

Abstract
Rho guanosine triphosphatases (GTPases) regulate multiple aspects of dendritic cell (DC) function, but what regulates the expression of Rho GTPases in DCs is unknown. Here, we show that the extracellular matrix protein mindin regulates the expression of Rho GTPases in DCs. Mindin−/− mice displayed defective CD4+ T-cell priming and impaired humoral immune responses to T-dependent antigens. Mindin−/− DCs had reduced expression of Rac1/2 and impaired priming capacity owing to inefficient engagement with T lymphocytes. Ectopic Rac1 expression restored the priming capability of Mindin−/− DCs. Furthermore, we show that DC adhesion to mindin matrix was blocked by antibodies to α4, α5 and β1 integrins. DCs lacking β1 integrin had reduced adhesion to mindin matrix, decreased expression of Rac1/2 and impaired priming capacity. These results suggest that mindin–integrin interactions play a key role in regulating Rho GTPase expression in DCs and DC priming of T lymphocytes.
Keywords: dendritic cells, integrin, mindin, rho GTPases
Introduction
As professional antigen-presenting cells, dendritic cells (DCs) are essential initiators of adaptive immunity (Banchereau and Steinman, 1998; Shortman and Liu, 2002). DCs have several special features that contribute to efficient antigen presentation including high expression levels of MHC and costimulatory molecules as well as specialized morphology (Banchereau and Steinman, 1998; Shortman and Liu, 2002). DCs have membrane processes that are several times the length of their cell body. These processes, referred to as dendrites, interact with T lymphocytes to establish stable DC–T cell contact (Benvenuti et al, 2004). The small guanosine triphosphatases (GTPases) of the Rho family regulate multiple functions in DCs. Inhibition of Rac1 or Cdc42 activation by their dominant negative mutants results in impaired antigen presentation and T-cell priming (Swetman et al, 2002; Kerksiek et al, 2005; Shurin et al, 2005). DCs lacking Rac1 and Rac2 expression fail to move toward T lymphocytes to establish a stable DC–T cell interaction (Benvenuti et al, 2004). These DCs are defective in priming CD4+ T cells in vivo and in vitro. In addition, several studies have shown that Rho GTPases are critically involved in DC endocytosis (Garrett et al, 2000; West et al, 2000; Kerksiek et al, 2005). However, it remains unclear how the expression of Rho GTPases is regulated in DCs.
Integrins are transmembrane receptors comprised of α and β heterodimers and recognize extracellular matrix (ECM) proteins including fibronectin, vitronectin, collagen and laminin as their ligands (Plow et al, 2000; Hynes, 2002). DCs express various types of integrins and some integrins have been used to define DC subsets (Kilshaw, 1993; Puig-Kroger et al, 2000; Pribila et al, 2004). Integrins such as α4, α5 and β1 are highly expressed by all mouse DCs (Pribila et al, 2004). Signaling through integrins activates Rho GTPases. For example, fibroblasts placed on fibronectin induced activation of Cdc42, Rac1 and Rho (Price et al, 1998; Ren et al, 1999). These studies suggest that an ECM protein/integrin/Rho GTPase signaling pathway may exist to regulate DC function. However, it is unknown which ECM protein–integrin interaction plays such a role in DCs.
Mindin (spondin 2) is a highly conserved ECM protein expressed abundantly in the spleen and lymph nodes (LNs) (He et al, 2004). It belongs to the mindin-F-spondin family that includes F-spondin and mindin (Klar et al, 1992; Higashijima et al, 1997; Umemiya et al, 1997; Feinstein et al, 1999). Mindin and F-spondin share three structural domains: an FS1 and FS2 (named for F-spondin) domain at the amino terminus as well as one or six thrombospondin repeats at the carboxyl terminus. Mindin matrix promotes neutrite outgrowth from embryonic hippocampal neurons (Feinstein et al, 1999). Mindin also functions as a pattern recognition molecule for microbial pathogens (He et al, 2004). Mindin−/− mice have an impaired capability to clear bacterial infection. Mindin−/− macrophages exhibit defective responses to a broad spectrum of microbial stimuli. Our recent data also show that mindin interacts with integrins αMβ2 and α4β1 on neutrophils and plays a critical role in inflammatory cell recruitment (Jia et al, 2005).
In this report, we show that mindin−/− mice exhibited defective CD4+ T-cell priming and humoral response to T-dependent (TD) antigens. Mindin−/− DCs failed to efficiently prime T lymphocytes in vitro owing to inefficient engagement with T cells and had reduced expression of Rac1, Rac2 and Cdc42. The defective priming capacity of mindin−/− DCs was restored by ectopic expression of Rac1. Furthermore, we found that mindin interacts with α4β1 and α5β1 integrins on DCs. DCs from β1 integrin-deficient mice also exhibited reduced expression of Rac1 and Rac2 and defective priming of T lymphocytes. Collectively, our results have identified mindin as a key regulator of Rho GTPase expression in DCs and suggest that mindin–integrin interactions are essential for the normal expression of Rho GTPases in DCs.
Results
Humoral immune response in mindin−/−mice
To examine the role of mindin in the adaptive immune response, we immunized mindin−/− mice with sheep red blood cells (SRBCs), a TD antigen commonly used to study antibody responsiveness. As expected, SRBC immunization of wild-type mice elicited a strong specific IgG response (Figure 1A). In contrast, mindin−/− mice exhibited an impaired anti-SRBC IgG production (Figure 1A). At the peak of the response 2–3 weeks after immunization, the anti-SRBC IgG amount in mindin−/− mice was only ∼5% of that of controls (Figure 1A). Furthermore, secondary immunization with SRBCs also displayed defective antibody production in mindin−/− mice (Figure 1A).
Figure 1.
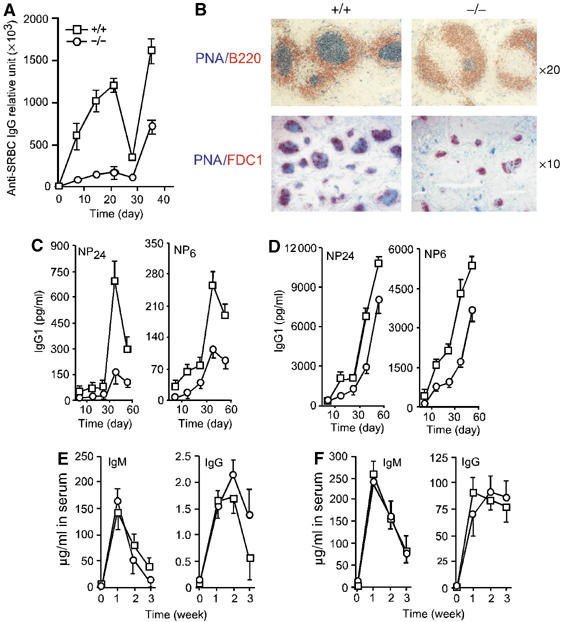
Impaired humoral immune response to TD antigens in mindin−/− mice. (A) Anti-SRBC-specific IgG in mindin−/− (−/−) (circle, all throughout) and control (+/+) (square, all throughout) mice. Mice (n=6) at 6–8 weeks of age were immunized intraperitoneally with 1 × 108 SRBC at day 0 and 28 and the sera were collected at the indicated time. Relative anti-SRBC IgG titers were determined by ELISA. (B) GC formation in SRBC-immunized mindin−/− (−/−) and control (+/+) mice. Spleens from mindin−/− and control mice 10 days after SRBC immunization were sectioned and stained as indicated. (C, D) Anti-NP-specific IgG1 in mindin−/− and control mice. Mice (n=5) at 6–8 weeks of age were immunized with NP24-KLH (50 μg) alone (C) or NP24-KLH (50 μg) plus alum (D) at days 0 and 28. Specific anti-NP IgG1 in the sera was determined by ELISA using NP24-BSA or NP6-BSA as substrates. (E, F). Antibody response to TI antigens. Mindin−/− and control mice (n=5) were immunized with TNP-LPS (50 μg) (E) or TNP-Ficoll (50 μg) (F) at day 0 and the sera were collected at a weekly interval. Serum anti-TNP antibody was determined by ELISA. Data are representative of 3–4 independent experiments.
We examined germinal center (GC) formation in the spleen of mindin−/− mice immunized with SRBCs. As expected, GCs were readily detected in control mice 10 days after SRBC immunization (Figure 1B). In contrast, the size and number of GCs in the spleen of mindin−/− mice were dramatically reduced as reflected by PNA and anti-FDC1 staining (Figure 1B). These results demonstrate that mindin plays a critical role in antibody production and GC formation after immunization with a TD antigen.
We also tested antibody production in mindin−/− mice using another TD antigen, NP24-KLH. As NP-KLH induces a strong IgG1 response (Croix et al, 1996), we tested the level of anti-NP IgG1 and the relative affinity for NP using different NP-BSA substrates. The total anti-NP24 IgG1 level in both primary and secondary responses in mindin−/− mice was reduced by 80% when compared to that of control mice (Figure 1C). Furthermore, although high-affinity anti-NP6 IgG1 was detected in the mutant mice, the level was only one-third of that found in controls (Figure 1C). Given that defective antibody response in some mutant mice can be restored by adjuvant (Wu et al, 2000), we tested antibody production in mindin−/− mice immunized with NP24-KLH precipitated with alum. Utilization of alum partially corrected the defective anti-NP antibody production in mindin−/− mice (Figure 1D).
We next determined humoral immune response to T-independent (TI) antigens in mindin−/− mice using the type I TI antigen TNP-LPS or the type 2 TI antigen TNP-Ficoll by i.p. injection. Antibody production was evaluated at different time points. Mindin−/− mice produced similar amounts of IgM and IgG antibodies against both types of TI antigens to those in control mice (Figure 1E and F). Together, these results demonstrate that antibody responses to TD but not TI antigens were impaired and suggest that B lymphocyte response is normal in mindin−/− mice.
Mindin expression in lymphocytes and DCs
The impaired TD antibody production in mindin−/− mice suggests a possible defect in the interactions between T–B lymphocytes and/or T–DCs. Our previous data showed that mindin is abundantly expressed in macrophages and mast cells (He et al, 2004). To characterize the role of mindin in TD humoral immunity, we examined mindin expression in lymphocytes and DCs. CD4+ T cells, CD8+ T cells and B220+ B cells were purified from the spleen and lymph node (LN) of wild-type mice by FACS sorting and subjected to semiquantitative RT–PCR analysis. Spondin2 (mindin) mRNA was readily detected in all subsets of lymphocytes (Figure 2A). In addition, mindin protein was readily detected in BM-derived DCs (BMDCs) from wild-type but not mindin−/− mice (Figure 2B).
Figure 2.
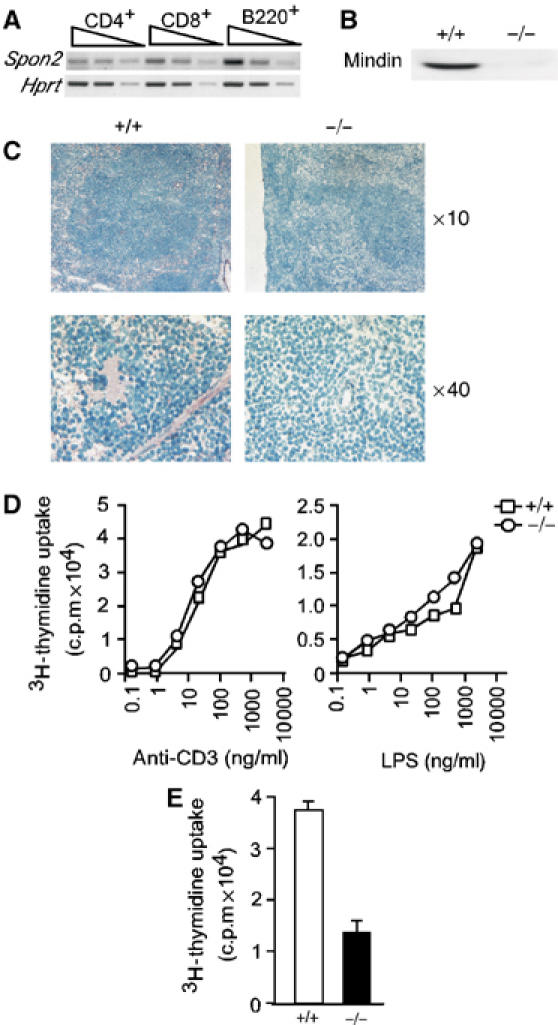
Impaired CD4+ T-cell priming in mindin−/− mice. (A) Spondin2 (mindin) mRNA expression in T and B lymphocytes. Total RNA from FACS sorted wild-type CD4+ and CD8+ T lymphocytes and B220+ B lymphocytes (>99%) was analyzed for Spondin2 mRNA expression in semiquantitative RT–PCR. Samples were tested in 1:5 serial dilutions. Hprt, hypoxanthine phosphoribosyltransferase, serves as loading control. (B) Mindin protein expression in BMDCs. Total cell lysates (10 μg protein) from BMDCs of wild-type (+/+) and mindin−/− (−/−) mice were blotted with a polyclonal anti-mindin antibody. (C) Mindin expression pattern in the spleen. Frozen spleen sections from wild-type (+/+) and mindin−/− (−/−) mice were stained with polyclonal anti-mindin followed by HRP-labeled anti-Rabbit Ig and 3-aminoethylcarbazole, and countered stained by methyl blue. (D) In vitro proliferation of T and B lymphocytes. Splenocytes from mindin−/− (circle) and wild-type (square) mice were stimulated with anti-CD3 or LPS at the indicated concentrations for 48 h and pulsed with 3H-thymidine in the last 8 h of the culture. Mean of triplicate determination with s.d. <5% of the means are shown. (E) Proliferation of in vivo primed CD4+ T cells from mindin−/− (−/−) and wild-type (+/+) mice. CD4+ T cells from mice immunized with Ova (50 μg) plus LPS (5 μg) in IFA for 10 days were stimulated with Ova (5 μg) presented by irradiated wild-type splenocytes for 3 days. Cells were pulsed with 3H-thymidine in the last 12 h of the culture. Mean+s.d. of triplicate determination are shown. Data are representative of three experiments.
We further examined the expression pattern of mindin protein in secondary lymphoid organs by immunohistochemical staining. Mindin protein was detected in a constant and diffuse pattern in wild-type spleen with positive signals seen across the whole tissue section including both white and red pulps (Figure 2C). As expected, no signal was detected in mindin−/− spleen after immunohistochemical staining (Figure 2C). These results demonstrate that mindin is expressed by mature lymphocytes and DCs in the secondary lymphoid organs.
In vivo CD4+ T-cell priming
The impaired humoral immune response to TD but not TI antigens in mindin−/− mice may be owing to defective T-cell activation and/or priming. We first examined T- and B-cell proliferation by stimulating splenocytes with anti-CD3 or LPS in vitro. The proliferation of mindin−/− T and B lymphocytes induced by anti-CD3 or LPS was essentially indistinguishable from that of control cells (Figure 2D), suggesting that mindin is not essential for T- or B-cell proliferation.
We then examined in vivo CD4+ T-cell priming in mindin−/− mice. CD4+ T cells from the spleen and LNs of mindin−/− and wild-type mice immunized with ovalbumin (Ova) plus LPS emulsified in incomplete Freund's adjuvant (Pasare and Medzhitov, 2004) were purified and restimulated in vitro with wild-type APCs. Proliferation of primed CD4+ T cells was measured by 3H-thymidine uptake. The proliferation of in vivo primed CD4+ T cells from mindin−/− mice was 60–70% lower than that of control CD4+ T cells (Figure 2E). These results indicate that efficient priming of CD4+ T cells in vivo depends on mindin.
Characterization of DCs in mindin−/− mice
The normal TCR-mediated proliferation but defective in vivo priming of mindin−/− T lymphocytes suggests that DCs may have impairment in development and/or function in the absence of mindin. We first characterized DC development in mindin−/− mice. The number of CD11c+ DCs in the spleen of mindin−/− mice was comparable to that in age-matched control mice (Supplementary Figure 1A). Furthermore, the major DC subsets as defined by the expression of CD4 and CD8 on CD11c+ DCs (Kamath et al, 2000) were similarly detected in mindin−/− and control mice (Supplementary Figure 1B). These results suggest that DC development is normal in mindin−/− mice.
Mindin−/− macrophages exhibit defective activation upon stimulation by microbial components (He et al, 2004). We examined whether mindin−/− DCs have defects in the upregulation of costimulatory molecules upon microbial component stimulation. Similar to control cells, both BM-derived and splenic DCs from mindin−/− mice readily upregulated CD40, CD80 and CD86 as well as MHC class II upon LPS stimulation (Supplementary Figure 1C). Furthermore, BMDCs from mindin−/− mice displayed similar survival kinetics with or without LPS stimulation (Supplementary Figure 1D). To determine whether mindin regulates DC endocytosis, we incubated immature BMDCs from mindin−/− and control mice with FITC-Ova. Mindin−/− DCs endocytosed FITC-Ova at a comparable level to that of control cells (Supplementary Figure 1E). Together, these results demonstrate that mindin is not involved in DC costimulatory molecule expression, survival or endocytosis. Given that mindin−/− macrophages exhibited defective phagocytosis of bacteria, we further examined the phagocytosis capacity of mindin−/− DCs. Consistent with previous results (He et al, 2004), mindin−/− DCs exhibited defective phagocytosis of Streptococcus pneumoniae and Salmonella typhimurium, but normal FcR-mediated phagocytosis of SRBCs (Supplementary Figure 2).
Impaired priming capacity by mindin−/− DCs
To directly test the priming capacity of mindin−/− DCs, we set up in vitro DC–T cell priming cultures by mixing splenic DCs with naïve CD4+ T cells in the presence of Ova. Wild-type DCs induced similarly high levels of proliferation in naïve CD4+ T cells from wild-type and mindin−/− mice (Figure 3A), suggesting that mindin−/− T cells have the ability to respond to DC priming. In contrast, mindin−/− DCs induced only low levels of proliferation in mindin−/− CD4+ T cells (Figure 3A). Interestingly, mindin−/− DCs induced an intermediate level of proliferation in wild-type T cells (Figure 3A), suggesting that mindin produced by wild-ype T cells may partially restore the priming function of mindin−/− DCs.
Figure 3.
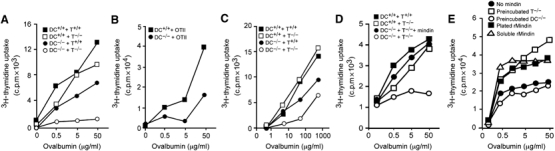
Impaired priming capacity of mindin−/− DCs. Purified naïve CD4+ T cells and mature DCs from either mindin−/− (−/−) or wild-type (+/+) mice were cocultured as indicated in the presence of Ova for 3–5 days. CD4+ T-cell proliferation was measured by 3H-thymidine uptake. The following cells were used: (A, B) splenic DCs from mindin−/− and wild-type mice; (C–E) BMDCs from mindin−/− and wild-type mice; (A, C–E) CD4+ T cells from mindin−/− and wild-type mice; (B) CD4+ T cells from OTII transgenic mice. (D) Restoration of priming capacity of mindin−/− DCs by exogenous mindin. DC–T cell coculture was set up as in (A). Exogenous mindin was added at 10 μg/ml. (E) Restoration of priming capacity of mindin−/− DCs by different ways of treatment with mindin. Mindin (10 μg/ml) was either preincubated with DCs or T cells at 37°C for 1 h and washed three times, precoated on the plates, or added to the culture medium. Mean of triplicate determinations with s.d. <10% of the means are shown and are representative of 3–5 experiments.
We then used OTII CD4+ T cells expressing a transgenic TCR recognizing an Ova peptide (Barnden et al, 1998) in the DC–T cell priming culture. Mindin−/− DCs induced an intermediate level of proliferation in OTII CD4+ T cells when compared to control DCs, consistent with the above results using wild-type T cells (Figure 3B). Furthermore, the defective priming capacity was also seen in BMDCs from mindin−/− mice (Figure 3C) and could be completely restored by exogenous mindin at 10 μg/ml (Figure 3D). These results demonstrate that mindin−/− DCs are defective in priming T lymphocytes in vitro.
We determined whether preincubating mindin with either DCs or T cells can restore the DC priming defect. Interestingly, preincubation of recombinant mindin with T cells or mindin coated on plates during DC:T cell culture restored the defective priming of mindin−/− DCs, whereas preincubation of mindin with DCs failed to do so (Figure 3E). These results suggest that mindin binds to T cells. The failure to restore DC priming after mindin preincubation with DCs may reflect that the bound mindin may be endocytosed by DCs.
Mindin−/− macrophages produce lower levels of IL-6 than wild-type cells when stimulated by microbial components (He et al, 2004). Recent studies suggest that IL-6 plays a critical role in T-cell priming by blocking the suppressive effect of Treg cells (Pasare and Medzhitov, 2003, 2004). In vivo depletion of Treg cells restored defective T-cell priming in MyD88−/− mice (Pasare and Medzhitov, 2004). We found that LPS-stimulated mindin−/− DCs produced only ∼40% of IL-6 and TNF-α when compared to control DCs (data not shown). To test whether a reduced production of IL-6 may account for the impaired priming capacity of mindin−/− DCs, we added exogenous IL-6 to in vitro DC–T cell culture. Excess amount of IL-6 in the DC–T cell priming culture failed to restore the priming capacity of mindin−/− DCs (Figure 4A). Furthermore, in vivo depletion of Treg cells in mindin−/− mice did not restore T-cell priming (Figure 4B). These results suggest that the defective antigen presentation of mindin−/− DCs is not solely due to reduced IL-6 production or an inability to overcome suppression by Treg cells in vivo.
Figure 4.
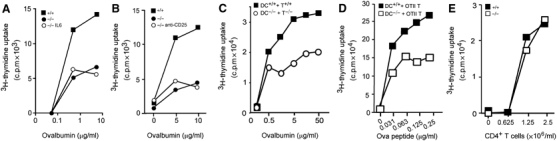
Mechanistic examination of the impaired priming capacity of mindin−/− DCs. (A) Effect of IL-6 on in vitro T-cell priming by mindin−/− DCs. CD4+ OTII T cells were in vitro primed by BMDCs from mindin−/− (−/−) or wild-type (+/+) mice with or without IL-6 (10 U/ml). (B) Effect of in vivo depletion of Treg cells on T- cell priming in mindin−/− mice. Mindin−/− mice treated with anti-CD25 mAb or control rat Ig at day –3 were immunized with Ova (50 μg) plus LPS (5 μg) in IFA at day 0. CD4+ T cells were isolated at day 10 and restimulated with Ova at the indicated concentration presented by irradiated wild-type splenocytes. Wild-type mice treated with rat Ig and similarly immunized serve as positive control for in vivo priming efficiency. (C) Priming capacity of mindin−/− DCs matured by TNF-α stimulation. BMDCs from mindin−/− (−/−) and control (+/+) mice were stimulated with TNF-α (50 ng/ml) for 16 h before used in in vitro priming. (D) Defective priming of T cells by mindin−/− DCs in the presence of Ova peptide. BMDCs from mindin−/− (−/−) and control (+/+) mice were cocultured with OTII CD4+ T cells in the presence of Ova323−339 peptides as indicated. (E) Mixed lymphocyte reaction induced by mindin−/− DCs. Irradiated BMDCs from mindin−/− (−/−) and control (+/+) mice (C57BL/6 background) (1 × 106/ml) were cultured with naïve CD4+ T cells from Balb/c mice at the indicated concentrations. The proliferation of CD4+ T cells were measured by H3-thymidine uptake. Mean of triplicate determinations with s.d. <10% of the means are shown and are representative of 3–4 experiments.
The defective priming capacity of mindin−/− DCs may be solely due to their impaired maturation induced by LPS. To address this, mindin−/− DCs matured by TNF-α stimulation were tested for their priming capacity. A similar priming defect of mindin−/− DCs matured by TNF-α to that of DCs matured by LPS was observed (Figure 4C). Furthermore, mindin−/− DCs failed to prime T cells efficiently even when Ova peptides were used as the antigen (Figure 4D), arguing against that the priming defect is owing to impaired antigen uptake, processing and MHC peptide loading. Interestingly, mindin−/− DCs stimulated allogeneic CD4+ T cells as efficiently as control DCs in a mixed lymphocyte reaction (Figure 4E), indicating that allostimulation by DCs does not depend on mindin.
Rac1/2 expression in mindin−/− DCs
Given the role of Rac1 and Rac2 in regulating DC priming capability (Benvenuti et al, 2004) and the fact that mindin serves as a ligand for integrins (Jia et al, 2005), we examined whether mindin regulates the expression of these small GTPases in DCs. Total cell lysates of purified splenic DCs from mindin−/− and wild-type mice were blotted for total Rac1, Rac2 and Cdc42 expression. Unstimulated splenic DCs from wild-type mice expressed abundant levels of these GTPases (Figure 5A). In contrast, the expression of Rac1, Rac2 and Cdc42 was much lower in mindin−/− splenic DCs (Figure 5A). Furthermore, whereas microbial component stimulation readily increased the expression levels of these Rho GTPases in wild-type DCs, less upregulation was observed in mindin−/− DCs (Figure 5A). Consistent with their defective priming capacity, the expression and upregulation of Rac1, Rac2 and Cdc42 were also impaired in mindin−/− BMDCs (Figure 5B).
Figure 5.
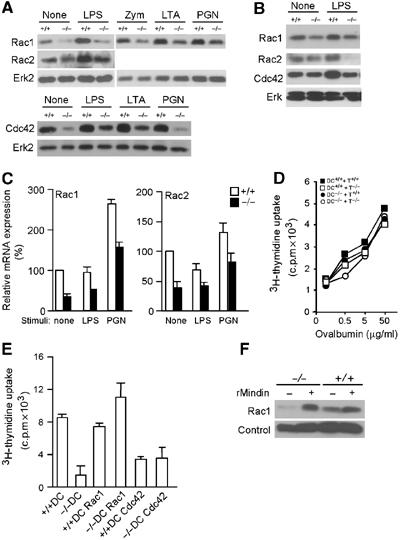
Reduced Rac1/2 expression in mindin−/− DCs. (A, B) Expression of Rac1, Rac2 and Cdc42 in splenic DCs (A) and BMDCs (B) from mindin−/− (−/−) and wild-type (+/+) mice. Total cell lysates from untreated DCs (None) or DCs stimulated with LPS (60 ng/ml), zymosan, LTA and PGN (all 10 μg/ml) for 12 h were blotted with the indicated antibodies. ERK protein expression serves as a loading control. (C) Rac1 and Rac2 mRNA expression in mindin−/− DCs. Total RNA from splenic DCs of mindin−/− (−/−) and wild-type (+/+) mice with or without stimulation were analyzed for Rac1/2 mRNA expression by real-time PCR. The relative mRNA levels of Rac1 and Rac2 were normalized for Gapdh mRNA expression and expressed as 100% for wild-type DCs. Data are mean+s.d. of triplicate determinations. (D) Restoration of priming capacity of mindin−/− DCs with round-bottom culture plates. DC–T cell priming was carried out as in Figure 4C except that round-bottom plates were used. (E) Restoration of priming capacity of mindin−/− DCs by retrovirally delivered Rac1. BMDCs expressing Rac1 or Cdc42 from mindin−/− (−/−) and wild-type (+/+) mice were sorted by FACS and used as APCs in DC–OT II CD4+ T-cell priming culture in the presence of Ova (5 μg/ml). The proliferation of OT II T cells was measured by 3H-thymiding uptake in the last 12 h of the culture. Mean+s.d. of triplicate determinations are shown. Data are representative of three experiments. (F) Restoration of Rac1 expression in mindin−/− DCs by recombinant mindin. BMDCs from mindin−/− (−/−) and control (+/+) mice were incubated with mindin (10 μg/ml) for 16 h and analyzed for Rac 1 expression by Western blot. Loading control is a nonspecific band on the same membrane.
We next determined whether the reduced Rac1/2 expression in mindin−/− DCs was owing to a lowered mRNA expression using quantitative RT–PCR. Expression of Rac1 and Rac2 mRNA in unstimulated splenic DCs from mindin−/− mice was only ∼40% of that found in wild-type DCs (Figure 5C). Furthermore, the lower levels of Rac1 and Rac2 mRNA expression in mindin−/− DCs was not restored by LPS or PGN (peptidoglycan) stimulation (Figure 5C), suggesting that mindin is required for normal expression of Rac1 and Rac2 mRNA in DCs.
It has been reported that DCs lacking Rac1/2 expression fail to prime T cells owing to inefficient DC–T cell contact and the defect can be restored by coculturing DCs and T cells in round-bottom plates (Benvenuti et al, 2004). If the reduced Rac1/2 expression in mindin−/− DCs is the primary defect, DC priming in round-bottom plates may restore T-cell proliferation. Indeed, T-cell priming by mindin−/− DCs was completely restored when round-bottom plates were used (Figure 5D). To further determine which Rho GTPase, Rac or Cdc42, accounts for the defective priming capacity of mindin−/− DCs, we ectopically expressed Rac1 or Cdc42 in BMDCs of mindin−/− and wild-type mice using the pMI retrovirus vector expressing human CD2 as a marker (He et al, 1998). Human CD2+ BMDCs were purified by FACS sorting and incubated with OTII CD4+ T cells in the presence of Ova. Expression of Rac1 (and human CD2) in wild-type DCs did not obviously affect their antigen presentation to OTII CD4+ T cells (Figure 5E). Expression of Rac1 in mindin−/− DCs completely restored T-cell priming (Figure 5E). In contrast, expression of Cdc42 in mindin−/− DCs failed to restore their priming capacity (Figure 5E). In addition, Cdc42 expression in wild-type DCs appeared to inhibit their antigen presentation to OTII CD4+ T cells (Figure 5E). Consistent with the restoration of mindin−/− DC priming by recombinant mindin, the expression of Rac1 and Cdc42 in mindin−/− DCs was also restored by exogenous mindin (Figure 5F and data not shown). Collectively, these results demonstrate that mindin plays a critical role in the normal expression and upregulation of Rho GTPases in DCs and that the reduced Rac expression accounts for the defective priming capacity in mindin−/− DCs.
Defective interaction between Mindin−/− DCs and T lymphocytes
To directly visualize the interaction between mindin−/− DCs and T lymphocytes in vitro, we performed real-time imaging of DC:T interaction using in vitro cell coculture. Wild-type DCs formed stable clusters with OTII CD4+ T cells as early as 2 h (Supplementary movie). Multiple T cells attached readily to wild-type DCs, and this attachment persisted for the observed period (Supplementary movie). In contrast, mindin−/− DCs only transiently interacted with OTII T cells and had markedly less OTII cells attached despite that these cells had normal appearance of dendrites (Supplementary movie). Furthermore, Mindin−/− DCs displayed faster movement (Supplementary movie). We then tracked the movement of DCs in the coculture. As shown in Figure 6A, wild-type DCs rapidly engaged with OTII T cells and essentially remained at the same spot for 6 h, whereas mindin−/− DCs failed to engage T cells and moved out of the imaging field within 90 min. The cumulative distance of mindin−/− DC movement was increased dramatically compared to that of control DCs (Figure 6B), indicating that mindin−/− DCs moved continuously whereas wild-type DCs stopped moving after engaging OTII T cells. Furthermore, the velocity of mindin−/− DCs was 5–6-fold higher than that of control DCs (Figure 6C). Taken together, these results demonstrate that mindin−/− DCs cannot engage T lymphocyte efficiently. The reduced movement and velocity of wild-type DCs are consistent with the observations that DCs form stable clusters with T cells during antigen presentation in vivo (Stoll et al, 2002; Bousso and Robey, 2003; Miller et al, 2004).
Figure 6.
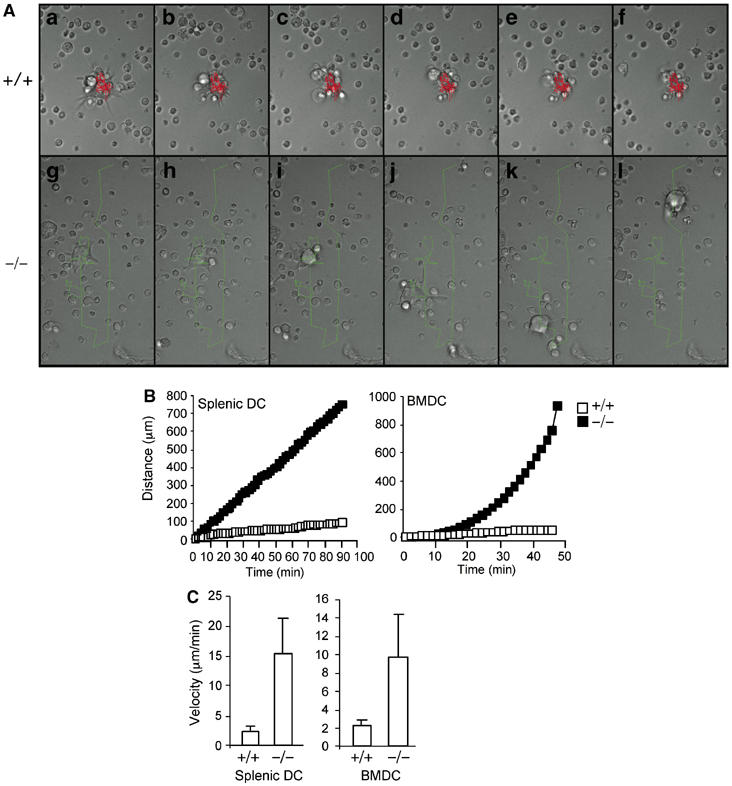
Real-time observation of the interaction between mindin−/− DC and T lymphocytes. (A) Representative cell centroid tracks of wild-type (+/+) (red) (a–f) and mindin−/− (green) (g–l) BMDCs. Strip chart time lapse movies to follow the centroid tracks of one DC each from wild-type and mindin−/− mice are shown. Time scale a–f is 354 min, g–l is 90 min, with each of the two strip charts divided into time points spaced at exact sixth of the total time in each experiment. Each individual panel is 86 μm wide. (B) Cumulative distance of DC movement in DC–T cell coculture. DCs from each group were randomly picked and tracked for their movement until the cells moved out of the imaging field. Wild-type DCs rarely moved out whereas mindin−/− DCs all moved out of the tracking field after 90 min. The mean values of 15 randomly tracked DCs from each group are shown. (C) Velocity of splenic and BM-derived DCs from wild-type and mindin−/− mice in DC–T cell coculture. Data are mean+s.d. calculated from the same 15 cells in (B) and representative of three independent experiments. P-values between +/+ and −/− groups are <0.001.
Mindin as a ligand for integrin on DCs
Our recent data show that mindin serves as a ligand for integrin αMβ2 and α4β1 on neutrophils (Jia et al, 2005). Various integrins are expressed on mouse DCs and can be used to define subsets of DCs (Kilshaw, 1993; Puig-Kroger et al, 2000; Pribila et al, 2004). Given the uniformly high expression of α4β1 and α5β1 integrins on all mouse DC subsets, we examined whether these integrins on DCs interact with mindin matrix. Immature and mature DCs readily adhered to mindin and fibronectin but not laminin matrix (Supplementary Figure 3A). Addition of anti-β1 integrin mAb reduced DC adhesion to mindin matrix by 50–70% (Supplementary Figure 3A). Addition of anti-α4 or anti-α5 integrin mAbs also reduced DC adhesion to mindin matrix, though combination of these antibodies did not further reduce mindin-mediated DC binding (Supplementary Figure 3A). As a negative control, anti-FcRII/III mAb did not affect DC adhesion to mindin matrix (Supplementary Figure 3A). These results suggest that DCs utilize integrins α4β1 and α5β1 to interact with mindin matrix.
We wondered whether mindin regulates DC function through modulating integrin expression levels. We determined the expression of integrin β1, α4 and α5 on mature DC by FACS analysis and found that the expression of these integrins on mature DCs from mindin−/− and control mice was almost identical (Supplementary Figure 3B), ruling out a role of mindin in regulating integrin expression on DCs.
Role of integrin β1 in DC priming
The above results suggest that mindin may regulate Rac1/2 expression in DCs by signaling through α4β1 and α5β1 integrins. To test this, we examined Rac1/2 expression in DCs lacking β1 integrin expression. To generate DCs lacking β1 expression, we crossed mice with β1 integrin alleles flanked with two loxP sites (β1f/f) (Raghavan et al, 2000) to Mx-cre transgenic mice (Kuhn et al, 1995) and induced β1 integrin deletion by repetitively injecting polyI:C. Mx-cre induced an efficient deletion of β1 integrin on ∼40–50% splenic DCs as assessed by FACS (Figure 7A). We purified β1low/neg splenic DCs from β1f/f Mx-cre mice by FACS sorting. Sorted splenic DCs from β1f/f mice similarly treated with polyI:C were used as a control (Figure 7A). We first tested their binding to mindin matrix. The binding of β1 integrin-deficient DCs to mindin was reduced by ∼40% when compared to that of control DCs (Figure 7B), suggesting that β1 integrin mediates mindin–DC interaction. We then examined Rac1 and Rac2 expression in these DCs. The expression levels of total Rac1 and Rac2 proteins in β1-deficient splenic DCs were reduced by ∼70 and ∼40%, respectively, when compared to control DCs (Figure 7C). BMDCs from β1f/f Mx-cre mice, which had efficient β1 integrin deletion on >90% of the cells, contained a dramatically reduced levels of Rac1 and Rac2 (Figure 7C). Furthermore, upregulation of Rac1 and Rac2 expression upon microbial component stimulation in β1-deficient splenic DCs and BMDCs was also impaired (Figure 7C). Correlated with the lower Rac1 and Rac2 protein expression in β1 integrin-deficient DCs, Rac1 and Rac2 mRNA expression levels were also reduced in these cells when compared to wild-type DCs (Figure 7D). These results indicate that β1 integrin-mediated signaling plays a critical role in Rac expression in DCs.
Figure 7.
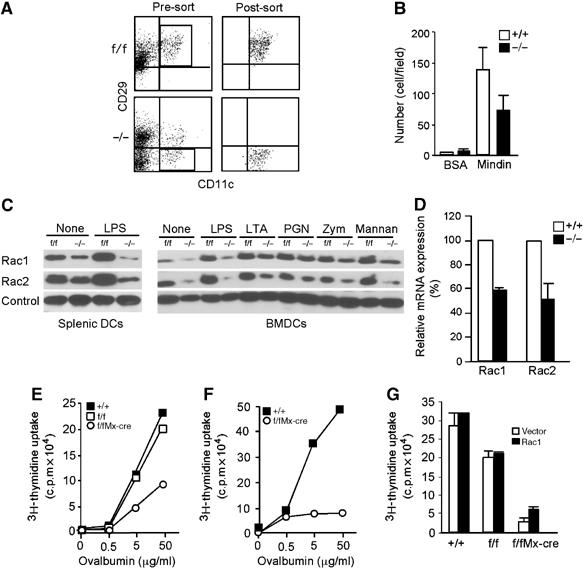
Impaired antigen presentation of β1 (CD29)-deficient DCs. (A) FACS profiles of sorted β1-deficient splenic DCs. Splenic DC preparations from β1f/fMx-cre (−/−) and β1f/f (f/f) mice treated with polyI:C were stained with anti-CD11c and anti-CD29 and subjected to FACS sorting. These sorted β1-deficient and β1+ splenic DCs were used in the experiments shown below. (B) Binding of β1-deficient splenic DCs to mindin matrix. LPS matured β1-deficient (−/−) and control (+/+) splenic DCs as shown in (A) were incubated with precoated mindin (10 μg/ml) at room temperature for 1 h, washed and counted. Data are mean+s.d. from at least four different fields. (C) Expression of Rac1/2 in β1-deficient DCs. Total cell lysates from splenic or BM-derived DCs of β1f/fMx-cre (−/−) and β1f/f (f/f) mice without treatment (None) or stimulated with LPS (60 ng/ml), LTA, PGN, zymosan, mannan (all 10 μg/ml) for 12 h were blotted with anti-Rac1 or anti-Rac2. Loading control is a nonspecific band from the same membrane. (D) Rac1 and Rac2 mRNA expression in β1 integrin-deficient DCs. Total RNA from sorted splenic DCs of β1f/fMx-cre (−/−) and wild-type (+/+) mice treated with polyI:C were analyzed for Rac1/2 mRNA expression by real-time PCR. The relative mRNA levels of Rac1 and Rac2 were normalized for Gapdh mRNA expression and expressed as 100% for wild-type DCs. Data are mean+s.d. of triplicate determinations. (E, F) Impaired antigen presentation by β1 integrin-deficient DCs. Integrin β1− or β1+ splenic (E) or BM-derived DCs (F) from β1f/fMx-cre (−/−), β1f/f (f/f) or β1+/+(+/+) mice treated with polyI:C were incubated with OTII CD4+ T cells in the presence of ovalbumin for 3 days. Mean of triplicate determinations with s.d. <10% of the mean are shown. (G) Effect of ectopic Rac1 expression on antigen presentation of β1 integrin-deficient DCs. BMDCs from β1f/fMx-cre, β1f/f or β1+/+ wild-type mice infected with pMIRac1 (Rac1) or pMI (Vector) retroviruses were sorted by FACS and incubated with OTII CD4+ T cells in the presence of ovalbumin (5 μg/ml). Mean+s.d. of triplicate determinations are shown. Data are representative of 2–4 experiments.
We then examined the priming capacity of these DCs in DC–T cell coculture. Similar to mindin−/− DCs, β1 integrin-deficient splenic DCs and BMDCs displayed defective priming of OTII CD4+ T cells in vitro (Figure 7E and F). The reduced expression of Rac proteins and impaired priming capability in β1 integrin-deficient DCs was not owing to a nonspecific effect caused by polyI:C treatment as wild-type and β1f/f mice were also treated with polyI:C. Given the reduced expression of Rac1/2 in β1-deficient DCs, we tested whether ectopically expressed Rac1 could restore the defective antigen presentation in these cells. Interestingly, Rac1 expression did not restore the defective antigen presentation of β1-deficient DCs (Figure 7G). Collectively, these results demonstrate that integrin β1 plays a critical role in the expression of Rac1/2 and antigen presentation capability of DCs.
Discussion
In this report, we show that mindin−/− mice have defective humoral immune responses to TD but not TI antigens. Mindin was first characterized as a pattern recognition molecule for microbial pathogens through carbohydrate recognition and is essential for the initiation of innate immune response to microbial pathogens (He et al, 2004). We have used nonpathogenic antigens to examine adaptive immune response in mindin−/− mice. Given that mindin is required for IL-6 and TNF-α production and inflammatory cell recruitment (He et al, 2004; Jia et al, 2005), it is likely that in vivo defects in migration of DCs or lymphocytes and/or lack of inflammatory cytokine production also contribute to the defective humoral immune response in mindin−/− mice. Nevertheless, our in vitro data supports a role for mindin in DC–T cell priming by maintaining Rho GTPase expression in DCs.
As professional APCs, DCs make close interaction with T lymphocytes to establish a stable contact (Benvenuti et al (2004) and Supplementary movie). Without this stable contact, DC priming of T lymphocytes is impaired. Recent reports have unequivocally demonstrated that Rho GTPases control DC priming capability (Benvenuti et al, 2004; Kerksiek et al, 2005). Several lines of evidence suggest that the impaired priming capacity of mindin−/− DCs is owing to reduced expression of Rac1 and Rac2. First, Rac 1 and Rac 2 expression levels were reduced in immature splenic, mature splenic and BM-derived DCs in mindin−/− mice. Ectopic expression of Rac1, but not Cdc42, restored the priming capability of mindin−/− DCs in vitro. Second, similar to DCs lacking both Rac1/2 expression (Benvenuti et al, 2004), coculture of mindin−/− DCs with T lymphocytes in round-bottom plates completely restored the impaired priming capacity of mindin−/− DCs. These results suggest that mindin−/− DCs cannot interact with T lymphocytes closely. Furthermore, real-time imaging studies demonstrate that mindin−/− DCs cannot engage T lymphocytes efficiently. Third, mindin−/− DCs did not display any obvious defect in the upregulation of costimulatory molecules, endocytosis or survival. The fact that mindin−/− DCs failed to efficiently prime T lymphocytes when Ova peptide was used as the antigen strongly argues against that the priming defect is caused by antigen uptake, processing and MHC peptide loading. Finally, mindin−/− DCs matured by TNF-α stimulation still exhibited defective priming capacity, suggesting that the defect is not solely owing to a requirement on LPS by mindin−/− DCs for their maturation.
There are notable differences in DC development and function between mindin−/− and Rac1/2-deficient mice. CD8α+ DC development was impaired and dendrite formation was abnormal in Rac1/2-deficient mice (Benvenuti et al, 2004). In contrast, CD8α+ DC development was normal in mindin−/− mice. Furthermore, we did not observe obvious defects in dendrite formation in mindin−/− DCs (Supplementary Movie). The normal dendrite formation in mindin−/− DCs may be owing to compensatory function from other ECM proteins such as fibronectin and collagens. The differences in CD8α+ DC development and dendrite formation in these two types of mice likely reflect the different expression levels of Rac1 and Rac2 in these mice. Our results suggest that the amounts of Rac1 and Rac2 in mindin−/− DCs are sufficient for CD8α+ DC development and dendrite formation, but insufficient for efficient priming.
Our data suggest that mindin regulates Rho GTPase expression in DCs by interacting with α4β1 and α5β1 integrins. We have recently shown that mindin serves as a ligand for αMβ2 and α4β1 integrins on neutrophils (Jia et al, 2005). Given the high level expression of α4β1 and α5β1 integrins on all DCs (Pribila et al, 2004) (Figure 7A), we focused our attention on these integrins. Our antibody blocking results indicate that DC adhesion to mindin matrix is mediated through α4β1 and α5β1 integrins. This is further supported by the observation that β1 integrin-deficient DCs exhibited reduced binding to mindin matrix. We also demonstrate that DCs lacking β1 integrin expression exhibited reduced expression of Rac1/2 and impaired antigen presentation to T lymphocytes. The similarity between mindin−/− and β1 integrin-deficient DCs suggests that mindin–β1 integrin interaction on DCs in vivo plays a key role in the expression of Rac1/2.
Our results indicate that DCs require constant interaction with environmental signals such as mindin to maintain normal function. At steady state, DCs are enmeshed in an extensive network, actively probing adjacent T lymphocytes with their dendrites in secondary lymphoid organs (Lindquist et al, 2004). Given the extensive expression of mindin in the extracellular matrix of secondary lymphoid organs, DCs must be in constant contact with mindin. This interaction is required for the normal expression of Rac proteins in DCs. This finding is surprising given that other α4β1 and α5β1 integrin ligands, such as fibronectin, are also expressed in these lymphoid organs (Liakka and Autio-Harmainen, 1992; Lo et al, 2003). The fact that the interaction between fibronectin and these integrins is not sufficient to compensate for mindin-mediated integrin signaling suggests that mindin as a ligand provides a unique signal. It has been shown that α4β1 and α5β1 integrins recognize RDG or other peptide motifs on fibronectin. It will be important to determine which motifs on mindin are recognized by these integrins.
We also show that a critical function of β1 integrin in DCs is to directly regulate Rac1 and Rac2 mRNA expression through interaction with mindin. Upon interaction with ECM ligands, integrins regulate Rho GTPases function in several ways: integrin signaling readily activates Rac and Cdc42 from GDP-bound inactive form to GTP-bound active form (Price et al, 1998; Ren et al, 1999). Integrin signaling also promotes Rho GTPases to interact with effector proteins and their membrane localization (del Pozo et al, 2000, 2002). Our results demonstrate that β1 integrin provides a critical signal to maintain normal expression levels of Rac1 and Rac2. This regulation occurs at transcription level as mRNA expression of Rac1 and Rac2 in both mindin−/− and β1 integrin-deficient DCs were lower than those found in wild-type DCs. However, it is possible that mindin–integrin interaction also regulates Rac expression at the protein level. Given the extensive studies on integrin-mediated signaling pathways (Hynes, 2002; Miranti and Brugge, 2002), it will be important to determine which signaling process mediated through β1 integrin regulates Rac mRNA expression in DCs. In summary, our results suggest that mindin/integrin β1/Rac signaling process plays a critical role in DC priming of T lymphocytes.
Materials and methods
Mice
All homozygous mindin−/− and wild-type mice were derived from breeding of heterozygous mindin−/− mice (He et al, 2004) after back-crossed to C57BL/6 for seven generations. The following mice were obtained from the Jackson Laboratory: CD29 (β1 integrin) floxed (CD29f/f) mice (Raghavan et al, 2000) and OTII TCR transgenic mice (Barnden et al, 1998). To generate β1 integrin-deficient DC, CD29f/f mice were crossed with Mx-cre transgenic mice (Kuhn et al, 1995). The CD29f/fMx-cre and CD29f/f control mice at 5 weeks of age were injected with 300μg of polyI:C (Sigma) every other day for three times. Mice were then injected with the same amount of polyI:C 2 more times at 1-week intervals (five injections in total). All mice were housed in a specific pathogen-free facility at the Duke University Vivarium and used at 6–12 weeks of age. Mice in different groups were sex and age-matched. Animal usage was carried out according to protocols approved by the Duke University Institutional Animal Care and Use Committee.
DC preparation and stimulation
To isolate splenic DCs, spleens were perfused with serum-free RPMI containing Liberase CI (400 μg/ml) (Rache Diagnostics) at 37°C for 30 min. The digested spleens were lysed of erythrocytes and resuspended (1 ml/spleen) in a 30% BSA solution (Sigma). Cold RPMI (4 ml) was placed over the BSA/splenocyte mixture to form an interface. After centrifugation at 7000 r.p.m. for 15 min, the interface was harvested for enriched splenic DCs. To further purify CD11c+ DCs, enriched splenic DCs were stained with anti-CD11c mAb and sorted by FACS to >98% pure. BMDCs were generated by culturing BM precursor cells in the presence of GM-CSF (200 U/ml) and IL-4 (5 U/ml) (PeproTech) for 6 days as described(Inaba et al, 1992; Zhang et al, 2004). At the end the culture, CD11c+ DCs accounted for 85–90% of the cells. All splenic DCs and BMDCs were pulsed with antigen, induced to undergo maturation by LPS stimulation (60 ng/ml) for 16 h and irradiated (3000 rads) before used for in vitro priming of T lymphocytes.
To express Rac1 or Cdc42 in DCs, the pMI retrovirus vector expressing human CD2 and mouse wild-type Rac1 and Cdc42 cDNA bicistronically were used to generate retroviruses as described (He et al, 1998). BMDCs were infected with retroviruses at day 2 and day 4 of the culture. Infected human CD2+ and CD11c+ BMDCs were sorted by FACS, pulsed with antigen and induced to mature with LPS before being used in T-cell priming. Analysis of DC phagocytic capability using FITC-Ova was performed as described (Zhang et al, 2004).
DCs (1–2 × 106 cells/ml) were stimulated with the following microbial component, LPS from Salmonella typhosa or Escherichia coli (Sigma), LTA from S. aureus (Sigma), zymosan and mannan from Saccharomyces cerevisiae (Sigma), and PGN from S. aureus (Fluka Chemical) at 37°C for 12 h. Total cell lysates were prepared for protein blot.
Supplementary Material
Supplementary materials for methods
Supplementary Movie
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Movie legend
Acknowledgments
We thank M Cook in the Flow Cytometry Facility of DUMC for FACS sorting, G Kelsoe for histology, and J Aliberti, J Poe and H Pua for critical review of this manuscript. This study was supported by NIH grants AI54685 and AI61364 to Y-W He.
References
- Banchereau J, Steinman RM (1998) Dendritic cells and the control of immunity. Nature 392: 245–252 [DOI] [PubMed] [Google Scholar]
- Barnden MJ, Allison J, Heath WR, Carbone FR (1998) Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol 76: 34–40 [DOI] [PubMed] [Google Scholar]
- Benvenuti F, Hugues S, Walmsley M, Ruf S, Fetler L, Popoff M, Tybulewicz VL, Amigorena S (2004) Requirement of Rac1 and Rac2 expression by mature dendritic cells for T cell priming. Science 305: 1150–1153 [DOI] [PubMed] [Google Scholar]
- Bousso P, Robey E (2003) Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol 4: 579–585 [DOI] [PubMed] [Google Scholar]
- Croix DA, Ahearn JM, Rosengard AM, Han S, Kelsoe G, Ma M, Carroll MC (1996) Antibody response to a T-dependent antigen requires B cell expression of complement receptors. J Exp Med 183: 1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Pozo MA, Kiosses WB, Alderson NB, Meller N, Hahn KM, Schwartz MA (2002) Integrins regulate GTP-Rac localized effector interactions through dissociation of Rho-GDI. Nat Cell Biol 4: 232–239 [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA (2000) Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J 19: 2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein Y, Borrell V, Garcia C, Burstyn-Cohen T, Tzarfaty V, Frumkin A, Nose A, Okamoto H, Higashijima S, Soriano E, Klar A (1999) F-spondin and mindin: two structurally and functionally related genes expressed in the hippocampus that promote outgrowth of embryonic hippocampal neurons. Development 126: 3637–3648 [DOI] [PubMed] [Google Scholar]
- Garrett WS, Chen LM, Kroschewski R, Ebersold M, Turley S, Trombetta S, Galan JE, Mellman I (2000) Developmental control of endocytosis in dendritic cells by Cdc42. Cell 102: 325–334 [DOI] [PubMed] [Google Scholar]
- He YW, Deftos ML, Ojala EW, Bevan MJ (1998) RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity 9: 797–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YW, Li H, Zhang J, Hsu CL, Lin E, Zhang N, Guo J, Forbush KA, Bevan MJ (2004) The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat Immunol 5: 88–97 [DOI] [PubMed] [Google Scholar]
- Higashijima S, Nose A, Eguchi G, Hotta Y, Okamoto H (1997) Mindin/F-spondin family: novel ECM proteins expressed in the zebrafish embryonic axis. Dev Biol 192: 211–227 [DOI] [PubMed] [Google Scholar]
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110: 673–687 [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM (1992) Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med 176: 1693–1702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia W, Li H, He YW (2005) The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood 106: 3854–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath AT, Pooley J, O'Keeffe MA, Vremec D, Zhan Y, Lew AM, D'Amico A, Wu L, Tough DF, Shortman K (2000) The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J Immunol 165: 6762–6770 [DOI] [PubMed] [Google Scholar]
- Kerksiek KM, Niedergang F, Chavrier P, Busch DH, Brocker T (2005) Selective Rac1 inhibition in dendritic cells diminishes apoptotic cell uptake and cross-presentation in vivo. Blood 105: 742–749 [DOI] [PubMed] [Google Scholar]
- Kilshaw PJ (1993) Expression of the mucosal T cell integrin alpha M290 beta 7 by a major subpopulation of dendritic cells in mice. Eur J Immunol 23: 3365–3368 [DOI] [PubMed] [Google Scholar]
- Klar A, Baldassare M, Jessell TM (1992) F-spondin: a gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell 69: 95–110 [DOI] [PubMed] [Google Scholar]
- Kuhn R, Schwenk F, Aguet M, Rajewsky K (1995) Inducible gene targeting in mice. Science 269: 1427–1429 [DOI] [PubMed] [Google Scholar]
- Liakka KA, Autio-Harmainen HI (1992) Distribution of the extracellular matrix proteins tenascin, fibronectin, and vitronectin in fetal, infant, and adult human spleens. J Histochem Cytochem 40: 1203–1210 [DOI] [PubMed] [Google Scholar]
- Lindquist RL, Shakhar G, Dudziak D, Wardemann H, Eisenreich T, Dustin ML, Nussenzweig MC (2004) Visualizing dendritic cell networks in vivo. Nat Immunol 5: 1243–1250 [DOI] [PubMed] [Google Scholar]
- Lo CG, Lu TT, Cyster JG (2003) Integrin-dependence of lymphocyte entry into the splenic white pulp. J Exp Med 197: 353–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MJ, Safrina O, Parker I, Cahalan MD (2004) Imaging the single cell dynamics of CD4+ T cell activation by dendritic cells in lymph nodes. J Exp Med 200: 847–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranti CK, Brugge JS (2002) Sensing the environment: a historical perspective on integrin signal transduction. Nat Cell Biol 4: E83–E90 [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R (2003) Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science 299: 1033–1036 [DOI] [PubMed] [Google Scholar]
- Pasare C, Medzhitov R (2004) Toll-dependent control mechanisms of CD4T cell activation. Immunity 21: 733–741 [DOI] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. J Biol Chem 275: 21785–21788 [DOI] [PubMed] [Google Scholar]
- Pribila JT, Itano AA, Mueller KL, Shimizu Y (2004) The alpha 1 beta 1 and alpha E beta 7 integrins define a subset of dendritic cells in peripheral lymph nodes with unique adhesive and antigen uptake properties. J Immunol 172: 282–291 [DOI] [PubMed] [Google Scholar]
- Price LS, Leng J, Schwartz MA, Bokoch GM (1998) Activation of Rac and Cdc42 by integrins mediates cell spreading. Mol Biol Cell 9: 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig-Kroger A, Sanz-Rodriguez F, Longo N, Sanchez-Mateos P, Botella L, Teixido J, Bernabeu C, Corbi AL (2000) Maturation-dependent expression and function of the CD49d integrin on monocyte-derived human dendritic cells. J Immunol 165: 4338–4345 [DOI] [PubMed] [Google Scholar]
- Raghavan S, Bauer C, Mundschau G, Li Q, Fuchs E (2000) Conditional ablation of beta1 integrin in skin. Severe defects in epidermal proliferation, basement membrane formation, and hair follicle invagination. J Cell Biol 150: 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Kiosses WB, Schwartz MA (1999) Regulation of the small GTP-binding protein Rho by cell adhesion and the cytoskeleton. EMBO J 18: 578–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shortman K, Liu YJ (2002) Mouse and human dendritic cell subtypes. Nat Rev Immunol 2: 151–161 [DOI] [PubMed] [Google Scholar]
- Shurin GV, Tourkova IL, Chatta GS, Schmidt G, Wei S, Djeu JY, Shurin MR (2005) Small rho GTPases regulate antigen presentation in dendritic cells. J Immunol 174: 3394–3400 [DOI] [PubMed] [Google Scholar]
- Stoll S, Delon J, Brotz TM, Germain RN (2002) Dynamic imaging of T cell–dendritic cell interactions in lymph nodes. Science 296: 1873–1876 [DOI] [PubMed] [Google Scholar]
- Swetman CA, Leverrier Y, Garg R, Gan CH, Ridley AJ, Katz DR, Chain BM (2002) Extension, retraction and contraction in the formation of a dendritic cell dendrite: distinct roles for Rho GTPases. Eur J Immunol 32: 2074–2083 [DOI] [PubMed] [Google Scholar]
- Umemiya T, Takeichi M, Nose A (1997) M-spondin, a novel ECM protein highly homologous to vertebrate F-spondin, is localized at the muscle attachment sites in the Drosophila embryo. Dev Biol 186: 165–176 [DOI] [PubMed] [Google Scholar]
- West MA, Prescott AR, Eskelinen EL, Ridley AJ, Watts C (2000) Rac is required for constitutive macropinocytosis by dendritic cells but does not control its downregulation. Curr Biol 10: 839–848 [DOI] [PubMed] [Google Scholar]
- Wu X, Jiang N, Fang YF, Xu C, Mao D, Singh J, Fu YX, Molina H (2000) Impaired affinity maturation in Cr2−/− mice is rescued by adjuvants without improvement in germinal center development. J Immunol 165: 3119–3127 [DOI] [PubMed] [Google Scholar]
- Zhang M, Tang H, Guo Z, An H, Zhu X, Song W, Guo J, Huang X, Chen T, Wang J, Cao X (2004) Splenic stroma drives mature dendritic cells to differentiate into regulatory dendritic cells. Nat Immunol 5: 1124–1133 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials for methods
Supplementary Movie
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Movie legend