along the sprouts. When applied before this response, cholesterol stabilized existing AChR pretzels at NMJs; when applied after massive sprouting had started, cholesterol treatment promoted maturation of sprout-induced AChR cluster to adopt adult-type pretzel shape. Cholesterol addition, at the time of agrin withdrawal, also stabilized AChR clusters in cultured src−/−;fyn−/− myotubes.
Conversely, sequestering cholesterol by MβCD treatment, leading to disruption of lipid microdomains, accelerated the disassembly of AChR pretzels in explants of soleus muscle, and it disrupted clusters of AChRs in wild-type myotubes in culture. These data establish cholesterol as a factor for the stabilization and maturation of the postsynaptic apparatus at the NMJ.
Interestingly, the mechanism of cholesterol action in postsynaptic stabilization does not involve a reactivation of those pathways that lead to the formation of the NMJ: we find that cholesterol addition to myotubes does not cause AChR clustering, activation of MuSK or phosphorylation of AChR β subunits. Since cholesterol nonetheless stabilizes AChR clusters, this shows that the pathways for stabilization of postsynaptic clusters are different from those for induction of cluster formation.
Cholesterol action in postsynaptic stabilization occurs via CRLMs
To investigate the mechanism of action of cholesterol, we used myotubes in culture. We found that AChRs, rapsyn, MuSK, SFKs, α-dystrobrevin-2, and β-dystroglycan were highly concentrated in CRLMs, and the degree of concentration, ca. 70% of total, was similar to cholesterol. Importantly, cholesterol addition to cultured src−/−;fyn−/− myotubes increased the CRLM association of AChRs and MuSK. It also augmented CRLM participation of cholesterol in these cells (see Figure 7E), suggesting that the cholesterol treatment increased the number of these microdomains.
These observations, together with the stabilizing effect of cholesterol on AChR clusters and the disassembly of clusters by the CRLM-disrupting agent MβCD, led to the conclusion that cholesterol and lipid microdomains are key players in stabilizing the postsynaptic apparatus of NMJs, by promoting the CRLM-association of postsynaptic proteins.
Cholesterol, lipid microdomains and SFKs: a core mechanism for stabilization of the postsynapse
We used src−/−;fyn−/− myotubes to investigate signaling pathways by which cholesterol and CRLMs stabilize clusters of AChRs. The following observations indicate that cholesterol and CRLMs act in concert with SFKs in postsynaptic stabilization. First, the absence of Src and Fyn (Smith et al, 2001; Marangi et al, 2002), like the disruption of CRLMs by MβCD (Figure 6), cause disassembly of AChR clusters in cultured myotubes. In vivo, SFKs maintain adult NMJs (Sadasivam et al, 2005), similar to the stabilizing action of cholesterol (Figures 1 and 2). Second, SFKs themselves are enriched in CRLMs, suggesting that their action in postsynaptic stabilization may occur through the microdomains. In cultured src−/−;fyn−/− myotubes, cytoskeletal linkage of AChRs is weakened and, following agrin withdrawal, AChR β phosphorylation and AChR–rapsyn interaction rapidly decrease (Sadasivam et al, 2005). We show here that addition of MβCD to agrin-treated wild-type myotubes produces the same effect, as it reduces AChR–rapsyn interaction and AChR β phosphorylation. Thus, CRLMs allow SFKs to act in the stabilization of AChR clusters by phosphorylating the AChR and maintaining its link with rapsyn and the cytoskeleton. Third, in src−/−;fyn−/− myotubes CRLM association of postsynaptic proteins is reduced due to both a decrease in the CRLM number and a loss of Src-Fyn-dependent recruitment of postsynaptic proteins into CRLMs. Cholesterol addition to src−/−;fyn−/− myotubes restores CRLM numbers, CRLM association of postsynaptic proteins and stability of AChR clusters.
Based on these results we propose a mechanism involving a reciprocal relationship between Src, Fyn and CRLMs in AChR cluster stabilization. In the first aspect of this dual mechanism, Src and Fyn mediate CRLM integrity and recruitment of postsynaptic apparatus components into the microdomains. CRLM integrity is known to involve an optimal balance between microdomain-lipids and -proteins: upon addition of excess ganglioside GM1 to cultured MDCK cells, CRLM proteins participate to a lesser degree into CRLMs, since the lipid–protein balance in the microdomains is disturbed (Simons et al, 1999). In a similar fashion, absence of the prominent CRLM components Src and Fyn, which contain both lipid (double acylation) and protein parts, may cause deranged lipid–protein ratios in CRLMs, leading to a decrease in functional microdomains. Treatment with cholesterol overcomes this, most likely because exogenous cholesterol is incorporated into the plasma membrane and restores lipid–protein ratios in the microdomains independently of Src and Fyn.
Recruitment of proteins into CRLMs is triggered by lipid modifications that act as anchors, a prominent example being GPI-linked proteins. SFKs are targeted to the microdomains through their fatty acyl groups (Resh, 1999). AChRs and MuSK lack such modifications but interact with SFKs (Fuhrer and Hall, 1996; Mohamed et al, 2001); AChRs associate with several postsynaptic proteins including utrophin (Fuhrer et al, 1999), which is linked to F-actin (Winder et al, 1995). These protein interactions are likely to form a basis for our observed SFK-dependent recruitment of postsynaptic proteins into CRLMs, and the interactions contribute to microdomain stabilization through link to the actin cytoskeleton.
In the second aspect of the dual mechanism for AChR cluster stabilization, CRLMs in turn create the required lipid and protein microenvironment for Src and Fyn to act in the NMJ stabilization pathway. Tyrosine kinases in muscle are known to be counteracted by phosphatases that control, for example, the end level of AChR and MuSK phosphorylation (Wallace, 1994; Madhavan et al, 2005). Incorporation of postsynaptic components in CRLMs may protect them from phosphatases, allowing kinases of the Src-family to act in NMJ stabilization. This action involves enhancement of protein modifications and of key protein interactions, such as phosphorylation of the AChR β subunit and interaction of AChR with rapsyn. The dual mechanism is likely to occur in a subpopulation of CRLMs, which are known to exist in various subtypes differing in lipid and protein composition. Such a subpopulation, affected by the lack of Src and Fyn, would create a strong effect on postsynaptic stability, while the decrease in overall CRLM association of postsynaptic proteins remains moderate when viewed at the level of all CRLMs.
Besides AChR and rapsyn, SFKs are likely to have other downstream target molecules in stabilizing the postsynapse, most likely cytoskeletal organizers (Sadasivam et al, 2005). CRLMs may participate in the regulation of such downstream pathways. In agreement with such a proposal, both SFKs and lipid microdomains are known in other cell types to promote signaling interactions that locally organize cytoskeletal elements, for example by favoring activation of Rho-like GTPases and promoting actin assembly (Golub et al, 2004; Rodgers et al, 2005), or by organizing microtubules (Cox and Maness, 1993; del Pozo et al, 2004; Palazzo et al, 2004). Thereby specialized domains can be formed at the cell surface as represented by focal adhesion sites, and these specializations are stabilized through many participating cytoskeletal elements. Such microdomain-dependent signaling processes occur at or close to the plasma membrane, consistent with our observation that changes in AChR turnover are not a major aspect of CRLM- and SFK-mediated cluster stability in cultured myotubes.
In summary, our work shows that CRLMs represent a microenvironment for postsynaptic NMJ proteins. The microdomains are formed due to SFK action and in turn allow these kinases to promote key phosphorylations and protein interactions for maintenance of the postsynaptic apparatus. The downstream substrates in this cascade remain to be investigated.
Materials and methods
In vivo and ex vivo experiments
Thy1-mGFPs and Thy1-spGFPmu reporter mice were as described elsewhere (De Paola et al, 2003); all treatments were initiated when mice were 1-month-old. Drugs were injected locally, subcutaneously (100 μl/mouse injection volumes). Botulinum toxin A (Allergan AG, Lachen, Switzerland) was applied at 0.02 U/g body weight every 4th day. Where indicated, cholesterol was applied daily (50 μM in injection solution). Nerve–muscle explants of soleus were maintained at 37°C in calcium-supplemented Ringer solution for 3 h, and then labeled with RITC-α-BT (2 μg/ml) for the analysis of AChR clusters. Where indicated, drugs in the incubation medium were 50 μM (cholesterol) and 10 mM (MβCD). Cryostat sections of unfixed muscles were postfixed (10 min, 3.5% formaldehyde in PBS) and labeled for immunocytochemistry as described before (Pun et al, 2002). Fluorescent data were imaged and acquired using an Olympus (BX61) confocal microscope, Fluoview 4.1 software, and identical settings for all experiments belonging to one set (denervation, nerve–muscle explants, paralysis experiments). NMJ labeling intensities (integral of RITC-α-BT signal at individual NMJs) were derived from z-stacks using ImageJ software. Only NMJs lying en-face with respect to the plane of imaging were included in the analysis. For paralysis experiments, we analyzed muscle innervation patterns using reporter mice expressing mGFP in neurons (Thy1-mGFPs) and whole mount preparations of muscles. Briefly, identified muscles were dissected, fixed in PBS with 3.5% formaldehyde (30 min, room temperature), washed and counterstained with RITC-α-BT (2 μg/ml).
Cell cultures and treatments
C2C12, src−/−;fyn−/− (clones DM11 and DM15), and their corresponding wild-type cells (clones SW5 and SW10) were propagated and fused to form myotubes as described earlier (Smith et al, 2001). To induce maximal AChR cluster formation, cells were treated with 1 nM recombinant neural agrin C-Ag12,4,8 (Fuhrer et al, 1999) overnight for 16 h. To withdraw agrin, cells were washed and incubated in agrin-free medium; this procedure removes the vast majority of agrin from cells (Mittaud et al, 2004). For cholesterol treatment, water-soluble cholesterol (Sigma; Fluka, Switzerland) was aliquoted in PBS, diluted in fusion medium immediately prior to use, and used at a final concentration of 50 μM if not specified otherwise. In MβCD treatments, MβCD (Sigma) was diluted in fusion medium at 100 mM and used at a final concentration of 10 or 5 mM. We confirmed effective depletion of cholesterol from the cell membrane after incubation with 5 mM MβCD for 40–60 min: total cellular cholesterol content was reduced to 50–65% of untreated controls (data not shown).
C2C12, SW5 and SW10 cells gave identical results in all assays and we refer to them commonly as wild-type cells. Likewise, DM11 and DM15 showed no clonal variation in all methods.
Preparation of CRLMs
We used a detergent-free method that was shown before to be efficient for preparing CRLMs from C2C12 myotubes (Song et al, 1996a, 1996b), with slight modifications. Briefly, cells grown in 10 cm dishes were washed two times with ice-cold PBS containing 1 mM Na-orthovanadate (NaO). After addition of 1.5 ml Buffer A (Na-carbonate 0.5 M, pH 11; inhibitors cocktail as follows: NaO 1 mM, phenylarsine oxide 50 μM, p-nitrophenylphosphate 10 mM, NaF 50 mM, aprotinin 25 μg/ml, leupeptin 25 μg/ml), cells were quickly scraped and suspended using the pipette tip, then homogenized two times in a dounce homogenizer and finally sonicated two times for 10 s. The inhibitors were always prepared freshly and added to buffers just before use. The total extract (final volume: 2 ml) was quickly mixed with 2 ml 90% sucrose in Buffer B (Mes 25 mM, pH 6.5, NaCl 150 mM+inhibitor cocktail as above) at the bottom of a 13-ml tube and overlaid with 4 ml of 35% sucrose in Buffer C (buffer B+Na-carbonate 250 mM) and then with 4 ml of 5% sucrose in Buffer C, for a total volume of 12 ml. Samples were centrifuged for 17 h at 37 000 rpm in a Sorvall TH-641 rotor at 4°C. 1 ml fractions were collected from the top and transferred into 3 ml ultraclear tubes (Beckman). Samples for protein determination (50 μl), cholesterol determination (30 μl) and ganglioside GM1 detection (2 μl) were taken before diluting each fraction with 2 ml Buffer C. Fractions were then centrifuged for 50 min at 100 000 r.p.m. in a Beckman TLA-100.3 rotor, supernatants were discarded and pellets were resuspended in Lämmli-buffer for SDS-Gel electrophoresis and Western blot.
Cell labeling and immunoprecipitation
For AChR stain, cells were incubated 40 min with rhodamine-coupled α-BT at 37°C, washed once in PBS at room temperature and then fixed in ice-cold methanol for 7 min at −20°C. Conventional fluorescence imaging was carried out using a Zeiss Axioskop 2 microscope equipped with a Hamamatsu Orcacam digital camera. For quantitation, compact clusters with intensities clearly higher than background and a minimal size of 5 μM were considered as detailed previously (Marangi et al, 2001, 2002). Clusters were counted in at least 15 fields and experiments were repeated at least three times.
For precipitation of MuSK or AChRs, cell lysates were prepared. MuSK antibodies followed by protein A-sepharose beads or biotin-α-BT followed by streptavidin-beads (Tox-P) were added as described before (Mittaud et al, 2001, 2004).
Protein and lipid detection
Antibodies against phosphotyrosine (mixture of PY20 and 4G10); the conserved C-terminus of Src, Fyn, and Yes (src-CT); MuSK; rapsyn (Rap-1); β-dystroglycan; the AChR β subunit (mAb124); and the AChR α subunit (mAb35) were all used in Western blots as described previously (Marangi et al, 2001; Mittaud et al, 2001; Moransard et al, 2003). Antibodies against α-dystrobrevin-2 were a gift from Dr D Blake (Oxford, UK). Anti-caveolin-3 (Santa Cruz Biotechnology) was used at 1:2000, anti-flotillin 2/ESA clone 29 (Transduction Laboratories) at 1:1000, and anti-α-tubulin clone DM1A (Sigma) at 1:500. Anti-p75 was as described before (Pun et al, 2002). Densitometric analysis of Western blot signals was performed as done earlier (Marangi et al, 2001) using the software Image J (NIH, USA).
For detection of ganglioside GM1, 2 μl of each fraction was applied to a nitrocellulose membrane, blocked with 5% milk in PBS and probed with horseradish peroxidase-coupled cholera toxin subunit B (10 ng/ml; Sigma; Hering et al, 2003). To measure cholesterol, 50 μl of each fraction was analyzed with the Amplex Red cholesterol assay kit (Molecular Probes, Eugene, OR; Hering et al, 2003) according to the manufacturer's instruction. Protein concentration was determined using the BCA protein assay kit (Pierce).
Statistical analysis
All values are given as mean±s.e.m. Significance was calculated with a t-test (two-tailed, unequal variance) and is indicated as *P<0.05 or **P<0.01. In the figure legends, n refers to the number of experiments.
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Data
Acknowledgments
We are grateful to Susanne Erb-Vögtli for cell culture. This work was supported by grants from the Swiss National Science Foundation, the Swiss Foundation for Research on Muscle Diseases, the Zürich Neuroscience Center (to CF), and the Novartis Research Foundation (to SP, AFS and PC).
References
- Barrantes FJ (1993) Structural–functional correlates of the nicotinic acetylcholine receptor and its lipid microenvironment. FASEB J 7: 1460–1467 [DOI] [PubMed] [Google Scholar]
- Borges LS, Ferns M (2001) Agrin-induced phosphorylation of the acetylcholine receptor regulates cytoskeletal anchoring and clustering. J Cell Biol 153: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14: 111–136 [DOI] [PubMed] [Google Scholar]
- Bruses JL, Chauvet N, Rutishauser U (2001) Membrane lipid rafts are necessary for the maintenance of the (alpha)7 nicotinic acetylcholine receptor in somatic spines of ciliary neurons. J Neurosci 21: 504–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Cory S (2002) The developing synapse: construction and modulation of synaptic structures and circuits. Science 298: 770–776 [DOI] [PubMed] [Google Scholar]
- Cox ME, Maness PF (1993) Tyrosine phosphorylation of alpha-tubulin is an early response to NGF and pp60v-src in PC12 cells. J Mol Neurosci 4: 63–72 [DOI] [PubMed] [Google Scholar]
- De Paola V, Arber S, Caroni P (2003) AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat Neurosci 6: 491–500 [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA (2004) Integrins regulate Rac targeting by internalization of membrane domains. Science 303: 839–842 [DOI] [PubMed] [Google Scholar]
- Fox MA, Umemori H (2006) Seeking long-term relationship: axon and target communicate to organize synaptic differentiation. J Neurochem 97: 1215–1231 [DOI] [PubMed] [Google Scholar]
- Fuhrer C, Gautam M, Sugiyama JE, Hall ZW (1999) Roles of rapsyn and agrin in interaction of postsynaptic proteins with acetylcholine receptors. J Neurosci 19: 6405–6416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer C, Hall ZW (1996) Functional interaction of Src family kinases with the acetylcholine receptor in C2 myotubes. J Biol Chem 271: 32474–32481 [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR (1996) Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 85: 525–535 [DOI] [PubMed] [Google Scholar]
- Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP (1995) Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 377: 232–236 [DOI] [PubMed] [Google Scholar]
- Glass DJ, Bowen DC, Stitt TN, Radziejewski C, Bruno J, Ryan TE, Gies DR, Shah S, Mattsson K, Burden SJ, DiStefano PS, Valenzuela DM, DeChiara TM, Yancopoulos GD (1996) Agrin acts via a MuSK receptor complex. Cell 85: 513–523 [DOI] [PubMed] [Google Scholar]
- Golub T, Wacha S, Caroni P (2004) Spatial and temporal control of signaling through lipid rafts. Curr Opin Neurobiol 14: 542–550 [DOI] [PubMed] [Google Scholar]
- Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR (2000) Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron 25: 279–293 [DOI] [PubMed] [Google Scholar]
- Hering H, Lin CC, Sheng M (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J Neurosci 23: 3262–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Cote PD, Rossi SG, Rotundo RL, Carbonetto S (2001) The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J Cell Biol 152: 435–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Huang YZ, Pitcher GM, Valtschanoff JG, Ma YH, Feng LY, Lu B, Xiong WC, Salter MW, Weinberg RJ, Mei L (2003) Ligand-dependent recruitment of the ErbB4 signaling complex into neuronal lipid rafts. J Neurosci 23: 3164–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavan R, Zhao XT, Ruegg MA, Peng HB (2005) Tyrosine phosphatase regulation of MuSK-dependent acetylcholine receptor clustering. Mol Cell Neurosci 28: 403–416 [DOI] [PubMed] [Google Scholar]
- Marangi PA, Forsayeth JR, Mittaud P, Erb-Vogtli S, Blake DJ, Moransard M, Sander A, Fuhrer C (2001) Acetylcholine receptors are required for agrin-induced clustering of postsynaptic proteins. EMBO J 20: 7060–7073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangi PA, Wieland ST, Fuhrer C (2002) Laminin-1 redistributes postsynaptic proteins and requires rapsyn, tyrosine phosphorylation, and Src and Fyn to stably cluster acetylcholine receptors. J Cell Biol 157: 883–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand S, Devillers-Thiery A, Pons S, Changeux JP, Cartaud J (2002) Rapsyn escorts the nicotinic acetylcholine receptor along the exocytic pathway via association with lipid rafts. J Neurosci 22: 8891–8901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauch DH, Nagler K, Schumacher S, Goritz C, Muller EC, Otto A, Pfrieger FW (2001) CNS synaptogenesis promoted by glia-derived cholesterol. Science 294: 1354–1357 [DOI] [PubMed] [Google Scholar]
- Mittaud P, Camilleri AA, Willmann R, Erb-Vogtli S, Burden SJ, Fuhrer C (2004) A single pulse of agrin triggers a pathway that acts to cluster acetylcholine receptors. Mol Cell Biol 24: 7841–7854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittaud P, Marangi PA, Erb-Vogtli S, Fuhrer C (2001) Agrin-induced activation of acetylcholine receptor-bound Src family kinases requires Rapsyn and correlates with acetylcholine receptor clustering. J Biol Chem 276: 14505–14513 [DOI] [PubMed] [Google Scholar]
- Mohamed AS, Rivas-Plata KA, Kraas JR, Saleh SM, Swope SL (2001) Src-class kinases act within the agrin/MuSK pathway to regulate acetylcholine receptor phosphorylation, cytoskeletal anchoring, and clustering. J Neurosci 21: 3806–3818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moransard M, Borges LS, Willmann R, Marangi PA, Brenner HR, Ferns MJ, Fuhrer C (2003) Agrin regulates rapsyn interaction with surface acetylcholine receptors, and this underlies cytoskeletal anchoring and clustering. J Biol Chem 278: 7350–7359 [DOI] [PubMed] [Google Scholar]
- Nishio M, Fukumoto S, Furukawa K, Ichimura A, Miyazaki H, Kusunoki S, Urano T (2004) Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J Biol Chem 279: 33368–33378 [DOI] [PubMed] [Google Scholar]
- Palazzo AF, Eng CH, Schlaepfer DD, Marcantonio EE, Gundersen GG (2004) Localized stabilization of microtubules by integrin- and FAK-facilitated Rho signaling. Science 303: 836–839 [DOI] [PubMed] [Google Scholar]
- Pike LJ (2006) Rafts defined. J Lipid Res 47: 1597–1598 [DOI] [PubMed] [Google Scholar]
- Pun S, Sigrist M, Santos AF, Ruegg MA, Sanes JR, Jessell TM, Arber S, Caroni P (2002) An intrinsic distinction in neuromuscular junction assembly and maintenance in different skeletal muscles. Neuron 34: 357–370 [DOI] [PubMed] [Google Scholar]
- Resh MD (1999) Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta 1451: 1–16 [DOI] [PubMed] [Google Scholar]
- Rhainds D, Bourgeois P, Bourret G, Huard K, Falstrault L, Brissette L (2004) Localization and regulation of SR-BI in membrane rafts of HepG2 cells. J Cell Sci 117: 3095–3105 [DOI] [PubMed] [Google Scholar]
- Riddell DR, Christie G, Hussain I, Dingwall C (2001) Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol 11: 1288–1293 [DOI] [PubMed] [Google Scholar]
- Rodgers W, Farris D, Mishra S (2005) Merging complexes: properties of membrane raft assembly during lymphocyte signaling. Trends Immunol 26: 97–103 [DOI] [PubMed] [Google Scholar]
- Sadasivam G, Willmann R, Lin S, Erb-Vogtli S, Kong XC, Ruegg MA, Fuhrer C (2005) Src-family kinases stabilize the neuromuscular synapse in vivo via protein interactions, phosphorylation, and cytoskeletal linkage of acetylcholine receptors. J Neurosci 25: 10479–10493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791–805 [DOI] [PubMed] [Google Scholar]
- Santos AF, Caroni P (2003) Assembly, plasticity and selective vulnerability to disease of mouse neuromuscular junctions. J Neurocytol 32: 849–862 [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31–39 [DOI] [PubMed] [Google Scholar]
- Simons M, Friedrichson T, Schulz JB, Pitto M, Masserini M, Kurzchalia TV (1999) Exogenous administration of gangliosides displaces GPI-anchored proteins from lipid microdomains in living cells. Mol Biol Cell 10: 3187–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Mittaud P, Prescott ED, Fuhrer C, Burden SJ (2001) Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J Neurosci 21: 3151–3160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP (1996a) Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 271: 9690–9697 [DOI] [PubMed] [Google Scholar]
- Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP (1996b) Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem 271: 15160–15165 [DOI] [PubMed] [Google Scholar]
- Taniuchi M, Clark HB, Johnson EM Jr (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA 83: 4094–4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, Baloh RH, Milbrandt J, Johnson EM Jr (2000) GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron 25: 611–623 [DOI] [PubMed] [Google Scholar]
- Wallace BG (1994) Staurosporine inhibits agrin-induced acetylcholine receptor phosphorylation and aggregation. J Cell Biol 125: 661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmann R, Fuhrer C (2002) Neuromuscular synaptogenesis: clustering of acetylcholine receptors revisited. Cell Mol Life Sci 59: 1296–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder SJ, Hemmings L, Maciver SK, Bolton SJ, Tinsley JM, Davies KE, Critchley DR, Kendrick-Jones J (1995) Utrophin actin binding domain: analysis of actin binding and cellular targeting. J Cell Sci 108 (Part 1): 63–71 [DOI] [PubMed] [Google Scholar]
- Zhang XL, Topley N, Ito T, Phillips A (2005) Interleukin-6 regulation of transforming growth factor (TGF)-beta receptor compartmentalization and turnover enhances TGF-beta1 signaling. J Biol Chem 280: 12239–12245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Data